

Abstract
Purpose of the review
We describe the history of passive immunization to provide context for the series of articles to follow. The history of passive immunization with antibodies to prevent or treat infectious diseases is a story of different eras. There was an extraordinary era of discovery and clinical implementation before the chemical nature of antibodies was even known. This empirical process provided the resources and reagents used to describe and characterize humoral immunity, better define the chemical properties and structure of antibodies, and extend the clinical use of immunoglobulin products to treat or prevent multiple viral and bacterial diseases over the ensuing several decades. The next distinct era came with the discovery of processes to produce monoclonal antibodies (mAb), and development of more specific therapies. Interestingly, mAb technology resulted in many products to treat autoimmune and allergic diseases, but only one common infectious disease, respiratory syncytial virus, and only in a restricted population of high-risk infants.
Recent findings
The current era began with a series of publications in 2008 demonstrating processes for rapidly producing human mAbs.
Summary
This technology combined with new sequencing technology, advances in structural biology, atomic-level molecular design, and increased capacity for synthetic biology, promises new opportunities to apply passive immunization to the prevention and treatment of infectious diseases.
Keywords: History, antibody, immunoglobulin, passive immunization, serum therapy
Introduction
Antibodies are critical for immunity against infectious diseases, and have been applied to the prevention and treatment of bacterial and viral infections for more than a century. There have been 5 Nobel Prizes awarded for discoveries related to treatment of infectious diseases with antibodies (1901), describing humoral immunity (1908), defining the chemical structure of antibodies (1972), production of monoclonal antibodies (mAbs) (1984), and explaining the mechanism for antibody diversity (1987). Here we will highlight some of the historical events that have guided the understanding and use of antibodies for preventing and treating infectious diseases since the end of the 19th century, and attempt to provide context for how the investigation and clinical use of antibodies has shaped current commercial capacity, regulatory practices, and the science of biologics in general. Because of a confluence of technological advances, including high throughput processes for human mAb isolation, the options for using passive antibody against infectious diseases to improve public health are still expanding.
Discovery of antibodies and the beginning of passive immunization
Emil von Behring was awarded the first Nobel Prize in Physiology or Medicine in 1901 for his discovery of serum therapy for diphtheria. He and Shibasaburo Kitasato showed that serum from rabbits immunized with tetanus toxin could prevent tetanus in rabbits. The same phenomenon was rapidly demonstrated for diphtheria toxin (1). This led to the term antitoxin and probably motivated the use of the term “antikörper” translated “antibody” by Paul Erlich in a 1891 paper (2). Erlich's work demonstrating that increasing doses of bacterial toxins could provide immunity against lethal doses of toxin was the basis for the serum therapy findings. His work also led to the concepts of active and passive immunization, and to his Nobel Prize in 1908 awarded for establishing the field of humoral immunity. It is fitting to highlight Erlich's contribution to the initial conception of passive immunization in 2015, the 100 year anniversary of his death.
In the 1890s von Behring and Erlich worked together to standardize production of serum for the treatment of diphtheria. The standardization of serum production in dairy cattle and horses led to the establishment of new companies or provided a new directions for existing pharmaceutical companies. For example, Erlich became associated with Hoechst, and von Behring established a company that eventually became Aventis Behring, both of which are now part of Sanofi Pasteur. Interestingly, other companies and organizations like Lederle (a successful pharmaceutical company that became part of Wyeth, then Pfizer) began in New York and Butantan (a state-owned and operated organization that still produces antivenoms, antitoxins, and vaccines in Sao Paulo, Brazil) originated on horse farms primarily for the purpose of making antiserum for bacterial toxins.
The new field of passive immunization resulted in a variety of events which have influenced the landscape of modern biologics. An incident involving diphtheria antitoxin contaminated with tetanus toxin in 1901 led to the 1902 Biologics Control Act, which gave responsibility for the regulation of biologics to the Hygienic Laboratory of the Public Health and Marine Hospital Service (Figure 1). The Hygenics Laboratory became the National Institute of Health in 1930, and part of the National Institutes of Health in 1948, where regulation of immunoglobulin products resided until 1972. At that time the responsibility was transferred to the Food and Drug Administration (FDA), and the FDA Center for Drug Evaluation and Research (CDER) is now responsible for the regulating immunoglobulins and monoclonal antibodies.
Fig 1. Collection of blood for production of anti-diphtheria horse serum.
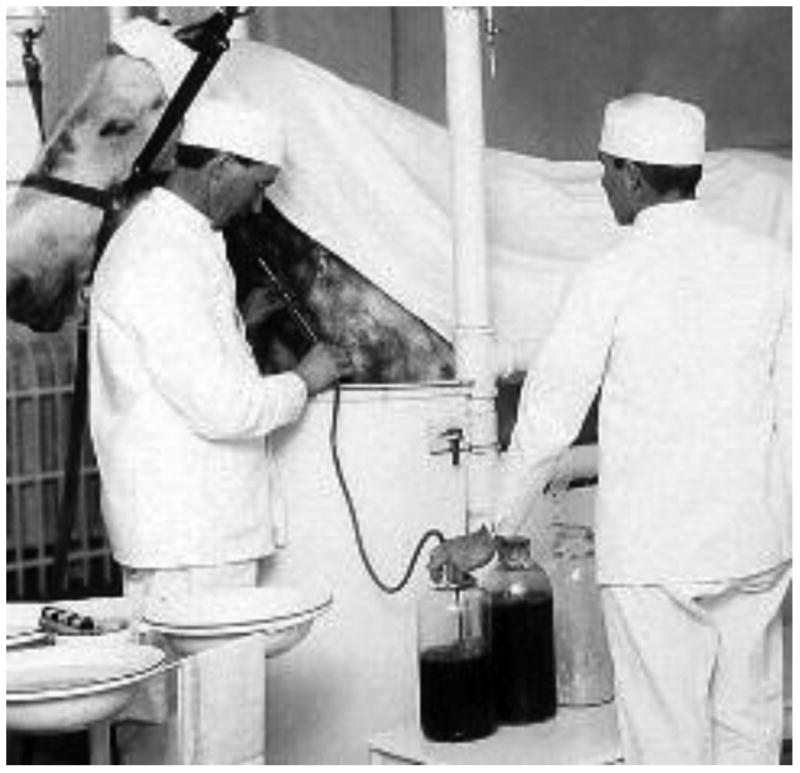
Jin was the horse associated with the deaths of 13 children treated with immune serum collected near the time of his death from tetanus in 1901. The 1902 Biologics Control Act established standards for the processing and labeling of biological products for human use. Source: National Archives and Records Administration
Another interesting by-product of the industrialization of serum therapy was that immunization of horses to make bacterial antitoxins led to the discovery of adjuvants. Noticing that the serum titers were higher in animals with infection at the site of toxin injection led the Frenchman, Gaston Ramon, to perform the initial studies on adjuvants derived from bacterial cell walls (3).
History of antibodies used for prevention of infectious diseases
Immune animal sera for treatment of diseases in the late 1800's was followed by an era of serum therapy for both viral and bacterial diseases. In fact, during the 1918 influenza pandemic, serum from recovered patients was used to successfully treat acutely ill patients (4). The role of serum therapy was expanded to many infections beyond influenza during the first half of the 20th century with clinical benefit demonstrated for other viral diseases like measles (5) and polio (6), and for invasive bacterial infections including pneumococcus, Haemophilus influenza B, and meningococcus (7, 8). These clinical programs in turn contributed to the basic knowledge of antibodies, which were then known to have specific antitoxin activity, and were associated with precipitins and agglutinins, but the underlying chemistry of antibodies was still not understood. This was addressed by the work of Michael Heidelberger and Oswald Avery in the 1920’s when it was common practice to treat pneumococcal pneumonia with type-specific antiserum derived from horses. First, using the specific antiserum as a reagent they showed that the antigen target was a carbohydrate that comprised the capsule of the pneumococcus (9). This was ground-breaking in itself, because it was assumed that all “antigens” were proteins. Using a relatively purified antibody-containing fraction from serum, and the new knowledge that the pneumococcal antigen was carbohydrate, they were able to prove that antibodies were protein (10). Heidelberger went on to find ways to quantify the precipitin and agglutinin reactions providing the basis for modern serological assays (11).
With the discovery of antibiotics by the 1940's, interest in treatment of bacterial infections with serum therapy waned. However, the field remained active for infections not treated with antibiotics, and was greatly advanced by Dr. Edwin Cohn's discovery of purified antibodies through ethanol fractionation of plasma (12). Many effective polyclonal antibody products were developed using this process that targeted either prevention of viral infections or treatment of bacterial toxin related diseases (Table 1). These products include: Diphtheria Immune Globulin, Tetanus Immune Globulin, Immune Serum Globulin, Rabies Immune Globulin, Hepatitis B Immune Globulin, Varicella Zoster Immune Globulin, CMV Globulin, and Botulinum Immune Globulin. The technology of ethanol fractionation of plasma resulted in successful product supply for these relatively small markets, however was ultimately limiting for the first large-scale infectious disease application.
Table 1. Licensed Polyclonal Antibody Products for Infectious Diseases.
| Product name | Polyclonal Product Description | Infection | Indication |
|---|---|---|---|
| BIG | Botulinum Immune Globulin | C. botulinum toxin | Treatment of infants |
| CMV-IGIV | Cytomegalovirus Immune Globulin | CMV | Prevention in Organ Transplants |
| HBIG | Hepatitis B Immune Globulin | Hepatitis B | Post Exposure Needle Stick Prevention in Organ Transplants |
| HRIG | Rabies Immune Globulin | Rabies | Post Exposure |
| ISG | Immune Serum Globulin | Hepatitis A Hepatitis B Measles |
Prevention in travelers/exposure Prevention in travelers/exposure Post Exposure |
| RSV-IGIV | Respiratory Syncytial Virus Immune Globulin | RSV | Prevention in High Risk Infants |
| TIG | Tetanus Immune Globulin | Tetanus | Post Exposure and Treatment |
| VIG | Vaccinia Immune Globulin | Vaccinia | Post Exposure |
| VZIG | Varicella Zoster Immune Globulin | Varicella Zoster | Post Exposure |
In 1996, RSV-IGIV (Respigam®), a polyclonal antibody product for prevention of respiratory syncytial virus (RSV), was licensed for use in high-risk infants. The pivotal trial included premature infants with bronchopulmonary dysplasia, and Respigam was shown to reduce the number of RSV hospitalizations by 41 percent and time in the hospital for these infants by 53 percent (13). Two years later with product demand quickly outstripping plasma availability, the humanized monoclonal antibody palivizumab (Synagis®) was licensed. Beyond the supply advantage, the monoclonal antibody technology offered increased consistency of the biologic, a reduced risk of infections due to blood borne pathogens and diminished number of adverse events during administration. Palivizumab was also 50-fold more potent than the polyclonal product resulting in reduced volume of administration and intramuscular use. These advantageous properties of monoclonal antibody technology would be soon exploited for application in other fields of medicine.
During the last three decades, thirty therapeutic monoclonal antibodies have been licensed, dramatically impacting cancer, ulcerative colitis, Crohn's disease, rheumatoid arthritis, multiple sclerosis, psoriasis, systemic lupus, allergy, as well as a few orphan diseases. Although palivizumab was one of the first licensed monoclonal antibodies, only two of the 30 therapeutic monoclonal antibodies are for infectious disease indications. The success of alternative methods to combat infectious disease such as cost effective vaccines and anti-viral small molecules partially explain the limited number of products. Although resistance of bacteria to antibiotics looms as a major public health threat, monoclonal antibodies have a limited success story in treating bacterial disease. Thus the current active areas of monoclonal development have focused on viral diseases without available vaccines (e.g. HIV, Ebola, SARS, MERS), viral diseases with limited effective anti-viral drugs (e.g. influenza, rabies), and bacterial toxin-mediated diseases (e.g. Anthrax, C. difficile colitis). The monoclonal antibody products currently licensed or progressing towards late stage development for infectious disease are listed in Table 2. We have not included programs that are inactive or have not yet advanced to Phase 2 studies. As the potential for antibodies to prevent and treat HIV are discussed in accompanying articles, we will comment here on the other areas of active development.
Table 2. Monoclonal Antibodies for Prevention or Treatment of Infectious Diseases Licensed or in Active Development.
| Infection | mAb Product Description | Product Name | Status | Indication |
|---|---|---|---|---|
| Anthrax | Human mAb to Anthrax Toxin | Anthrin®(raxibacumab)* | Licensed 2012 |
Post Exposure |
| C. difficile | Human mAbs to C. difficile Toxins (two mAbs combined) | MK-3415a | Phase 3 | Prevention of recurrence of diarrhea |
| Ebola | Humanized mAbs to Ebola Zaire (three mAbs combined) | zMAPP | Phase 2/3 | Post Exposure |
| Hepatitis C | Human mAb to hepatitis C | MBL-HCV-1 | Phase 2 | Prevention for Liver Transplants |
| HIV-1 | Human HIV mAb | VRC01 | Phase 2 | Prevention of infection and potentially therapy |
| Influenza | Human mAbs to influenza A (two mAbs combined) | CR8020 CR6261 |
Phase 2 | Treatment of Severe Disease |
| Rabies | Human mAb to rabies virus | SII-RMab | Phase 2/3 | Post Exposure |
| Rabies | Human mAbs to rabies virus (two mAbs combined) | CL184 | Phase 2a | Post Exposure |
| RSV | Humanized mAb to RSV F protein | Synagis® (palivizumab) |
Licensed 1998 |
Prevention for High Risk Infants |
Convention for naming mAbs: -omab (murine), -ximab (chimeric), –zumab (humanized), -umab (human)
History of antibodies used for post-exposure treatment of infectious diseases
While antibodies are typically considered to be designed to prevent infection, in diseases that produce toxins that target organs distant from the site of infection, have a long incubation period, or have prolonged replication, post-exposure treatment with antibodies can be highly effective. For example, hepatitis immune globulin can protect against hepatitis B even when given after exposure. Rabies is another example where post-exposure treatment with immunoglobulin is effective, and also illustrates the importance of using monoclonal antibody technology to replace polyclonal products that are expensive, in limited supply, and often less potent. In 2014, there are still over 60,000 deaths due to rabies worldwide with over half of the victims being children. Although vaccine administration combined with Human Rabies Immune Globulin (HRIG) is 100% effective in prevention of rabies following exposure, the expense of polyclonal antibody limits its use. There are two products in development that could remedy this situation; CL184 consists of two monoclonal antibodies combined and is in a Phase 2 trial, SII-RMab consists of a single human monoclonal antibody and has just completed enrollment in the pivotal trial in India. It is projected that the monoclonal antibody products could be widely available in low-income countries due to the lowering of the cost of goods through process development activities and local manufacturing.
Bacterial toxins also offer attractive targets for monoclonal antibody development as suggested by some of the earliest effective polyclonal antibody products that neutralized toxins (Diphtheria and Tetanus Immune Globulins). A recent example is the licensure of monoclonal antibody directed to anthrax toxin, raxibacumab, licensed in 2012 for treatment of inhalation anthrax. Of note, this was the first biologic to be licensed by FDA according to the animal rule in contrast to establishing efficacy in clinical field trials. The stability of monoclonal antibody for long-term storage and the immediacy of neutralization make this product uniquely suitable for this indication. C. difficile colitis is another example of a bacterial toxin mediated disease with an active monoclonal product in late stage development. MK-3415A consists of two antibodies, one directed to toxin A and one directed to toxin B. This product is now in Phase 3 studies to evaluate efficacy in prevention of recurrent disease. It is interesting to consider that this passively administered monoclonal antibody therapeutic trial has implications for vaccine development. Efficacy of passively administered antibody would validate antigen selection and guide the atomic level design of candidate vaccines. Furthermore, such trials could establish specific biomarkers of protection for vaccine studies utilizing the efficacious monoclonal antibodies. The approach of therapeutic monoclonal antibody trials identifying vaccine targets has appeal for other infectious diseases perhaps most notably HIV.
There are some examples of immune serum being used to treat viral diseases during the symptomatic phase of illness, even when viremia may have peaked. As noted, convalescent serum was used to treat people during the 1918 influenza epidemic (4), during the SARS epidemic (14), and has been explored for patients with Ebola infection (15), but the reports are anecdotal. Another example is the treatment of the Argentine hemorrhagic fever, caused by an arenavirus (Junin virus), in which patients were shown to benefit from immune serum administered within one week after symptoms began (16-18). The basis for the therapeutic effect was shown to be associated with neutralizing activity (NT), which did not correlate well with the other methods of antibody quantification, complement fixation or immunofluorescence (19). NT antibody was able to effectively control the viremia and a NT unit needed per kg of patient body weight was established for treatment protocols (17). Interestingly, there is a late neurological syndrome in about 10% of patients treated with immune serum that does not occur in untreated survivors (17). The syndrome begins several weeks after apparent resolution of the acute infection and includes fever and symptoms of cerebellar and cranial nerve damage. The pathogenesis is not well understood, but presumably involves either the failing to clear virus from the CNS or facilitating the transport of virus into the CNS.
Evolution of technologies to identify and produce monoclonal antibodies
As noted by the content of this issue, we are in the midst of another history-making era in the field of passive immunization. This is in large part due to the development of technologies to rapidly isolate human monoclonal antibodies. The publication by Milstein and Köhler describing the method for producing murine monoclonal antibodies (mAbs) by immortalizing B cells as hybridomas (20) was a landmark paper and led to their Nobel Prize in 1984. After that a variety of approaches to produce human monoclonal antibodies evolved (21-27), but in 2003 a transformative technology was reported in which the heavy and light chain immunoglobulin (Ig) genes were amplified from single human B cells and cloned into expression vectors (28). In 2008 a more high throughput version of this approach was reported (29). This advance along with innovations in cell sorting and cloning has allowed the discovery of new human mAbs to occur on a large scale. This has been particularly valuable to study infectious diseases for which new interventions were needed, as described in this issue. The availability of multiple human antibodies has opened up new avenues for understanding disease pathogenesis and vaccine-induced immunity. When considered in the context of new sequencing technologies and structure-guided atomic level design, the new harvest of human mAbs is not only providing new products with therapeutic potential, but has made new investigations possible including: 1) defining the antigenic topography of glycoprotein ectodomains, and sites of neutralization sensitivity, 2) isolation of antibodies with particular functional properties, and using those sequences to characterize the ontogeny of antibody responses after encountering an immunogen, 3) molecular engineering of antibodies to improve potency, specificity, or half-life, and 4) understanding the role of post-translational modifications of antibodies.
Conclusion
The advent of synthetic antibodies with higher potency and longer half-lives that can be made in mammalian cells, yeast, plants, and transgenic animals, fosters the hope and promise that passive immunization options will be available for a growing number of infectious diseases. New manufacturing and delivery approaches should also make the development more rapid and cost-effective, so that passive immunization approaches can be more readily applied to emerging infectious disease threats and available in low resource settings to improve public health.
Key points.
Passive administration of antibody to prevent or treat infectious diseases has been done successfully for more than 100 years.
With the advent of regulations for biologics manufacturing, the use of immunoglobulin and antibody products has an outstanding record of safety and efficacy.
The recent revolution in technologies for isolating and characterizing human monoclonal antibodies promises a new era of progress and options for managing infectious disease threats to the public health.
Acknowledgments
None
Financial support and sponsorship: This work was supported by intramural funding from the National Institute of Allergy and Infectious Diseases.
Abbreviations
- mAb
monoclonal antibody
- CMV
cytomegalovirus
- CNS
central nervous system
- HIV
human immunodeficiency virus
- MERS
Middle East Respiratory Syndrome coronavirus
- NT
neutralizing activity
- RSV
respiratory syncytial virus
- SARS
Severe Acute Respiratory Syndrome coronavirus
Footnotes
Conflicts of interest: Dr. Ambrosino is an inventor on patents involving monoclonal antibodies for rabies and C. difficile toxin. The remaining author has no conflicts of interest.
References
- 1.Winau F, Winau R. Emil von Behring and serum therapy. Microbes and infection / Institut Pasteur. 2002;4(2):185–8. doi: 10.1016/s1286-4579(01)01526-x. [DOI] [PubMed] [Google Scholar]
- 2.Lindenmann J. Origin of the terms ‘antibody’ and ‘antigen’. Scandinavian Journal of Immunology. 1984;19(4):281–5. doi: 10.1111/j.1365-3083.1984.tb00931.x. [DOI] [PubMed] [Google Scholar]
- 3.Ramon G. Sur l'augmentation anormale de l'antitoxine chez les chevaux producteurs de serum antidiptherique. Bull Soc Centr Med Vet. 1925;101:227. [Google Scholar]
- 4.Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Annals of Internal Medicine. 2006;145(8):599–609. doi: 10.7326/0003-4819-145-8-200610170-00139. [DOI] [PubMed] [Google Scholar]
- 5.Janeway CA. Use of concentrated human gamma globulin in the prevention and attenuation of measles. Bull NY Acad Med. 1945;21:202–2. [PMC free article] [PubMed] [Google Scholar]
- 6.Hammon WM, Coriell LL, Wehrle PF, Stokes J., Jr Evaluation of Red Cross gamma globulin as a prophylactic agent for poliomyelitis. IV. Final report of results based on clinical diagnoses. J Am Med Assoc. 1953;151(15):1272–85. [PubMed] [Google Scholar]
- 7.Alexander HE, Leidy G, et al. Hemophilus influenzae meningitis treated with streptomycin. J Am Med Assoc. 1946;132:434–40. doi: 10.1001/jama.1946.02870430014005. [DOI] [PubMed] [Google Scholar]
- 8.Casadevall A, Scharff MD. Serum therapy revisited: animal models of infection and development of passive antibody therapy. Antimicrobial Agents and Chemotherapy. 1994;38(8):1695–702. doi: 10.1128/aac.38.8.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heidelberger M, Avery OT. The Soluble Specific Substance of Pneumococcus. The Journal of Experimental Medicine. 1923;38(1):73–9. doi: 10.1084/jem.38.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heidelberger M, Avery OT. The Soluble Specific Substance of Pneumococcus : Second Paper. The Journal of Experimental Medicine. 1924;40(3):301–17. doi: 10.1084/jem.40.3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heidelberger M, Kendall FE. A Quantitative Study of the Precipitin Reaction between Type Iii Pneumococcus Polysaccharide and Purified Homologous Antibody. The Journal of Experimental Medicine. 1929;50(6):809–23. doi: 10.1084/jem.50.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kendrick DB. Blood Program in World War II. Washington, DC: US Government Printing Office; 1964. p. 922. [Google Scholar]
- 13.Reduction of respiratory syncytial virus hospitalization among premature infants and infants with bronchopulmonary dysplasia using respiratory syncytial virus immune globulin prophylaxis. The PREVENT Study Group. Pediatrics. 1997;99(1):93–9. doi: 10.1542/peds.99.1.93. [DOI] [PubMed] [Google Scholar]
- 14.Yeh KM, Chiueh TS, Siu LK, Lin JC, Chan PK, Peng MY, et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. The Journal of Antimicrobial Chemotherapy. 2005;56(5):919–22. doi: 10.1093/jac/dki346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mupapa K, Massamba M, Kibadi K, Kuvula K, Bwaka A, Kipasa M, et al. Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. International Scientific and Technical Committee. The Journal of Infectious Diseases. 1999;179(Suppl 1):S18–23. doi: 10.1086/514298. [DOI] [PubMed] [Google Scholar]
- 16.Maiztegui JI, Fernandez NJ, de Damilano AJ. Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet. 1979;2(8154):1216–7. doi: 10.1016/s0140-6736(79)92335-3. [DOI] [PubMed] [Google Scholar]
- 17.Enria DA, Briggiler AM, Sanchez Z. Treatment of Argentine hemorrhagic fever. Antiviral Research. 2008;78(1):132–9. doi: 10.1016/j.antiviral.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enria DA, Maiztegui JI. Antiviral treatment of Argentine hemorrhagic fever. Antiviral research. 1994;23(1):23–31. doi: 10.1016/0166-3542(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 19.Enria DA, Briggiler AM, Fernandez NJ, Levis SC, Maiztegui JI. Importance of dose of neutralising antibodies in treatment of Argentine haemorrhagic fever with immune plasma. Lancet. 1984;2(8397):255–6. doi: 10.1016/s0140-6736(84)90299-x. [DOI] [PubMed] [Google Scholar]
- 20.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–7. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 21.Steinitz M, Klein G, Koskimies S, Makel O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature. 1977;269(5627):420–2. doi: 10.1038/269420a0. [DOI] [PubMed] [Google Scholar]
- 22.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348(6301):552–4. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 23.Green LL, Hardy MC, Maynard-Currie CE, Tsuda H, Louie DM, Mendez MJ, et al. Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nature Genetics. 1994;7(1):13–21. doi: 10.1038/ng0594-13. [DOI] [PubMed] [Google Scholar]
- 24.Lonberg N, Taylor LD, Harding FA, Trounstine M, Higgins KM, Schramm SR, et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature. 1994;368(6474):856–9. doi: 10.1038/368856a0. [DOI] [PubMed] [Google Scholar]
- 25.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298(5601):2199–202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 26.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453(7195):667–71. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X, Tsibane T, McGraw PA, House FS, Keefer CJ, Hicar MD, et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455(7212):532–6. doi: 10.1038/nature07231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 29*.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. Journal of Immunological Methods. 2008;329(1-2):112–24. doi: 10.1016/j.jim.2007.09.017. This is an important paper because it provides a step-by-step non-biased method for cloning immunoglobulin genes from individual B cells (for repertoire analysis) and expressing them in eukaryotic cells to produce human mAbs (for functional analysis). Variations of this method have been used to characterize many of the antibodies described in this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]