

Abstract
Melanoma is the deadliest form of skin cancer, and BRAFV600E is a driver mutation that promotes melanoma growth and survival. PLX4032 is the first effective compound in clinical use for the treatment of patients with mutant BRAFV600. However, resistance to PLX4032 develops quickly within months. Activation of a series of receptor tyrosine kinases, including the platelet-derived growth factor receptor (PDGFR), has been identified to be the underlying mechanism for development of resistance to PLX4032. In this work, we investigated the anticancer activity of tyrphostin AG1296, a PDGFR inhibitor, in melanoma, especially PLX4032-resistant melanoma. We found that tyrphostin AG1296 could effectively reduce the viability of both PLX4032-sensitive and PLX4032-resistant melanoma cells. There is an additive effect between tyrphostin AG1296 and PLX4032 in reducing cell viability. Tyrphostin AG1296 induced dramatic apoptosis in PLX4032-resistant cells, and also dramatically inhibited migration of PLX4032-resistant cells. Importantly, tyrphostin AG1296 significantly suppressed A375R tumor growth in vivo. This is the first report on the anticancer activity of tyrphostin AG1296 in melanoma. Tyrphostin AG1296 is a promising compound in the treatment of melanoma, especially for those who have developed resistance towards BRAF inhibitors, and might shed new light on melanoma therapy.
Keywords: melanoma, PLX4032, resistance, viability, apoptosis, migration
Introduction
Melanoma is a highly aggressive form of skin cancer for which effective treatment has been limited.1–5 Identification of a frequent mutation (>50%) at valine position 600 of the B-isoform of RAF kinase (BRAFV600) in melanoma has held great promise for therapeutic intervention.6 Recent clinical trials with BRAF-targeting agents such as GSK2118436 (dabrafenib) and PLX4032 (vemurafenib) have shown impressive clinical responses in melanoma patients with mutant BRAFV600.7–9 The US Food and Drug Administration has approved PLX4032 for the treatment of BRAFV600E mutation-positive, inoperable, or metastatic melanoma. PLX4032 has been identified as a potent and selective chemical inhibitor of mutant BRAF signaling.7 Recent Phase I–III trials with PLX4032 demonstrated that the majority of melanoma patients, selected for mutant BRAF positivity, showed tumor regression.7–9 The Phase III trial in previously untreated patients additionally showed improved overall survival and progression-free survival.7 However, tumor regression in most patients was only temporary, and the average efficient treatment period for PLX4032 was only around 7 months. Additionally, 19%–52% of patients in the Phase I–III trials did not show tumor regression by Response Evaluation Criteria in Solid Tumors (RECIST) criteria.7–9 These results indicate that intrinsic and acquired resistance is a crucial problem in melanoma therapy when using PLX4032, which needs to be addressed and resolved.
Drug resistance presents a challenge in the treatment of cancer patients, including those with melanoma.10–12 Activa tion of receptor tyrosine kinases (RTKs), including platelet-derived growth factor receptor (PDGFR), insulin-like growth factor-1 receptor, hepatocyte growth factor, AKT3, fibroblast growth factor receptor 3, and COT1 has been identified, either in clinical or preclinical models, as conferring resistance to BRAF inhibitors.13–18 Therefore, inhibition of single, dual, or multiple RTKs might provide a potential therapeutic strategy for melanomas, especially PLX4032-resistant melanomas. In this work, we investigated the anticancer activity of tyrphostin AG1296, a PDGFR inhibitor, in melanoma, especially PLX4032-resistant melanoma, both in vitro and in vivo.
Materials and methods
Materials
PLX4032 was purchased from AdooQ BioScience (Irvine, CA, USA). Rabbit anti-phospho-PDGFR-α, anti-phospho-PDGFR-β, anti-PDGFR-α, anti-PDGFR-β, anti-glyceraldehyde 3-phosphate dehydrogenase antibodies, and secondary horseradish peroxidase-conjugated antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Tyrphostin AG1296 and other reagents and chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).
Cells and cell culture
Human melanoma cell lines (A375 and SK-MEL-5) were purchased from the American Type Culture Collection (Rockville, MD, USA). PLX4032-resistant cell lines (A375R and SK-MEL-5R) were obtained by culturing A375 and SK-MEL-5 cells with gradually increasing doses of PLX4032. All the cells were cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamine. The cells were cultured in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Cell viability assay
Melanoma cells were seeded in 96-well white plates at a density of 5×103 cells per well. The compounds were added and incubated for 72 hours. A CellTiter-Glo kit (Promega, Madison, WI, USA) was then used to examine cell viability as follows: a cell lysis/ATP detection reagent was added (30 μL/well), shaken for 10 minutes, and the luminescence was measured using a plate reader (Molecular Devices, Sunnyvale, CA, USA). IC50 values were determined using CompuSyn software (ComboSyn, Inc., Paramus, NJ, USA).
Cell migration assays
Transwell® assay
Twenty-four-Transwell Boyden chambers (Costar, Bedford, MA, USA) with a polystyrene membrane (6.5 mm diameter, 10 μm thickness, and 8 μm pore size) were used to measure cell migration. A375R cells were seeded in the upper compartment of the well in serum-free medium (5×104 cells/well). Next, 600 μL of serum-free medium supplemented with 20 μg/mL fibronectin was added to the lower compartment. After treatment for 8 hours, the cells were fixed and stained with 0.1% crystal violet. The non-migrating cells on the upper surface of the membrane were removed, and the cells that had migrated to the lower side were photographed with a microscope (Nikon, Tokyo, Japan). The migrated cells were then lysed with 10% acetic acid and absorbance was measured at 595 nm.
Wound healing assay
A375R cells were seeded into fibronectin-coated 96-well plates (2×104 cells/well). After reaching confluence, the cell monolayers were scratched with a pipette tip. Fresh serum-free medium was added, and the cells were treated with the study compounds for 8 hours. The cells were then photographed with a microscope. The width of the wounded cell monolayers was measured.
Nuclei staining
A375R cells were seeded on coverslips and treated with the study compounds. The cells were then fixed with 4% paraformaldehyde for 30 minutes, permeabilized with 0.1% Triton X-100 for 20 minutes, blocked with 5% normal serum for 30 minutes, and incubated with Hoechst for 10 minutes. Images were then obtained using a BX51 microscope (Olympus Tokyo, Japan).
Flow cytometry analysis
A375R cells were seeded in six-well plates (5×105 cells/well) and treated with the compounds. The cells were then harvested, fixed with 70% ethanol, and stained with propidium iodide 5 mg/L in the presence of RNase 1 g/L, 1 g/L sodium citrate, and 0.5% Triton X-100 (v/v) for 30 minutes in the dark. Cells were then collected for analysis of apoptosis using FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA), and the percentage of hypodiploidy was analyzed using ModFIT LT software (Verity Software House Inc, Topsham, ME, USA).
Immunohistochemistry
Tumor tissues were fixed in phosphate-buffered formalin, embedded in paraffin, cut to a 4 μm thickness, and applied to the slides. The slides were deparaffinized in xylene using three changes for 5 minutes each, and hydrated gradually through graded alcohols: 100% ethanol twice for 10 minutes each, 95% ethanol twice for 10 minutes each, and then deionized water for one minute with stirring. For antigen unmasking, the slides were placed in a container, covered with 10 mM sodium citrate buffer (pH 6.0), and heated in a convection steamer for one hour. The slides were washed in deionized water three times for 2 minutes each, and blocked with 5% normal goat blocking serum for 30 minutes. Deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was performed using a FragEL kit (Calbiochem, San Diego, CA, USA) according to the manufacturer’s instructions. The slides were then incubated with Hoechst for 10 minutes. The slides were analyzed and photographed using a fluorescent microscope.
Suppression of subcutaneous tumor growth in vivo
The nud/nud mice were purchased from Slack Company (Shanghai, People’s Republic of China), and housed in a BL2 laboratory. All care and treatment of experimental animals was in accordance with Animal Care and Use Committee guidelines. A375R cells were injected subcutaneously into the axillary regions of the nud/nud mice (3×106 cells/100 μL/mouse). After one week, when the tumor volumes had reached about 50 mm3, the mice were randomized to a control group, a tyrphostin AG1296 40 mg/kg group, or a tyrphostin AG1296 80 mg/kg group, with six mice per group (vehicle, 10% 1-methyl-2-pyrrolidinone and 90% polyethylene glycol 300). The tumors were measured every other day with a microcaliper and body weight was measured every other day with a scale. Tumor volumes were calculated as follows: mm3 = width × width × length ×0.5. Two weeks later, the mice were euthanized using CO2 and the tumors were harvested and analyzed.
Statistical analysis
The Student’s t-test and analysis of variance were performed using StatView (SAS Institute, Cary, NC, USA). P<0.05 was considered to indicate a statistically significant difference. The data shown are the mean values of triplicate measurements, with error bars corresponding to the standard deviation.
Results
Tyrphostin AG1296 reduces viability of PLX4032-sensitive and PLX403-resistant melanoma cells
In order to get a PLX4032-resistant cell line, we cultured A375 and SK-MEL-5 cells with gradually increasing doses of PLX4032, a selective BRAF inhibitor. After 6 months of culture, we examined and compared the sensitivity of PLX4032-sensitive (A375 and SK-MEL-5) and PLX4032-resistant (A375R and SK-MEL-5R) cell lines towards PLX4032. The results show an approximately 100-fold increase in IC50 for resistant compared with matched parental cells, suggesting that PLX4032-resistant cell lines were successfully generated (Figure 1A and B).
Figure 1.

Tyrphostin AG1296 suppresses viability of PLX4032-resistant melanoma cells.
Notes: (A, B) A375 and SK-MEL-5 cells were induced to become PLX4032-resistant cells (A375R and SK-MEL-5R) by incubation with gradually reducing doses of PLX4032. PLX4032 sensitivity was then examined by cell viability assay. (C, D) Both PLX4032-sensitive and PLX4032-resistant cells were treated with tyrphostin AG1296 (0.625–20 μM) for 72 hours, followed by measurement of cell viability. (E) A375 cells were treated with tyrphostin AG1296 together with PLX4032 for 72 hours, followed by measurement of cell viability.
Abbreviations: T, tyrphostin AG1296; P, PLX4032.
PDGFR has been shown to play a critical role in promoting the viability of melanoma cells.19,20 Moreover, RTKs including PDGFR are known to mediate generation of PLX4032 resistance.13–18 Therefore, we next examined the effect of tyrphostin AG1296 on the viability of both PLX4032-sensitive and PLX4032-resistant melanoma cells. As shown in Figure 1C and D, tyrphostin AG1296 significantly reduced the viability of both PLX4032-sensitive and PLX4032-resistant cell lines. We also explored the possibility of an additive effect between tyrphostin AG1296 and PLX4032. We found that there is an obvious additive effect between tyrphostin AG1296 and PLX4032 (Figure 1E).
Tyrphostin AG1296 induces apoptosis of A375R cells
PDGFR-mediated signaling plays a crucial role in inhibiting apoptosis and increasing cell viability,19,20 so we next examined tyrphostin AG1296-induced apoptosis in A375R cells. Figure 2A and B shows that the fraction of cells with a subG1 DNA content increased significantly after treatment with tyrphostin AG1296 (P<0.01). We also observed the changes in nuclear morphology in A375R cells after treatment with tyrphostin AG1296. After treatment with tyrphostin AG1296, the nuclei of A375R cells showed a condensed and fragmented morphology, which is a characteristic of apoptosis (Figure 2C and D), suggesting that tyrphostin AG1296 could induce dramatic apoptosis in A375R cells. These data are consistent with the viability suppression results shown above.
Figure 2.

Tyrphostin AG1296 induces apoptosis in PLX4032-resistant cells.
Notes: (A, B) A375R cells were treated with tyrphostin AG1296 at the indicated concentrations for 48 hours, followed by propidium iodide staining and flow cytometry analysis. (C) A375R cells were incubated with tyrphostin AG1296 for 48 hours. The nuclei were stained with Hoechst, and analyzed using a fluorescent microscope. The representative images are shown. (D) Number of cells with condensed/fragmented nuclei was quantitated by counting in five random fields and the inhibition was calculated.
Tyrphostin AG1296 inhibits PDGFR phosphorylation in A375R cells
We next observed the effect of tyrphostin AG1296 on phosphorylation of PDGFR in A375R cells. We found that tyrphostin AG1296 could significantly inhibit phosphorylation of both PDGFR-α and PDGFR-β. There were no significant changes in total protein levels (Figure 3).
Figure 3.
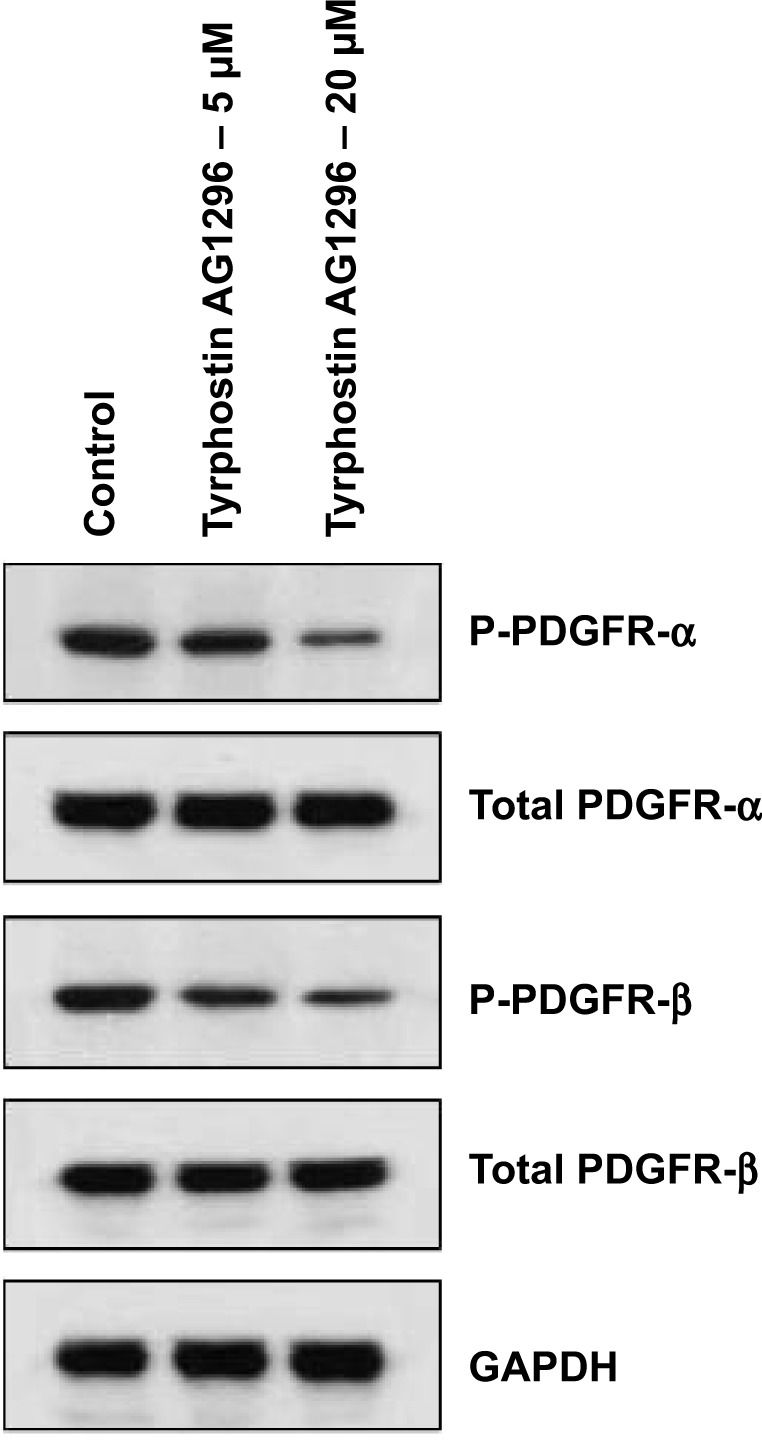
Tyrphostin AG1296 inhibits PDGFR phosphorylation in A375R cells. A375R cells were treated with tyrphostin AG1296 (5 and 20 μM) for 2 hours, followed by Western blot analysis with the indicated antibodies.
Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PDGFR, platelet-derived growth factor receptor.
Tyrphostin AG1296 inhibits migration of A375R cells
PDGFR-mediated signaling plays a critical role in the migration of cancer cells.21,22 Therefore, we next investigated the effect of tyrphostin AG1296 on migration of A375R cells. Figure 4A and B shows that tyrphostin AG1296 inhibited migration of A375R cells in a Tran-swell model. Similar inhibition of cell migration was also observed in a wound healing model (Figure 4C and D). These data suggest that, in addition to viability-suppressing activity, tyrphostin AG1296 could also suppress migration of melanoma, especially PLX4032-resistant cells.
Figure 4.

Tyrphostin AG1296 inhibits cell migration of PLX4032-resistant cells.
Notes: (A) A375R cells were treated with tyrphostin AG1296 (0.0625, 0.25 or 1 μM) for 8 hours. The non-migrated cells on the upper surface of the filter were removed, and the migrated cells on the lower side were stained and photographed. The representative images are shown. Cells were then lysed and colorimetric determination was done at 595 nm. (B) Quantitation of the inhibition from Transwell assay. (C) A scratch was introduced into a monolayer of A375R cells, followed by treatment with tyrphostin AG1296 (0.0625, 0.25, or 1 μM) for 8 hours. The width of the wounded cell monolayer was measured in five random fields, and representative images are shown. (D) Quantitation of inhibition from wound healing assay.
Tyrphostin AG1296 suppresses A375R tumor growth in vivo
We next treated nud/nud mice with tyrphostin AG1296 (40 and 80 mg/kg), and examined the effect of tyrphostin AG1296 (administered intraperitoneally) on A375R tumor growth in mice. The results show that the low dose of tyrphostin AG1296 (40 mg/kg) led to an intermediate level of tumor growth suppression, whereas the high dose of tyrphostin AG1296 (80 mg/kg) led to significant inhibition of A375R tumor growth (Figure 5A). Consistent with the in vitro apoptosis data shown above, tyrphostin AG1296 induced dramatic apoptotic characteristics in A375R tumors, as examined using TUNEL and nuclei staining assays (Figure 5B). At the same time, administration of tyrphostin AG1296 was well tolerated by healthy mice without significant signs of overt toxicity or weight loss (P>0.05, Figure 5C). Taken together, these results suggest that tyrphostin AG1296 could induce apoptosis and reduce the viability of PLX-4032-resistant melanoma cells in vitro and suppress resistant tumor growth in vivo.
Figure 5.

Tyrphostin AG1296 suppresses A375R tumor growth in vivo.
Notes: (A) After inoculation of A375R cells, tyrphostin AG1296 (40 and 80 mg/kg) was injected in to the mice every day. The tumors were measured every other day and tumor volumes are shown. (B) Tyrphostin AG1296 induced apoptosis of A375R tumor cells in vivo examined with TUNEL assay (green), and the nuclei were stained with Hoechst (blue). (C) Tyrphostin AG1296 had no significant cytotoxic effects on body weight of mice during the treatments.
Discussion
Melanoma is the deadliest form of skin cancer, and the number of new cases of melanoma increases every year. Advanced melanoma is associated with resistance to conventional chemotherapies, and the 5-year survival rate for patients with metastatic melanoma remains very low.1–5 The most common mutation in BRAF is a V600E substitution in the activation domain that causes its kinase function to remain constitutively active. Overwhelming evidence shows that BRAFV600E is a driver mutation that promotes growth and survival of melanoma.6 PLX4032 has been shown to be an effective compound in melanoma patients with mutant BRAFV600. However, the duration of response has been limited because resistance to BRAF inhibitors develops quickly within months following initiation of treatment.7 Activation of a series of RTKs including PDGFR has been identified to be the underlying mechanism for development of resistance to BRAF inhibitors.13–18 These clinical observations make it imperative to explore new targets in the treatment of melanoma, especially PLX-4032-resistant melanomas. Inhibition of RTKs might be a promising therapeutic strategy for this.10–12
Considering the critical roles of PDGFR in tumor growth and metastasis as well as development of PLX4032 resistance,13,19–22 we have investigated the anticancer activity of tyrphostin AG1296, a PDGFR inhibitor, in melanoma, especially PLX4032-resistant melanoma both in vitro and in vivo. We found that tyrphostin AG1296 could effectively suppress viability of both PLX4032-sensitive and PLX4032-resistant melanoma cells. Moreover, there is an additive effect between tyrphostin AG1296 and PLX4032 in reducing cell viability. Consistently, tyrphostin AG1296 induced significant apoptosis in PLX4032-resistant cells. In addition, tyrphostin AG1296 also dramatically inhibited migration of PLX4032 resistant cells. Importantly, tyrphostin AG1296 significantly suppressed A375R tumor growth in vivo. This is the first report on tyrphostin AG1296 in melanoma cells and tumors. Imatinib is an orally bioavailable multi-target inhibitor of v-Abl, c-Kit, and PDGFR. However, there is a report showing that imatinib failed to inhibit melanoma tumor growth in vivo. The difference in in vivo effects between tyrphostin AG1296 and imatinib might be due to the differences in their potential targets, in vivo metabolism, and underlying signaling mechanisms.23 PLX4032 is the first effective compound in melanoma therapy for patients with mutant BRAFV600. However, resistance to PLX4032 develops quickly within only months.7 Therefore, overcoming or attenuating PLX4032 resistance becomes an urgent issue. The anticancer activity of tyrphostin AG1296 in PLX4032-resistant cells make it a promising compound for the treatment of melanoma, especially melanoma that has already developed resistance to BRAF inhibitors. Moreover, tyrphostin AG1296 also has strong inhibitory activity on migration of melanoma cells, while the current anti-melanoma compounds in clinical use do not have this activity.
In summary, we have demonstrated for the first time that tyrphostin AG1296, a specific inhibitor of PDGFR, is a potential therapeutic compound in melanoma, especially melanomas with resistance to BRAF inhibitors. This might open up another avenue for overcoming BRAF inhibitor resistance in the treatment of melanoma
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 3.Shakhova O. Neural crest stem cells in melanoma development. Curr Opin Oncol. 2014;26(2):215–221. doi: 10.1097/CCO.0000000000000046. [DOI] [PubMed] [Google Scholar]
- 4.Korman JB, Fisher DE. Developing melanoma therapeutics: overview and update. Wiley Interdiscip Rev Syst Biol Med. 2013;5(3):257–271. doi: 10.1002/wsbm.1210. [DOI] [PubMed] [Google Scholar]
- 5.Pretto F, Neri D. Pharmacotherapy of metastatic melanoma: emerging trends and opportunities for a cure. Pharmacol Ther. 2013;139(3):405–411. doi: 10.1016/j.pharmthera.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 7.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauschild A, Agarwala SS, Trefzer U, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27(17):2823–2830. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 10.Tentori L, Lacal PM, Graziani G. Challenging resistance mechanisms to therapies for metastatic melanoma. Trends Pharmacol Sci. 2013;34(12):656–666. doi: 10.1016/j.tips.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Hartsough E, Shao Y, Aplin AE. Resistance to RAF inhibitors revisited. J Invest Dermatol. 2014;134(2):319–325. doi: 10.1038/jid.2013.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das Thakur M, Stuart DD. The evolution of melanoma resistance reveals therapeutic opportunities. Cancer Res. 2013;73(20):6106–6110. doi: 10.1158/0008-5472.CAN-13-1633. [DOI] [PubMed] [Google Scholar]
- 13.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yadav V, Zhang X, Liu J, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem. 2012;287(33):28087–28098. doi: 10.1074/jbc.M112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70(16):6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Gu X, Liu Y, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478(7369):349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kundra V, Escobedo JA, Kazlauskas A, et al. Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature. 1994;367(6462):474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- 22.Sieg DJ, Hauck CR, Ilic D, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 23.McGary EC, Onn A, Mills L, et al. Imatinib mesylate inhibits platelet-derived growth factor receptor phosphorylation of melanoma cells but does not affect tumorigenicity in vivo. J Invest Dermatol. 2004;122(2):400–405. doi: 10.1046/j.0022-202X.2004.22231.x. [DOI] [PubMed] [Google Scholar]