

Abstract
We herein report the development of crosslinked polyether particles as a reactive platform for the preparation of functional microgels. Thiol-ene crosslinking of poly(allyl glycidyl ether) in miniemulsion droplets - stabilized by a surface active, bio-compatible polyethylene glycol block copolymer - resulted in colloidal gels with a PEG corona and an inner polymeric network containing reactive allyl units. The stability of the allyl groups allows the microgels to be purified and stored before a second, subsequent thiol-ene functionalization step allows a wide variety of pH- and chemically-responsive groups to be introduced into the nanoparticles. The facile nature of this synthetic platform enables the preparation of microgel libraries that are responsive to different triggers but are characterized by the same size distribution, surface functionality, and crosslinking density. In addition, the utilization of a crosslinker containing cleavable ester groups renders the resulting hydrogel particles degradable at elevated pH or in the presence of esterase under physiological conditions.
Introduction
The triggered release of active agents from polymeric carriers such as micelles, nanoparticles or nano-/microgels is a rapidly developing and highly versatile concept that promises to be a key approach to next generation therapeutics.1–7 Within these materials, microgels8 offer a number of advantages due to their unique combination of colloidal size and internal network structure.9–13 In contrast to polymeric micelles and capsules, which predominantly exhibit a burst release, controlling the degree of swelling of the colloidal networks offers the ability to precisely tune loading and release profiles.11–16 Since the swelling behaviour is dictated by the mesh size of the gel, it can either be controlled by (reversibly) varying the crosslink density or by introducing stimuli-responsive groups in the network-forming polymer. Both approaches enable swelling and degradation to be tuned in response to external triggers17 such as pH,18–24 enzymes,25–27 temperature28–32 and light.33–38
In addition to a specific release profile, the suitability of such stimuli-responsive particles for therapeutic applications also requires control over size and surface composition. As these materials are characterized by an extremely high surface/volume ratio, the size distribution as well as the surface properties of microgels critically determine their bio-compatibility, circulation time and (targeted) cellular uptake.39–43
Regarding the synthesis of stimuli-responsive microgels, traditional routes are based on the crosslinking of polymeric or monomeric starting materials using dispersion or precipitation based methods.10, 44–51 A major drawback of these strategies is that the production of different microgels containing distinct functional groups requires their independent preparation. This leads to batch-to-batch variability in the microgels’ crosslinking density, size distribution and surface morphology.
Therefore, the development of a simple procedure to prepare a master batch of precursor particles as a general platform for secondary functionalization would be a major advance. This strategy would allow for the generation of structurally equivalent microgels that vary only in their functionality.
In addressing this need, we report a facile approach for the preparation of responsive microgels using crosslinked reactive precursor nanoparticles that can be functionalized via simple click chemistry in a second subsequent step. By introducing different stimuli-responsive groups into the network after the particle synthesis, one batch of starting particles can be used to prepare a multitude of microgels that each exhibits a different response profile. A key feature of this strategy is the decoupling of responsive properties for the microgels from the initial structural parameters that are traditionally defined by the polymeric or monomeric starting materials.
To demonstrate the versatility of this approach towards the development of microgels for biomedical applications, reactive precursor nanoparticles were prepared by controlled crosslinking of poly(allyl glycidyl ether), PAGE,52 in miniemulsion droplets using poly(ethylene oxide)-b-poly(allyl glycidyl ether) (PEO-b-PAGE) as a reactive non-ionic surfactant.53 Pentaerythritoltetrakis(3-mercaptopropionate) (PTMP) was used as the crosslinker to allow for degradability of the microgels. This allows for a variety of groups to be introduced via thiol-ene addition to unreacted AGE units remaining in the precursor microgels.54–57 By introducing acidic (anionic) or basic (cationic) functional groups, secondary microgels with opposite swelling profiles can be achieved. Furthermore, the use of combinations thereof allows the preparation of zwitterionic particles with unique swelling profiles and phase transitions, thereby demonstrating the power and modular nature of this approach.
Results and Discussion
The successful development of a platform strategy for the synthesis and secondary functionalization of microgels requires the following key aspects to be addressed. First, a facile and scalable method for the formation of the precursor particles is required to ensure control over particle morphology, size distribution and crosslinking density. Once formed, efficient chemistry for the secondary modification of the particles is needed. This chemistry should allow for the introduction of a wide range of functional groups without side reactions leading to a decrease or increase in crosslinking density.58–59
Reactive precursor microgels
To address these requirements, allyl units and thiol-ene chemistry were chosen as versatile and modular synthetic platforms for constructing the microgel particles. For this purpose, a miniemulsion was formed by dispersing a toluene solution of poly(allyl glycidyl ether) homopolymer and a tetra-functional, small molecule crosslinker such as pentaerythritol tetrakis(3-mercaptopropionate). To this mixture were added 2,2-dimethoxy-2-phenyl-acetophenone as photoinitiator and PEO-b-PAGE block copolymer as a functional surfactant to covalently stabilize the particles during light-activated thiol-ene crosslinking (Scheme 1). This strategy has a number of advantages when compared to traditional precipitation or emulsion based methods. For example, the minimization of diffusion of monomers or the nucleation and growth of particles allows for greater chemical homogeneity.60–62 As a result, tuning the feed ratio of the respective building blocks adjusts the structural composition and surface chemistry of the reactive precursor particles. In addition, the utilization of an amphiphilic PAGE-b-PEO block copolymer as a reactive non-ionic surfactant allows covalent functionalization of the microgel surface with bio-compatible PEO chains. The mesh size is also determined by the relative amount of crosslinker to allyl groups in the dispersed phase and the cleavable ester groups of PTMP render the crosslinking points, and therefore the whole particle network, degradable in response to pH and biological triggers such as enzymes.
Scheme 1.
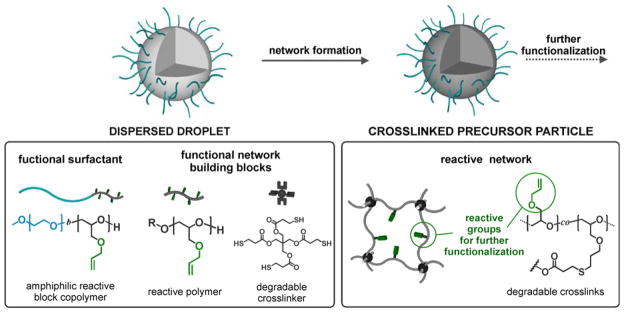
Microgel formation via crosslinking of PAGE and PAGE-b-PEO using light induced thiol-ene reaction with PTMP in dispersed droplets.
To use these precursor particles for the formation of structurally equivalent microgels with different functionalities, it is crucial to ensure that the crosslinking density is not influenced by the post-formation functionalization. Since both reactions – crosslinking and functionalization – are based on thiol-ene additions, the first reaction should be complete to avoid uncontrolled crosslinking in the presence of the thiols used for secondary functionalization.
In order to investigate the conversion of the crosslinking reaction, the influence of irradiation time on particle formation was studied by varying the reaction time from 2 to 24 hours while keeping the crosslinker concentration at 2mol-%. The dispersions were then purified by repeated centrifugation/redispersion in Milli-Q water and analysed by dynamic light scattering (DLS) and scanning electron microscopy (SEM). For crosslinking times of 2 hours or less, macroscopic phase separation of the emulsion was observed indicating unsuccessful particle formation due to insufficient crosslinking. For irradiation times of 4 hours and longer, stable spherical microgels with similar average hydrodynamic diameters (300–350 nm obtained by DLS) and low polydispersity were obtained (Figure 1 and Figure S-1 in the Supporting Information). The observation that particle size is not controlled by irradiation time is significant as it demonstrates that this miniemulsion based method is a reliable platform approach for the preparation of well-defined microgels.
Fig. 1.

Representative SEM image of precursor microgels MG-A24.
Having demonstrated successful particle formation for tirr > 4h, the qualitative influence of reaction time on the crosslinking density and mesh size was investigated by determining the respective swelling ratios of the microgels. For this, the purified microgel particles were transferred into THF (a good solvent for linear PAGE), the average diameters of the swollen microgels MG-A4, -A6, -A8 and -A10 were measured by DLS, and the swelling ratios Q were calculated as Q = Vswollen/Vnon-swollen.
The dependency of Q on the irradiation time (Figure 2) clearly illustrates that the degree of swelling sharply decreases between 4 and 6 hours. In contrast, the observed decrease of Q between 6 and 24 hours is negligible and the swelling degree can be seen as constant regarding the experimental error. Since a decreased swelling ability corresponds to an increased crosslinking density, it is assumed that after 6 hours of irradiation, the crosslinking reaction is close to completion. This is an important parameter in avoiding unwanted side reactions during the secondary functionalization step.
Fig. 2.

Swelling degree of microgels MG-A4, -A6, -A8, -A10, and -A24 in THF calculated from the hydrodynamic diameters determined by DLS.
To further tune the molecular characteristics of these microgel particles, the relationship between mesh size and feed ratio of crosslinker was investigated at constant irradiation time. To ensure completion of the crosslinking reaction, an irradiation time of 8 hours was studied with crosslinker concentrations of 0.2, 0.5, 1.0, 1.5 and 2.0 mol-percent, respectively. At crosslinker concentrations of 0.5 mol-% or lower, stable microgels were not formed while at higher concentrations (1.0, 1.5 and 2.0 mol-%) stable microgels particles were obtained. These particles [MG-A8 (containing 2 mol-% crosslinker), -B8 (containing 1.5 mol-% crosslinker), C8 (containing 1 mol-% crosslinker)] were then investigated with respect to their diameter and size distribution by DLS. In analogy to the above reaction time study, DLS measurements in water revealed a slight variation in particle size from 320 to 370 nm (see Figure S-2 in the Supporting Information).
To demonstrate the effect of the crosslinker feed ratio, the microgels were swollen in THF or MeOH and the swelling ratios calculated from the average hydrodynamic diameters determined by DLS. As can be seen in Figure 3, for an irradiation time of 8 hours, a strong correlation between Q and crosslinker concentration was observed in THF. In MeOH a similar trend is observed but the overall degree of swelling is reduced due to a less favourable interaction between the network forming PAGE polymer and the solvent. As expected, increasing the feed ratio of crosslinker from 1.0 to 1.5 and 2.0 mol-% leads to decreased swelling ratios and mesh sizes, implying increased crosslinking densities.
Fig. 3.

Influence of crosslinker feed ratio on the swelling ratios of MG-A8 precursor microgels from 8 hours irradiation time in THF and methanol.
Similar trends were observed for analogous particles that were prepared with irradiation times of 10 hours (see Supporting Information) therefore further supporting the assumption that the crosslinking reaction has reached maximum conversion after an irradiation time of 8 hours. Overall, this facile tunability of the network parameters clearly demonstrates the modular and controlled nature of this approach to reactive precursor microgels that have a well-defined size distribution, surface chemistry and crosslinking density/mesh size.
Microgel functionalization via thiol-ene click reactions
The observed swelling behaviour of the microgels is critically important for secondary functionalization reactions on preformed particles, as it allows access to the reactive AGE units contained within the particle network. Having demonstrated the ability to control mesh size and swellability for crosslinked PAGE nanoparticles, the stability and presence of unreacted allyl groups in the core of the microgels enables functionalization by secondary thiol-ene coupling. Anionic and cationic thiol derivatives were therefore introduced to illustrate the dramatically different properties and swelling profiles obtainable from a common precursor particle (see Scheme 2).
Scheme 2.
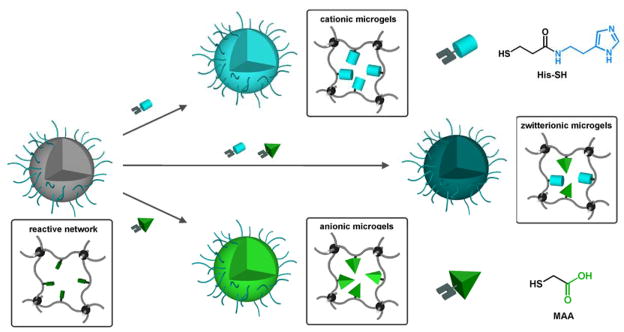
Preparation of pH responsive microgels through the network functionalization of precursor particles via thiol-ene click reaction
To demonstrate the modular nature of this post-functionalization approach, precursor microgels were reacted with mercaptoacetic acid (MAA) or a thiol-functionalized histamine derivative (His). Functionalization reactions with MAA were carried out in THF with an excess of thiol by irradiation with 365 nm light. Due to the limited solubility of the histamine-based thiol (His) in THF, all reactions employing His were performed in methanol. Following the light-induced thiol-ene click reaction, the microgels were extensively washed with THF (or MeOH for His) in order to remove unreacted thiols and photoinitiator from the network. The modified particles were then transferred into an aqueous environment and the resulting dispersions were stable without additional surfactant due to steric repulsion of the covalently attached PEO corona. Analysis of the particles using infrared spectroscopy revealed the appearance of characteristic bands for the functional groups of MAA and His in the respective microgels and the concomitant loss of peaks due to the allyl units (see Supporting Information).
Tailored functionalization of the particles is expected to result in tunability of particle swelling when exposed to a variety of different external environments and stimuli. To investigate this feature in detail, the pH-dependent swelling profiles for acidic, basic, and zwitterionic particles were studied by DLS. Significantly, for all levels of crosslinking, the acid-functionalized microgels MG-A8-MAA, MG-B8-MAA and MG-C8-MAA show a sharp volume phase transition (VPT) from a collapsed to a swollen state with increasing pH. As expected, the deprotonation/protonation of the carboxylic acid groups translates to the VPT of the microgel with this transition being observed in the biologically relevant pH range between pH 7 and 8 (Figure 4 and Supporting Information). It is also important to note that the structural parameters of the network – established during the synthesis of the initial precursor particles – clearly translate to the structure of the final responsive microgels. For example, the particle sizes of the functionalized microgels (collapsed at pH 2) do not significantly deviate from the size of their respective precursor particles. Additionally, the MAA functionalized particles with different crosslinking degrees demonstrate a decreasing relationship between Q in their fully swollen state (at pH 10) and crosslinker concentration analogous to their swollen precursor gels in THF (see Supporting Information).
Fig. 4.

Characterization of the pH-dependent swelling profiles of functionalized microgels MG-C8.
In contrast to the anionic MAA-functionalized microgels, the basic imidazole units in the histamine-functionalized microgels are expected to result in the opposite swelling profile. To investigate this assumption, pH-dependent DLS measurements of MG-A8-His and MG-C8-His were performed and the observed swelling ratios demonstrate that the protonation of the imidazole moieties does result in an inverse shape for the swelling profile with swollen particles being observed for pH < 5 with stable particle sizes being found for pH values of 6 and above (see Figure 4 and Supporting Information). This sharp volume phase transition correlates to the pKa of the histamine functionality, demonstrating the successful transfer of the basic character for the small molecule introduced in the second functionalization step to the final polymeric network. This translation of the pKa for the small molecule to the swelling behaviour of the final nanoscale object is clearly seen when the microgels MG-C8-MAA and MG-C8-His are compared (Figure 4). Regarding the maximum swelling ratio of the functionalized particles, it was found that the microgel MG-C8-His swells significantly more compared to MG-C8-MAA (Figure 4). It is noteworthy that at neutral pH, the histamine-functionalized microgels exhibit a slightly larger volume than MG-A8-MAA and MG-C8-MAA; this may be due to the larger size of the histamine thiol compared to mercaptoacetic acid as well as different hydrophilic character for the respective neutral species.
To further highlight the versatility and modularity of this approach, a mixture of the functional thiols MAA and His was used to modify the microgels’ interior. Since the two functional groups are characterized by dramatically different pKa values, their combined incorporation is expected to yield microgels with a novel zwitterionic network structure, resulting in a distinctive pH-dependent swelling behaviour. As shown in Figure 5, the hydrodynamic diameters of the resulting ‘mixed’ microgels MG-A8-His+MAA are strongly pH-dependent and follow the anticipated trend of a zwitterionic network.
Fig. 5.
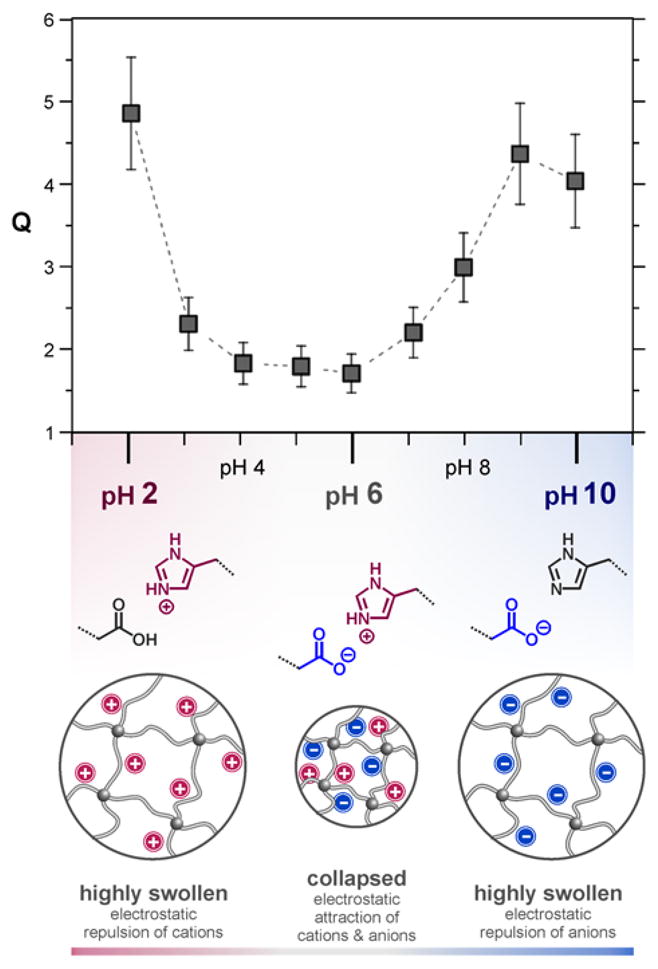
Characterization of zwitterionic microgels from the modification with histamine and acetic acid moieties: pH dependent swelling degrees of MG-A8-His+MAA and schematic illustration of the charge distribution in the bifunctional microgels at different pH values.
At acidic pH (< 3), the microgels are swollen due to the cationic nature of the protonated histamine groups and the neutral character of the protonated carboxylic acid groups. In contrast, for basic pH values (ca. 10), the network is negatively charged due to the deprotonated acid groups and the neutral imidazole units. Of most interest are the physiologically relevant values (pH 3 to 8) where the particles are less swollen with Q values of approximately 2. This behaviour is likely the result of the zwitterionic functionality creating ionic crosslinks between the histamine and acetic acid groups (Figure 5). Therefore, not only is the swelling reduced but the pH values of the phase transitions are also shifted to lower and higher pH values compared to their purely cationic or anionic counterparts, respectively.
These results clearly demonstrate the power of this two-step approach to functional microgel particles that allows a variety of different, and in some cases novel, materials to be obtained from a common precursor particle through a simple secondary functionalization strategy.
Degradation experiments
The potential of these materials as delivery systems was then examined through a series of degradability experiments which focused on ester cleavage of the small molecule PTMP crosslinker. To examine the degradability of the functionalized microgels, the anionic microgel MG-A10-MAA was placed in either a pH 10 solution at room temperature, or incubated with porcine liver esterase at 37°C under physiological conditions (PBS buffer pH 7.4). As shown in Figure 6, for both conditions, the observed particle diameters initially increase by ca. 10 %, followed by a sharp decrease in particle size to constant values of ca. 20 nm (pH 10) and 75 nm (esterase). This degradation profile can be explained by initial cleavage of crosslinking points upon ester hydrolysis where the number of crosslinks decreases but the overall particle retains intact. As a result, the microgels initially swell due to the lower crosslinking density. However, as the number of crosslinking points is further reduced the particles disintegrate into smaller fragments and linear polymers (Figure 6). In both cases, control experiments at neutral pH and in the absence of esterase showed no change in particle size by DLS. This highlights the stability of the microgels under neutral conditions and that efficient degradation can be “switched on” by either chemical or biological triggers, further exemplifying the ability to design materials for biomedical applications.
Fig. 6.

Degradation experiments of MG-A10-MAA in Milli-Q water with pH 10 at room temperature, in PBS buffer with pH 7.4 in the presence of porcine liver esterase at 37 °C and in pure Milli-Q water (control). The hydrodynamic diameters (≥90 % by number) were followed over time.
Conclusions
A new synthetic platform for the preparation of functional polyether-based microgels has been developed. Reactive precursor particles are formed by thiol-ene crosslinking of poly(allyl glycidyl ether) (PAGE) in miniemulsion droplets stabilized by PEO-b-PAGE as a non-ionic reactive surfactant. A key feature of these precursor particles is the presence of stable, unreacted allyl groups after the initial crosslinking step that are then available for a secondary, functionalization reaction. As a result, fundamental characteristics such as morphology, surface structure, and size distributions can be defined in the first crosslinking step and from a common batch of precursor particles, a diverse range of microgel systems bearing different functional groups can be prepared. This versatility is illustrated by tuning physical characteristics such as swelling behaviour through both the crosslinker concentration, and the covalent attachment of pH sensitive groups. Of particular note is the preparation of mixed, zwitterionic systems that combine the swelling profiles of anionic and cationic materials into a single system. Together with a bio-compatible PEO corona and biodegradable crosslinking points, the ability to tailor these microgels leads to opportunities to engineer novel systems for biomedical applications that incorporate and release guests in response to external triggers.
Experimental Section
Materials
Chemicals were purchased from Sigma Aldrich unless specified as otherwise. Tetrahydrofuran, toluene, acetonitrile and methanol were obtained in analytical pure grades from Fisher Scientifics. Tetrahydrofuran and methanol were filtered through a syringe filter (PTFE, pore size 0.45 μm) prior to use in DLS measurements. Potassium naphthalenide was prepared by dissolving recrystallized naphthalene (10 g) in dry THF (250 mL) and then adding potassium metal (3 g) under positive argon pressure. The system was closed with a septum and allowed to stir overnight with a glass-coated stir-bar at room temperature. For the preparation of N-(2-(1H-imidazol-4-yl)ethyl)-4-mercapto-butanamide (His), histamine and γ-thiobutyrolactone were reacted according to a procedure published in a previous study.63 The polymerization of poly(allyl glycidyl ether) (PAGE) was carried out neat at room temperature using the procedure described by Lee et al.52 The molecular weight of the polymer was 3100 g/mol as determined by 1H NMR spectroscopy. The polydispersity index was 1.1 as determined by size exclusion chromatography.
Methods
1H NMR spectroscopy was carried out on a Bruker AC 500 spectrometer in deuterated chloroform. Size exclusion chromatography (SEC) was performed on a Waters chromatograph with four Viscotek columns (two IMBHMW-3078, I-series mixed bed high molecular weight columns and two I-MBLMW-3078, I-series mixed bed low-molecular weight columns) connected to a Waters 2414 differential refractometer and a 2996 photodiode array detector. Chloroform with 0.1% triethylamine at room temperature was used as the mobile phase.
Scanning electron microscopy
Samples were prepared via drop-casting of an aqueous suspension of MG-A24 on a silicon wafer. After drying, a carbon film was sputtered on top of the samples and microscopy was performed on a FEI XL40 SEM.
Dynamic light scattering
The samples were freshly prepared on the day of use and allowed to stand for two hours prior to measurements. For each sample of the pH-dependent measurements, the same amount of microgel dispersion was placed in water that was previously adjusted to the desired pH by use of hydrochloric acid or potassium hydroxide solution. For particle characterization and swelling experiments, dynamic light scattering (DLS) experiments were carried out on a BI-200SM (Brookhaven Instruments) multi-angle detector system with a 632.8 nm (HeNe continuous wave) laser. The temperature was controlled using a circulating bath. Measurements of the intensity autocorrelation, g(q,s), were made at a temperature of 25 °C and scattering angles of 70°, 80°, 90°, and 110°. The average hydrodynamic diameter for each angle was obtained by a cumulate analysis. Subsequently, the apparent diffusion coefficients Dapp were calculated and plotted against the quadratic scattering vector q2 (see Supporting Information). The z-averaged diffusion coefficient Ds was obtained by extrapolation to the y-intercept and used to calculate the actual hydrodynamic radius Rh of the microgels (see Supporting Information).
For degradation experiments, DLS measurements were performed on a more concentrated dispersion. Therefore, the particle sizes were measured at a fixed detection angle of 173° in backscattering mode on a Malvern Zetasizer Nano ZS ZEN 3600 at a temperature of 25 °C with a laser wavelength of 633 nm. The solutions were placed in a glass cuvette with a layer thickness of 1 cm. The non-negative-least-squares algorithm in general-purpose mode was used for interpretation. For analysis, the results by number were taken into account and mean values of at least five measurements were calculated.
Syntheses
Synthesis of poly(allyl glycidyl ether)-block-poly(ethylene oxide) (PAGE-b-PEO)
All polymerizations were carried out on a Schlenk line in custom thick-walled glass reactors fitted with ACE-threads under argon atmosphere. The reactor was fitted with a burette containing THF from a dry solvent system, a flexible connector attached to a burette containing ethylene oxide (EO) at 0 °C, and an addition port capped with a septum. 0.96 μL (0.93 mmol) BzOH was first charged into the reactor followed by titration with potassium naphtalenide until a light green colour persisted. Subsequently 4.7 g (41 mmol) of allyl glycidyl ether (AGE) was charged into the reactor and the reaction was stirred with a glass-coated stir bar at room temperature overnight. 150 mL THF was then added, a sample collected and analyzed by 1H-NMR to confirm full conversion of AGE before addition of 18.6 g (422 mmol) of EO. The reactor was then heated to 45 °C (oil bath) and the reaction was allowed to proceed overnight. To terminate the reaction 2 mL degassed MeOH was added and the resulting polymer was precipitated in 2 L of hexanes, filtered and dried under vacuum. The purified polymer (22.7 g, 97% yield) was then analyzed by GPC and 1H-NMR. GPC (CHCl3) Mn = 21700 g/mol, PDI = 1.09. The degree of polymerization of the two blocks was determined by comparing the integration values for the two benzylic protons at 4.53 ppm with the values for the two allylic protons at 5.28-5.14 ppm from the PAGE block and the backbone protons at 3.80-3.40 ppm resulting from both PAGE and PEO blocks. Mn (Tot) = 26100 g/mol, Mn (AGE) = 4700 g/mol, Mn (EO) = 21400 g/mol.
Synthesis of native microgels
Aqueous continuous phase: The PAGE5k-b-PEO20k block copolymer (100.0 mg) was dissolved in Milli-Q water (10.0 mL). Dispersed phase: PAGE3.1k (500.0 mg), 2,2-dimethoxy-2-phenylacetophenone (2.0 mg, 0.008 mmol) and pentaerythritol tetrakis(3-mercaptopropionate) (PTMP) (see Table 1) were dissolved in 0.5 mL toluene. Both phases were combined in a 20 mL glass vial and pre-mixed by sonication in an ultrasonic bath for 2 minutes. Full dispersion was achieved by ultrasonication with a maximum amplitude of 70 % (Branson Digital Sonifier W-450 D equipped with a microtip; pulse duration: 15 seconds, pause duration: 10 seconds, total duration: 2 minutes) The dispersions were then transferred into round-bottom flasks, placed under an UV lamp (Black-Ray UV BenchLamp equipped with two Sylvania 15 W 350 Blacklight fluorescent tubes, λ=365 nm, distance lamp-flask approx. 10 cm) and continuously stirred. Depending on the intended degree of crosslinking, the samples were irradiated for 4, 6, 8, 10 and 24 hours, respectively. Afterwards, the dispersions were centrifuged (20 minutes, 10 000 rpm) and the supernatant solvent decanted. The solid was then washed with Milli-Q water (3× 10 mL), methanol (3× 10 mL), Milli-Q water (3× 10 mL) and centrifuged after each washing step. The purified microgels were dispersed in Milli-Q water for storage.
Table 1.
Crosslinker feed amounts in microgel synthesis.
| microgel | PTMP per AGE units [mol %] | mass of PTMP [mg] |
|---|---|---|
| MG-A | 2.0 | 52.0 |
| MG-B | 1.5 | 39.0 |
| MG-C | 1.0 | 26.0 |
Functionalization of native microgels with mercaptoacetic acid
2.0 mL aqueous microgel dispersion (c = approx. 15 mg/mL) were centrifuged and washed with THF (3 × 10 mL). Afterwards, the microgels were dissolved in THF (2.0 mL); then, DMPA (3.0 mg, 0.012 mmol) and mercaptoacetic acid (25.0 mg, 0.27 mmol) were added. Following purging with argon for 15 minutes, the dispersion was continuously stirred for 2 hours under an UV lamp (Black-Ray UV BenchLamp equipped with two Sylvania 15 W 350 Blacklight fluorescent tubes, λ=365 nm, distance lamp-flask approx. 10 cm). Subsequently, the dispersion was centrifuged, washed with methanol (6 × 10 mL) and Milli-Q water (3 × 10 mL). The functionalized microgels were stored in Milli-Q water.
Functionalization of native microgels with N-(2-(1H-imidazol-4-yl)ethyl)-4-mercaptobutanamide
2.0 mL aqueous microgel dispersion (c = approx. 15 mg/mL) were centrifuged and washed with methanol (3 × 10 mL). Afterwards, the microgels were dissolved in methanol (2.0 mL); then, DMPA (3.0 mg, 0.012 mmol) and N-(2-(1H-imidazol-4-yl)ethyl)-4-mercaptobutanamide (53.0 mg, 0.25 mmol) were added. After purging with argon for 15 minutes, the dispersion was continuously stirred for 2 hours under an UV lamp (Black-Ray UV BenchLamp equipped with two Sylvania 15 W 350 Blacklight fluorescent tubes, λ=365 nm, distance lamp-flask approx. 10 cm). Subsequently, the dispersion was centrifuged, washed with methanol (6 × 10 mL) and Milli-Q water (3 × 10 mL). The functionalized microgels were stored in Milli-Q water.
Functionalization of native microgels with N-(2-(1H-imidazol-4-yl)ethyl)-4-mercaptobutanamide and mercaptoacetic acid
2.0 mL aqueous microgel dispersion (c= approx. 15 mg/mL) were centrifuged and washed with methanol (3 × 10 mL). Afterwards, the microgels were dissolved in 2.0 mL methanol; then, DMPA (3.0 mg, 0.012 mmol), mercaptoacetic acid (25.0 mg, 0.27 mmol) and N-(2-(1H-imidazol-4-yl)ethyl)-4-mercaptobutanamide (53.0 mg, 0.25 mmol) were added. After purging with argon for 15 minutes, the dispersion was continuously stirred for 2 hours under an UV lamp (Black-Ray UV BenchLamp equipped with two Sylvania 15 W 350 Blacklight fluorescent tubes, λ=365 nm, distance lamp-flask approx. 10 cm). Subsequently, the dispersion was centrifuged, washed with methanol (6 × 10 mL) and Milli-Q water (3 × 10 mL). The functionalized microgels were stored in Milli-Q water.
Degradation experiments
For the degradation experiment employing esterase, MG-A10-MAA (50.0 μL dispersion, c = approx. 15 mg/mL) was placed in 30.0 mL PBS buffer (pH 7.40). After the addition of porcine liver esterase (4 mg, 80 U), the dispersion was constantly stirred at 37°C. For the investigation of the degradation at pH 10, 30.0 mL Milli-Q water was adjusted to pH 10 by use of potassium hydroxide. After the addition of MG-A10-MAA (50.0 μL dispersion, c = approx. 15 mg/mL), the mixture was continuously stirred at room temperature. For both experiments, samples of 1.0 mL were taken after regular time intervals and DLS measurements were conducted without further dilution. The average results by number were consulted for particle size analysis.
Supplementary Material
Acknowledgments
This work was supported by the Institute for Collaborative Biotechnologies through grant W911NF-09-0001 from the U.S. Army Research Office (D.K. and C.J.H.), the U.S. Army Basic Research Program and Edgewood Chemical Biological Center (K.L.K.), and the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN268201000046C (D.K., and C.J.H.). The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. P. L. would like to thank the Wenner-Gren foundations and Bengt Lundqvist memorial foundation for financial support. Characterization work was enabled by MRL Central Facilities supported by the MRSEC Program of the National Science Foundation under Award No. DMR 1121053 (C.F., J.G. and P.L.).
Footnotes
Electronic Supplementary Information (ESI) available: Interpretation of dynamic light scattering experiments. See DOI: 10.1039/b000000x/
Contributor Information
Craig J. Hawker, Email: hawker@mrl.ucsb.edu.
Daniel Klinger, Email: klinger@mrl.ucsb.edu.
Notes and references
- 1.Freiberg S, Zhu X. Int J Pharm. 2004;282(1–2):1–18. doi: 10.1016/j.ijpharm.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Brannon-Peppas L, Blanchette JO. Adv Drug Del Rev. 2012;64:206–212. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Couvreur P. Adv Drug Del Rev. 2013;65(1):21–23. doi: 10.1016/j.addr.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Kataoka K, Harada A, Nagasaki Y. Adv Drug Del Rev. 2001;47(1):113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 5.Naahidi S, Jafari M, Edalat F, Raymond K, Khademhosseini A, Chen P. J Controlled Release. 2013;166(2):182–194. doi: 10.1016/j.jconrel.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Rosler A, Vandermeulen GWM, Klok HA. Adv Drug Del Rev. 2012;64:270–279. [Google Scholar]
- 7.O’Reilly RK, Joralemon MJ, Hawker CJ, Wooley KL. Chem Eur J. 2006;12:6776–6786. doi: 10.1002/chem.200600467. [DOI] [PubMed] [Google Scholar]
- 8.Saunders BR, Laajam N, Daly E, Teow S, Hu X, Stepto R. Adv Colloid Interface Sci. 2009;147–48:251–262. doi: 10.1016/j.cis.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Klinger D, Landfester K. Polymer. 2012;53(23):5209–5231. [Google Scholar]
- 10.Nayak S, Lyon LA. Angew Chem Int Ed. 2005;44(47):7686–7708. doi: 10.1002/anie.200501321. [DOI] [PubMed] [Google Scholar]
- 11.Hamidi M, Azadi A, Rafiei P. Adv Drug Del Rev. 2008;60(15):1638–1649. doi: 10.1016/j.addr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Vinogradov SV. Curr Pharm Des. 2006;12(36):4703–4712. doi: 10.2174/138161206779026254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zha L, Banik B, Alexis F. Soft Matter. 2011;7(13):5908–5916. [Google Scholar]
- 14.Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. Prog Polym Sci. 2008;33(4):448–477. [Google Scholar]
- 15.Hoare T, Pelton R. Langmuir. 2008;24(3):1005–1012. doi: 10.1021/la7024507. [DOI] [PubMed] [Google Scholar]
- 16.Kabanov AV, Vinogradov SV. Angew Chem Int Ed. 2009;48(30):5418–5429. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smeets NMB, Hoare T. J Polym Sci, Part A: Polym Chem. 2013;51(14):3027–3043. [Google Scholar]
- 18.Das M, Mardyani S, Chan WCW, Kumacheva E. Adv Mater. 2006;18(1):80–83. [Google Scholar]
- 19.Eichenbaum GM, Kiser PF, Dobrynin AV, Simon SA, Needham D. Macromolecules. 1999;32(15):4867–4878. doi: 10.1021/ma970897t. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Mardyani S, Chan WCW, Kumacheva E. Biomacromolecules. 2006;7(5):1568–1572. doi: 10.1021/bm050912z. [DOI] [PubMed] [Google Scholar]
- 21.Chan Y, Bulmus V, Zareie MH, Byrne FL, Barner L, Kavallaris M. J Controlled Release. 2006;115(2):197–207. doi: 10.1016/j.jconrel.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 22.Goh SL, Murthy N, Xu MC, Frechet JMJ. Bioconjugate Chem. 2004;15(3):467–474. doi: 10.1021/bc034159n. [DOI] [PubMed] [Google Scholar]
- 23.Jhaveri SB, Carter KR. Macromolecules. 2007;40(22):7874–7877. [Google Scholar]
- 24.Murthy N, Thng YX, Schuck S, Xu MC, Frechet JMJ. J Am Chem Soc. 2002;124(42):12398–12399. doi: 10.1021/ja026925r. [DOI] [PubMed] [Google Scholar]
- 25.Gu Z, Dang TT, Ma ML, Tang BC, Cheng H, Jiang S, Dong YZ, Zhang YL, Anderson DG. ACS Nano. 2013;7(8):6758–6766. doi: 10.1021/nn401617u. [DOI] [PubMed] [Google Scholar]
- 26.De Geest BG, De Koker S, Demeester J, De Smedt SC, Hennink WE. Polymer Chemistry. 2010;1(2):137–148. [Google Scholar]
- 27.Klinger D, Aschenbrenner E, Weiss CK, Landfester K. Polymer Chemistry. 2012;3(1):204–216. [Google Scholar]
- 28.Gao HF, Yang WL, Min K, Zha LS, Wang CC, Fu SK. Polymer. 2005;46(4):1087–1093. [Google Scholar]
- 29.Lee Y, Park SY, Kim C, Park TG. J Controlled Release. 2009;135(1):89–95. doi: 10.1016/j.jconrel.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Nayak S, Lee H, Chmielewski J, Lyon LA. J Am Chem Soc. 2004;126(33):10258–10259. doi: 10.1021/ja0474143. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Ballard N, Coleman OD, Hands-Portman IJ, Bon SAF. J Polym Sci, Part A: Polym Chem. 2014;52:1745–1754. [Google Scholar]
- 32.Vihola H, Laukkanen A, Tenhu H, Hirvonen J. J Pharm Sci. 2008;97(11):4783–4793. doi: 10.1002/jps.21348. [DOI] [PubMed] [Google Scholar]
- 33.Klinger D, Landfester K. Macromolecules. 2011;44(24):9758–9772. [Google Scholar]
- 34.Klinger D, Landfester K. Macromol Rapid Commun. 2011;32(24):1979–1985. doi: 10.1002/marc.201100493. [DOI] [PubMed] [Google Scholar]
- 35.Bian S, Zheng J, Yang W. J Polym Sci, Part A: Polym Chem. 2014;52:1676–1685. [Google Scholar]
- 36.Rodriguez-Fernandez J, Fedoruk M, Hrelescu C, Lutich AA, Feldmann J. Nanotechnology. 2011;22(24) doi: 10.1088/0957-4484/22/24/245708. [DOI] [PubMed] [Google Scholar]
- 37.He J, Tong X, Zhao Y. Macromolecules. 2009;42(13):4845–4852. [Google Scholar]
- 38.Fomina N, McFearin C, Sermsakdi M, Edigin O, Almutairi A. J Am Chem Soc. 2010;132(28):9540–9542. doi: 10.1021/ja102595j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, Chen YB, Nan JY, Wang CP, Chu FX. J Appl Polym Sci. 2012;124(6):4678–4685. [Google Scholar]
- 40.Shi LJ, Khondee S, Linz TH, Berkland C. Macromolecules. 2008;41(17):6546–6554. [Google Scholar]
- 41.Eichenbaum GM, Kiser PF, Shah D, Simon SA, Needham D. Macromolecules. 1999;32(26):8996–9006. doi: 10.1021/ma970897t. [DOI] [PubMed] [Google Scholar]
- 42.Wells LA, Sheardown H. Eur J Pharm Biopharm. 2007;65(3):329–335. doi: 10.1016/j.ejpb.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 43.Alarcon CDH, Pennadam S, Alexander C. Chem Soc Rev. 2005;34(3):276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 44.Boyko V, Richter S, Pich A, Arndt KF. Colloid Polym Sci. 2003;282(2):127–132. [Google Scholar]
- 45.Debord JD, Lyon LA. Langmuir. 2003;19(18):7662–7664. [Google Scholar]
- 46.Hoare T, Pelton R. Langmuir. 2004;20(6):2123–2133. doi: 10.1021/la0351562. [DOI] [PubMed] [Google Scholar]
- 47.a) Mecerreyes D, Lee V, Hawker CJ, Hedrick JL, Wursch A, Volksen W, Magbitang T, Huang E, Miller RD. Adv Mater. 2001;13:204–208. [Google Scholar]; b) Beck JB, Killops KL, Kang T, Sivanandan K, Bayles A, Mackay ME, Wooley KL, Hawker CJ. Macromolecules. 2009;42:5629–5635. doi: 10.1021/ma900899v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kratz K, Hellweg T, Eimer W. Colloids Surf Physicochem Eng Aspects. 2000;170(2–3):137–149. [Google Scholar]
- 49.McPhee W, Tam KC, Pelton R. J Colloid Interface Sci. 1993;156(1):24–30. [Google Scholar]
- 50.Nayak S, Gan DJ, Serpe MJ, Lyon LA. Small. 2005;1(4):416–421. doi: 10.1002/smll.200400089. [DOI] [PubMed] [Google Scholar]
- 51.Pich A, Karak A, Lu Y, Ghosh AK, Adler HJP. Macromol Rapid Commun. 2006;27(5):344–350. [Google Scholar]
- 52.Lee BF, Kade MJ, Chute JA, Gupta N, Campos LM, Fredrickson GH, Kramer EJ, Lynd NA, Hawker CJ. J Polym Sci, Part A: Polym Chem. 2011;49(20):4498–4504. doi: 10.1002/pola.24891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee A, Lundberg P, Klinger D, Lee BF, Hawker CJ, Lynd NA. Polymer Chemistry. 2013;4(24):5735–5742. doi: 10.1039/C3PY00747B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.a) Campos LM, Killops KL, Sakai R, Paulusse JMJ, Damiron D, Drockenmuller E, Messmore BW, Hawker CJ. Macromolecules. 2008;41(19):7063–7070. [Google Scholar]; b) Campos LM, Meinel I, Guino RG, Schierhorn M, Gupta N, Stucky GD, Hawker CJ. Adv Mater. 2008;20:3728–3733. [Google Scholar]
- 55.Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49(9):1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 56.a) Kade MJ, Burke DJ, Hawker CJ. J Polym Sci, Part A: Polym Chem. 2010;48(4):743–750. [Google Scholar]; b) Hawker CJ, Fokin VV, Finn MG, Sharpless KB. Aust J Chem. 2007;60:381–383. [Google Scholar]
- 57.Gupta N, Lin BF, Campos L, Dimitriou MD, Hikita ST, Treat ND, Tirrell MV, Clegg DO, Kramer EJ, Hawker CJ. Nat Chem. 2010;2:138–145. doi: 10.1038/nchem.478. [DOI] [PubMed] [Google Scholar]
- 58.Tiwari R, Hönders D, Schipmann S, Schulte B, Das P, Pester CW, Klemradt U, Walther A. Macromolecules. 2014 doi: 10.1021/ma402530y. [DOI] [Google Scholar]
- 59.van Berkel KY, Hawker CJ. J Polym Sci, Part A: Polym Chem. 2010;48:1594–1606. doi: 10.1002/pola.23917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landfester K. Angew Chem Int Ed. 2009;48(25):4488–4507. doi: 10.1002/anie.200900723. [DOI] [PubMed] [Google Scholar]
- 61.Landfester K, Musyanovych A. Hydrogels in Miniemulsions. In: Pich ARW, editor. Chemical Design of Responsive Microgels. Vol. 234. 2010. pp. 39–63. [Google Scholar]
- 62.Landfester K, Willert M, Antonietti M. Macromolecules. 2000;33(7):2370–2376. [Google Scholar]
- 63.Lundberg P, Lynd NA, Zhang YN, Zeng XH, Krogstad DV, Paffen T, Malkoch M, Nystrom AM, Hawker CJ. Soft Matter. 2013;9(1):82–89. doi: 10.1039/C2SM26996A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.