

Abstract
Selenium is an essential micronutrient in humans due to the important roles of the selenocysteine-containing selenoproteins. Organoselenium metabolites are generally found to be substrates for the biochemical pathways of their sulfur analogs, and the redox chemistry of selenomethionine and some other metabolites have been previously reported. We now report the first synthesis and characterization of Se-adenosylselenohomocysteine selenoxide (SeAHO) prepared via hydrogen peroxide oxidation of Se-adenosylselenohomocysteine (SeAH). The selenoxide SeAHO, in contrast to its corresponding sulfoxide S-adenosylhomocysteine (SAHO), can form hydrate, has an electrostatic interaction between the α-amino acid moiety and the highly polar selenoxide functional group, and readily oxidizes glutathione (GSH) and cysteine thiols.
Keywords: organoselenium, Se-adenosylselenohomocysteine selenoxide, Se-adenosylselenohomocysteine, selenomethionine, oxidation
1. Introduction
Selenium is an essential micronutrient for all animals and many other living organisms.[1-10] However, a high level of selenium is toxic, thus, selenium metabolites should be maintained within a fairly narrow concentration range of adequacy for the biosynthesis of the over 25 human selenoproteins to balance deficiency and toxicity.[5,11] Organoselenium metabolites are only present in trace amounts,[6,12] relative to the well-known sulfur analogs that include the amino acids L-methionine (4, X = S) and L-cysteine (8, X = S),[13] the biological methyl donor S-adenosyl-L-methionine (1, X = S; AdoMet or SAM),[14-17] and the byproduct of methylation S-adenosyl-L-homocysteine (2, X = S; AdoHcy or SAH)[17-19] (see Scheme 1). Oxidations of S-adenosylhomocysteine (2, X = S; SAH) are reported to give the sulfoxide SAHO 3 (X = S)[18,20-23] and the corresponding sulfone.[22-24] These have not been detected as metabolites in vivo, but as close structural analogs of AdoMet 1 (X =S), these analogs are methyltransferase enzyme inhibitors in vitro. As examples, sulfoxide SAHO 3 (X = S) is an inhibitor of catechol-O-methyltransferase (COMT),[22,25] phenylethanolamine N-methyltransferase (PNMT),[22] histamine N-methyltransferase (HMT),[22] protein methyltransferase II,[26] viral mRNA methyltransferases,[27] and E. coli cyclopropane fatty acid synthase.[23]
Scheme 1.

α-Amino acids L-methionine (4, X = S) and L-selenomethionine (4, X = Se), the corresponding oxidation products sulfoxide 7 (X = S) and selenoxide 7 (X = Se), and the corresponding methylation substrates S-adenosylmethionine (1, X = S; AdoMet; SAM) and Se-adenosylselenomethionine (1, X = Se) are shown. The byproducts of biological methylations, S-adenosylhomocysteine (2, X = S; SAH) and Se-adenosylselenohomocysteine (2, X = Se; SeAH), can undergo oxidation to the corresponding sulfoxide 3 (X = S; SAHO) and selenoxide 3 (X = Se; SeAHO), or well-understood further metabolism via homocysteine (5, X = S) and selenohomocysteine (5, X = Se), respectively.
The redox biochemistry of methionine (4, X = S) is well understood.[28-30] The selenium analog of methionine (4, X = Se) is also easily oxidized with biological oxidants such as hydrogen peroxide to give a mixture of selenoxide 7 (X = Se) and hydrate in the neutral pH range.[28,31,32] NMR data is pH dependent, and only single compounds are seen at low pH[28,33] and at high pH.[28,32] These data are consistent with studies of other selenoxides.[34,35] Selenomethionine selenoxide (7, X = Se) is homogeneous by HPLC and stable at ambient temperature.[36,37] Homocysteine (5, X = S),[38] selenohomocysteine (5, X = Se),[39,40] cysteine (8, X = S),[30,41-44] and selenocysteine (8, X = Se)[30,45] also undergo oxidations.
We have previously reported our studies of sulfur[46-51] and selenium chemistry,[39,52] and our work with methyltransferases.[53-56] Interestingly, very little is known about the biochemical activity and redox chemistry[57] of Se-adenosylselenohomocysteine (2, X = Se; SeAH),[8,9,58-60] which is structurally related to its methyl analog selenomethionine (4, X = Se)[6,12,28,31,32] and to its corresponding sulfur analog S-adenosylhomocysteine (2, X = S; SAH).[17,18] Thus, we now detail the synthesis and characterization of both the organoselenide 2 (X = Se; SeAH) and its oxidation product Se-adenosylselenohomocysteine selenoxide (3, X = Se; SeAHO).
2. Results and Discussion
The previously unreported selenoxide SeAHO 3 (X = Se) was prepared in 21% overall yield (see Scheme 2). Adenosine (6) was first converted to 5′-chloro-5′-deoxyadenosine (9, 5′-Cl-5′-dA) by the reported method[61] in 88% yield. Reaction of 5′-Cl-5′-dA 9 with L-selenohomocysteine (5, X = Se), prepared by our previously reported method,[39,59] gave SeAH 2 (X = Se) that was isolated in 24% yield after two recrystallizations. Partial protonation or a conformational difference in the adenosine moiety may account for the two sets of aromatic and glycosidic protons seen in pH 3 D2O phosphate buffer that was not seen in deuterated acetic acid solution by proton NMR. The oxidations of both this selenium analog SeAH 2 (X = Se) and the commercially available sulfur analog SAH 2 (X = S) were studied under the same conditions.
Scheme 2.

Synthesis of Se-adenosylselenohomocysteine selenoxide (3, X = Se; SeAHO).
For the sulfur analog, hydrogen peroxide oxidation of SAH 2 (X = S) in acetic acid solution according to a literature report[23] gave nearly equal ratios of the R and S isomers at the newly chiral sulfur center for the product SAHO 3 (X = S). These two diastereomers had distinctly different resonances in the proton NMR for the diastereotopic α, γ, 2′, 3′, 4′, 5′, and for one aromatic resonance (see Table 1). The diastereomeric (at sulfur) mix of sulfoxides SAHO 3 (X = S) had 975 and 998 cm-1 bands in the IR that are characteristic of sulfoxide stretching.[20,21] For example, our IR of dimethylsulfoxide (DMSO, data not shown) showed strong absorptions at 1018 and 1042 cm-1.
Table 1.
1H Chemical shifts of SAH 2 (X = S), SeAH 2 (X = Se), and SAHO 3 (X = S) in CD3CO2D; and, of SeAHO 3 (X = Se) in 97:3 CD3CO2D/30% aqueous H2O2.
| Proton assign. | SAH 2 (X = S) |
SAHO 3 (X = S) (1:1 mix) |
SeAH 2 (X = Se) |
SeAHO 3 (X = Se) |
|---|---|---|---|---|
| α | 4.18 | 4.15, 4.17 | 4.16 | 4.58-4.67 |
| βa | 2.27-2.34 | 2.49-2.58 | 2.32-2.39 | 2.74-2.83 |
| βb | 2.16-2.23 | 2.44-2.53 | 2.22-2.30 | 2.60-2.69 |
| γa | 2.76-2.85 | 3.28-3.37, 3.28-3.37 | 2.74-2.84 | 3.84-3.95 |
| γb | 2.76-2.85 | 3.19-3.25, 3.28-3.37 | 2.74-2.84 | 3.84-3.95 |
| 1′ | 6.16 | 6.19 | 6.16 | 6.20 |
| 2′ | 4.84 | 4.88, 4.93 | 4.86 | 4.94 |
| 3′ | 4.51 | 4.64, 4.67 | 4.49 | 4.72 |
| 4′ | 4.35 | 4.57-4.62, 4.60-4.65 | 4.38 | 4.58-4.67 |
| 5a′ | 3.05 | 3.54, 3.61 | 3.05 | 3.84-3.95 |
| 5b′ | 3.00 | 3.54, 3.55 | 3.03 | 3.65 |
| Ar | 8.43 | 8.42 | 8.44 | 8.45 |
| Ar | 8.48 | 8.43, 8.46 | 8.50 | 8.48 |
The corresponding hydrogen peroxide oxidation of the selenium analog SeAH 2 (X = Se) to SeAHO 3 (X = Se) was first performed in acetic acid solution. Vacuum transfer of an acetic acid solution gave a solid sample of SeAHO 3 (X = Se) that was characterized by melting point and IR. Some carbonyl stretch was observed at 1777 cm-1 in the IR spectrum, characteristic of α-amino carboxylic acids at low pH.[62] The selenoxide SeAHO 3 (X = Se) also showed characteristic Se=O stretching bands[63] in the IR at 847 and 878 cm-1. The 1H NMR of selenoxide SeAHO 3 (X = Se) in acetic acid-d4/H2O/H2O2 600:9:1, an organic acidic environment near the pKa's of the carboxylic acid group and the adenine residue, was clean and assignable for the selenoxide SeAHO 3 (X = Se). The selenoxide SeAHO 3 (X = Se) has a distinctly different conformation than the corresponding sulfoxide SAHO 3 (X = S) as evidenced by the dramatic downfield shift of the α-proton in the NMR (see Table 1). The hydrolysis of the C1′-adenine bond of SeAHO 3 (X = Se) in the acetic acid/water was followed over several hours by 1H NMR (see Supplementary data, Figure S13).
Characterization of selenoxide SeAHO 3 (X = Se) in an aqueous environment was of greater biological significance. The selenoxide SeAHO 3 (X = Se) was completely stable to hydrolysis of the C1′-adenine bond in phosphate buffered aqueous solutions for at least three hours at ambient temperature over the wide pH range of 3-12 as no elimination or other degradation products were seen by HPLC or proton NMR. The SeAHO 3 (X = Se) prepared in phosphate buffers at pHs 3, 7 and 12 each gave homogeneous chromatograms by reversed-phase HPLC (see Supplementary data, Figure S2), although some decomposition was observed by HPLC after 2 weeks or when prepared in pure deionized water.
The selenoxide SeAHO 3 (X = Se) was readily reduced back to selenide SeAH 2 (X = Se) at ambient temperature by glutathione (GSH) (see Figure 1) as observed by C18 HPLC (150 × 4.6 mm, 260 nm, 0.1% formic acid, 2:98 acetonitrile/water, 1 mLmin-1). The selenoxide SeAHO 3 (X = Se) was also reduced by cysteine (8, X = S), but not by thioethers methionine (4, X = S) or SAH 2 (X = S) (see Supplementary data, Figure S6). The sulfoxide SAHO 3 (X = S) analog was not reduced under these biological conditions with glutathione (GSH) or cysteine (8, X = S). The reduction of sulfoxides generally requires more forcing conditions.[64,65]
Figure 1.
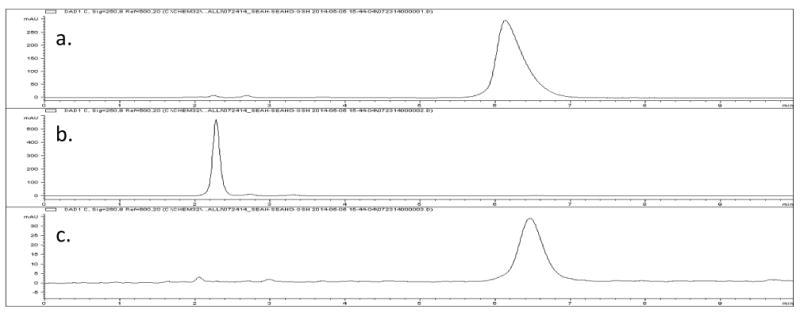
HPLC analysis of the reduction of SeAHO 3 (X = Se, retention time 6.2 min) to SeAH 2 (X = Se, retention time 2.3 min) by glutathione (GSH). a. SeAH 2 (X = Se), 13.4 mM in 50 mM K3PO4 (pH 12). b. SeAHO 3 (X = Se) from oxidation with 360 mole percent H2O2 for 10 min, followed by treatment with bovine catalase (EC 1.11.1.6) at ambient temperature (pH 8) to decompose the excess H2O2, and ultrafiltration through a 30 kDa membrane to remove the catalase. c. SeAH 2 (X = Se) from reduction of the above sample by treatment with 150 mole percent of glutathione (GSH) for 5 min.
The selenoxide SeAHO 3 (X = Se) generally appeared to be a 60:40 mixture by 1H NMR in 50 mM phosphate buffers of D2O at measured pH's of 3 and 7. High resolution NMR at pH 7 showed that 40% of the material had the α-proton shifted downfield, and that the selenoxide SeAHO 3 (X = Se) was also a 50:50 mixture, likely to be a mix of selenoxide and hydrate, analogous to the reported data for selenomethionine selenoxide (7, X = Se).[28,31,32] The selenoxide SeAHO 3 (X = Se) was presumably mostly hydrate at pH 3 and mostly in the selenoxide form at pH12 where only a very small amount of decomposition was observed.
The coordination of the α-amino acid moieties with the selenoxide functional group of the Se-methyl analog, selenomethionine selenoxide (7, X =Se), in aqueous solutions have already been proposed at acidic,[32] neutral,[31,32] and basic[28,32] pHs. The protonation, hydration, and racemization of the selenoxide functional group at low pH is also well-known.[34,35] These data correlate well with our NMR data for Se-adenosylselenohomocysteine selenoxide (3, X = Se; SeAHO) in the acidic organic (acetic acid-d4) and in the phosphate buffered aqueous environments at pHs 3, 7, and 12. The characteristic coordination of the selenoxide 3 (X = Se; SeAHO) can be intermolecular or intramolecular (as shown in Figure 2). The interaction of the α-amino acid moiety of SeAHO 3 (X = Se) with the selenoxide / hydrate group results in deshielding of the α-proton completely in acetic acid-d4 and to the extent of about 40% in acidic and neutral buffered aqueous solutions.
Figure 2.

Hydration and racemization of SeAHO 3 (X = Se). The selenoxide 3 (SeAHO) must have intermolecular or intramolecular (as shown) coordination of the acidic protons of the ammonium (as shown) or carboxylic acid groups with the selenoxide oxygen, and can exist in a selenoxide R1R2Se=O (3a) or hydrate R1R2Se(OH)2 (3f) form in acidic or neutral aqueous solutions. A very homogeneous form (3c) was observed in acetic acid-d4 solution.
The mass spectra of selenoxide SeAHO 3 (X = Se) were obtained by LC-MS (ToF) and LCQ-MS (ion trap) in an acidic environment (acetonitrile/water/0.1% formic acid), as well as by MALDI (matrix of α-cyano-4-hydroxycinnamic acid and trifluoroacetic acid). The molecular ion of the selenoxide SeAHO 3 (X = Se) m/z 449 (M + H)+ was observed by the LCQ-MS and MALDI techniques, and hydrated [-Se+(OH)-][-OH] and/or dihydroxyseleno -Se(OH)2- ion with an m/z 467 (M + H2O + H)+ was also seen by the softer MALDI ionization technique. The MS-MS fragmentations of the m/z 449 and 467 ions were distinct from the fragmentation of the base peak m/z 431 seen by the LC-MS, LCQ-MS, and MALDI techniques. Under the various ionization conditions, cyclic analogs and/or eliminations to give [-Se+=] species can account for the m/z 431. MALDI MS-MS of the m/z 431 ion gives the m/z 250 ion (5′-adenosyl cation) resulting from cleavage of the selenium-C5′ bond and also m/z 136 (adenine + H)+ ion.
3. Conclusions
Se-Adenosylselenohomocysteine selenoxide (3, X = Se; SeAHO) was synthesized from adenosine (6) by a method that did not require any extractions or column chromatography. Selenoxide SeAHO 3 (X = Se) was stable in buffered aqueous environments with no evidence of glycosidic hydrolysis or electrocyclic eliminations over a wide (3-12) pH range at ambient temperature. This selenoxide 3 (X = Se; SeAHO) has not yet been characterized from biological samples, perhaps due to low abundance in cellular reducing environments and a weak molecular ion in the MS. Selenoxide SeAHO 3 (X = Se) is quite distinct from its sulfoxide 3 (X = S; SAHO) analog. Selenoxide SeAHO 3 (X = Se) is readily reduced by biological reductants glutathione (GSH) and cysteine (8, X = S) thiols, it undergoes hydration at the larger more polarizable selenium, and is racemized at the selenium center at low pH. The greater conformational flexibility of the selenoxide analog 3 (X = Se; SeAHO) was seen in the proton NMR chemical shift of the α-proton, likely due to electrostatic interactions of the amino acid moieties with the selenoxide functional group. Due to the close structural similarity to the sulfoxide 3 (X = S; SAHO) analog, the selenoxide SeAHO (3, X = Se) should also be an inhibitor and/or activator for S-adenosylmethionine-dependent methyltransferases, other enzymes, and proteins.[66]
4. Experimental
4.1. General data
TLC was carried out on plastic-backed silica gel 60, PE SIL G Whatman Plates, UV254. HPLC used an Agilent/Hewlett Packard Series 1100. NMR spectra were recorded on a Bruker 700 MHz spectrometer. IR spectra were recorded on a Bruker Alpha-P FT IR with OPUS software. Mass spectrometry instrumentation used included: 1. LC-MS: Waters LCT Premier ToF MS; 2. LCQ-MS: Finnigan LCQ ESI ion trap MS; 3. MALDI-MS: AB SCIEX 5800 TOF/TOF; and 4. HRMS: Waters Q-TOF Ultima ESI.
4.2. Syntheses
4.2.1. Synthesis of Se-adenosyl-L-selenohomocysteine (2, X = Se; SeAH; CAS 4053-91-2)[8,9,58-60]
Se-Adenosyl-L-selenohomocysteine (2, X = Se; SeAH) was prepared by a variation of previous literature via L-selenohomocysteine (5, X = Se, CAS 29475-60-3).[59,67] Ammonia (30 mL) was added by condensation using a dry ice condenser from a cylinder of anhydrous ammonia to a round bottom flask that was prepared with inert atmosphere and L-selenomethionine (4, X = Se, Chem-Impex Int'l, CAS 3211-76-5) (700 mg, 3.57 mmol) in a -80°C dry ice/acetone bath. This reaction was stirred magnetically and small pieces of sodium metal were added until the reaction remained blue (260 mg, 11.3 mmol). The reaction was stirred in darkness for an additional 1 hour, and then ammonium chloride (590 mg, 11.0 mmol) was added slowly to neutralize any sodium amide present. The reaction was then removed from the dry ice/acetone both and nitrogen was blown over the stirred mixture to remove solvent. The residue dried under vacuum to give L-selenohomocysteine (5, X = Se)[39,59,60] as a white solid that was dissolved in water (4 mL). To this solution was added 5′-chloro-5′-deoxyadenosine[61,68] (9, 5′-Cl-5′-dA, CAS 892-48-8, 926 mg, 3.25 mmol). To this mixture was then added 2.5 mL of a 10% aqueous sodium hydroxide solution and an additional 5 mL of water. This reaction mixture was stirred magnetically for 1.5 hours at 80°C. The solution remained cloudy for 15 minutes before becoming homogeneous. Acetic acid (∼1.25 mL) was then added dropwise until the solution pH was between 5 and 6. Solvent was removed under vacuum to give a yellowish white solid. This solid was dissolved in an ethanol/toluene solution, the solvents removed by rotary evaporation, and the residue dried under vacuum to remove all moisture. Two hot filtration/recrystallizations from methanol gave 330 mg (0.764 mmol, 24% yield) of SeAH 2 (X = Se) as a grey amorphous solid. Trace amounts of residual methanol were removed by dissolving SeAH 2 (X = Se) at a concentration of 5 mg/mL in deionized water (pH 6), freezing with dry ice, and lyophylization to give a white solid. MP 204-205 °C. TLC 12:1:3 isopropanol:water:acetic acid: 5′-Cl-5′-dA 9 Rf 0.80, selenomethionine (4, X = Se) Rf 0.73, and SeAH 2 (X = Se) Rf 0.44. TLC 12:5:3 isopropanol:water:acetic acid: SeAH 2 (X = Se) (Rf 0.72). diode array detector UV spectrum (DAD) λmax of 258 nm. 1H NMR (700 MHz, CD3CO2D) δ 2.22-2.30 (m, 1 H, βb), 2.32-2.39 (m, 1 H, βa), 2.74-2.84 (m, 2 H, γ), 3.03 (dd, J = 13.2, 6.2 Hz, 1 H, 5b′), 3.05 (dd, J = 13.2, 5.7 Hz, 1 H, 5a′), 4.16 (m, 1 H, α), 4.38 (ddd, J = 4.3, 5.7, 6.2 Hz, 1 H, 4′), 4.49 (dd, J = 4.3, 5.4 Hz, 1 H, 3′), 4.86 (dd, J = 4.4, 5.4 Hz, 1 H, 2′), 6.16 (d, J = 4.4 Hz, 1 H, 1′), 8.44 (s, 1 H), 8.50 (s, 1 H, Ar). IR: 1670 (sh), 1638, 1598, 1577 cm-1. LC-MS (M + H)+ calc'd for C14H21N6O5Se m/z 433.07 (Lit.[2,69]), observed 433.01, 297.96 (loss of adenine), 181.90 (loss of adenosine), 136.00 (adenine cation). LCQ MS m/z 433.0. LCQ MS-MS of 433.0: m/z 298.07 (loss of adenine), 182.27 (loss of adenosine), 136.25 (adenine cation). MALDI MS 433.10 (M + H)+, 250.09 (5′-adenosyl cation).
4.2.2. Synthesis of Se-adenosylselenohomocysteine selenoxide (3, X = Se; SeAHO)
Se-Adenosylselenohomocysteine (2, X = Se; SeAH) (6 mg, 1.4 × 10-5 mol) in a 4 mL vial was suspended in acetic acid (0.2 mL) and stirred magnetically. Excess 30% hydrogen peroxide solution (65 μL) was added, the suspended grey solid became soluble giving a homogeneous, clear, and colorless solution within 5 min. TLC was used to monitor the reaction and showed almost instantaneous complete formation of the oxidation product 3 (X = Se; SeAHO). After 10 min, the reaction mixture was frozen by shell freezing with dry ice and the solvent removed by vac transfer into a clean trap over an hour to give a quantitative yield of Se adenosylselenohomocysteine selenoxide (3, X = Se; SeAHO) as a grey solid. MP 115-120 °C (dec.). TLC 12:1:3 isopropanol:water:acetic acid: SeAHO 3 (X = Se) (Rf 0.27). TLC 12:5:3 isopropanol:water:acetic acid: SeAHO 3 (X = Se) (Rf 0.45). diode array detector UV spectrum (DAD) λmax of 254 nm. 1H NMR (700 MHz, 97:3 CD3CO2D/30% aqueous H2O2) δ 2.60-2.69 (m, 1 H, βb), 2.74-2.83 (m, 1 H, βa), 3.65 (br d, J = 12.0 Hz, 1 H, 5b′), 3.84-3.95 (m, 3 H, 5a′, γ), 4.58-4.67 (m, 2 H, 4′, α), 4.72 (dd, J = 5.9, 4.7 Hz, 1 H, 3′), 4.94 (dd, J = 4.7, 3.8 Hz, 1 H, 2′), 6.20 (d, J = 3.8 Hz, 1 H, 1′), 8.45 (s, 1 H), 8.48 (s, 1 H, Ar). 13C NMR (700 MHz, 97:3 CD3CO2D/30% aqueous H2O2) δ 32.9 (β), 44.6 (γ), 49.1 (5′), 73.9 (3′), 74.5 (2′), 80.7 (4′, α), 91.0 (1′), 142.7 (Ar), 149 (Ar) (the carbon assignments of 2′, 3′, 4′, and α from the HSQC are tentative due to hydrolysis of the C1′-adenine bond during the course of the experiment). IR: 1777, 1688 (sh), 1644, 1598, 1575, 878, 847 cm-1. LC-MS (M - OH)+ calc'd for C14H19N6O5Se+ m/z 431.06, observed 431.01, 250.02 (5′-adenosyl cation), 136.0 (adenine cation). LCQ MS m/z 449.0 (M + H)+, 431.0 (M - OH)+. LCQ MS-MS of 449.0: m/z 431.0 (M - 18)+. LCQ MS-MS of 431.0: m/z 250.1 (5′-adenosyl cation), 136.0 (adenine cation). MALDI MS m/z 467.0 (M + H2O + H)+, 449.1 (M + H)+, 431.1 (M - OH)+, 250.1 (5′-adenosyl cation). MALDI MS-MS of 431.1: m/z 250.2 (5′-adenosyl cation), 136.1 (adenine cation). HRMS (ESI+): Calculated for C14H19N6O6Se M+: 447.0531, found 447.0548. [α]D24 +140° (c, 3.0, 97:3 CH3CO2H/30% aqueous H2O2).
Supplementary Material
Acknowledgments
The authors are grateful to Wanlu Qu, Shanshan Liu, and William Devine for assistance with mass spectrometry, and to Jason Guo and Roger Kautz for assistance with NMR. We gratefully acknowledge the general assistance of Tianzhu “Indi” Zang, Kun Zhang, and Michael Pollastri.
Funding: This work was supported by NIH NIGMS under grant 1R01GM101396 to Z.S.Z.
Footnotes
Supplementary data: Supplementary data associated with this article, including characterizations of compounds 2 (X = S), and 3 (X = S), and 9 can be found in the online version, at http://.
Contributor Information
Richard I. Duclos, Jr., Email: r.duclos@neu.edu.
Dillon C. Cleary, Email: cleary.di@husky.neu.edu.
Kalli C. Catcott, Email: catcott.k@husky.neu.edu.
References
- 1.Mudd SH, Cantoni GL. Selenomethionine in enzymatic transmethylations. Nature. 1957;180:1052. doi: 10.1038/1801052a0. [DOI] [PubMed] [Google Scholar]
- 2.Wrobel K, Wrobel K, Caruso JA. Selenium speciation in low molecular weight fraction of Se-enriched yeasts by HPLC-ICP-MS: detection of selenoadenosylmethionine. J Anal At Spectrom. 2003;17:1048–54. [Google Scholar]
- 3.Thomson CD. Assessment of requirements for selenium and adequacy of selenium status: a review. Eur J Clin Nutr. 2004;58:391–402. doi: 10.1038/sj.ejcn.1601800. [DOI] [PubMed] [Google Scholar]
- 4.Tinggi U. Selenium toxicity and its adverse health effects. Rev Food Nutr Tox. 2005;4:29–55. [Google Scholar]
- 5.Reilly C. Selenium in Food and Health. 2nd. New York: Springer; 2006. [Google Scholar]
- 6.Gammelgaard B, Gabel-Jensen C, Sturup S, Hansen HR. Complementary use of molecular and element-specific mass spectrometry for identification of selenium compounds related to human selenium metabolism. Anal Bioanal Chem. 2008;390:1691–706. doi: 10.1007/s00216-007-1788-8. [DOI] [PubMed] [Google Scholar]
- 7.Lenz M, Lens PNL. The essential toxin: the changing perception of selenium in environmental sciences. Sci Total Environ. 2009;407:3620–33. doi: 10.1016/j.scitotenv.2008.07.056. [DOI] [PubMed] [Google Scholar]
- 8.Ogra Y, Kitaguchi T, Ishiwata K, Suzuki N, Toida T, Suzuki KT. Speciation of selenomethionine metabolites in wheat germ extract. Metallomics. 2009;1:78–86. [Google Scholar]
- 9.Rao Y, McCooeye M, Windust A, Bramanti E, D'Ulivo A, Mester Z. Mapping of selenium metabolic pathway in yeast by liquid chromatography-orbitrap mass spectrometry. Anal Chem. 2010;82:8121–30. doi: 10.1021/ac1011798. [DOI] [PubMed] [Google Scholar]
- 10.Rayman MP. Selenium and human health. Lancet. 2012;379:1256–68. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 11.Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39:112–20. doi: 10.1016/j.tibs.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.B'Hymer C, Caruso JA. Selenium speciation analysis using inductively coupled plasma-mass spectrometry. J Chromatogr A. 2006;1114:1–20. doi: 10.1016/j.chroma.2006.02.063. [DOI] [PubMed] [Google Scholar]
- 13.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–37. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 14.Loenen WAM. S-adenosylmethionine: jack of all trades and master of everything? Biochem Soc Trans. 2006;34:330–3. doi: 10.1042/BST20060330. [DOI] [PubMed] [Google Scholar]
- 15.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, McCann PP. S-Adenosylmethionine and methylation. FASEB J. 1996;10:471–80. [PubMed] [Google Scholar]
- 16.Struck AW, Thompson ML, Wong LS, Micklefield J. S-Adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem. 2012;13:2642–55. doi: 10.1002/cbic.201200556. [DOI] [PubMed] [Google Scholar]
- 17.Stolowitz ML, Minch MJ. S-Adenosyl-L-methionone and S-Adenosyl-L-homocysteine, an NMR study. J Am Chem Soc. 1981;103:6015–9. [Google Scholar]
- 18.Duerre JA. Preparation and properties of S-adenosyl-L-homocysteine, S-adenosyl-L-homocysteine sulfoxide and S-ribosyl-L-homocysteine. Arch Biochem Biophys. 1962;96:70–6. doi: 10.1016/0003-9861(62)90453-8. [DOI] [PubMed] [Google Scholar]
- 19.Biastoff S, Teuber M, Zhou ZS, Drager B. Colorimetric activity measurement of a recombinant putrescine N-methyltransferase from Datura stramonium. Planta Med. 2006;72:1136–41. doi: 10.1055/s-2006-947191. [DOI] [PubMed] [Google Scholar]
- 20.Duerre JA, Salisbury L, Miller CH. Preparation and characterization of sulfoxides of S-adenosyl-L-homocysteine and S-ribosyl-L-homocysteine. Anal Biochem. 1970;35:505–15. doi: 10.1016/0003-2697(70)90213-7. [DOI] [PubMed] [Google Scholar]
- 21.Rekunova VN, Rudakova IP, Yurkevich AM. Synthesis of potential inhibitors of transmethylases. Tetrahedron Let. 1973;14:3811–4. [Google Scholar]
- 22.Borchardt RT, Wu YS. Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 1. modification of the amino acid portion of S-adenosylhomocysteine. J Med Chem. 1974;17:862–8. doi: 10.1021/jm00254a016. [DOI] [PubMed] [Google Scholar]
- 23.Guerard C, Breard M, Courtois F, Drujon T, Ploux O. Synthesis and evaluation of analogues of S-adenosyl-L-methionine, as inhibitors of the E. coli cyclopropane fatty acid synthase. Bioorg Med Chem Lett. 2004;14:1661–4. doi: 10.1016/j.bmcl.2004.01.051. [DOI] [PubMed] [Google Scholar]
- 24.Hickey SF, Hammond MC. Structure-guided design of fluorescent S-adenosylmethionine analogs for a high-throughput screen to target SAM-I riboswitch RNAs. Chem Biol. 2014;21:345–56. doi: 10.1016/j.chembiol.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coward JK, D'Urso-Scott M, Sweet WD. Inhibition of catechol-O-methyltransferase by S-adenosylhomocysteine and S-adenosylhomocysteine sulfoxide, a potential transition-state analog. Biochem Pharmacol. 1972;21:1200–3. doi: 10.1016/0006-2952(72)90114-1. [DOI] [PubMed] [Google Scholar]
- 26.Gillet L, Looze Y, Deconinck M, Leonis J. Binding capacities of various analogues of S-adenosyl-L-homocysteine to protein methyltransferase II from human erythrocytes. Experientia. 1979;35:1007–9. doi: 10.1007/BF01949909. [DOI] [PubMed] [Google Scholar]
- 27.Pugh CS, Borchardt RT. Effects of S-adenosylhomocysteine analogues on vaccinia viral messenger ribonucleic acid synthesis and methylation. Biochemistry. 1982;21:1535–41. doi: 10.1021/bi00536a011. [DOI] [PubMed] [Google Scholar]
- 28.Zainal HA, Wolf WR, Waters RM. An NMR spectroscopic investigation of the oxidation reactions of DL-selenomethionine. J Chem Technol Biotechnol. 1998;72:38–44. [Google Scholar]
- 29.Schoneich C. Redox processes of methionine relevant to β-amyloid oxidation and Alzheimer's disease. Arch Biochem Biophys. 2002;397:370–6. doi: 10.1006/abbi.2001.2621. [DOI] [PubMed] [Google Scholar]
- 30.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–58. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 31.Block E, Birringer M, Jiang W, Nakahodo T, Thompson HJ, Toscano PJ, Uzar H, Zhang X, Zhu Z. Allium chemistry: synthesis, natural occurrence, biological activity, and chemistry of Se-alk(en)ylselenocysteines and their γ-glutamyl derivatives and oxidation products. J Agric Food Chem. 2001;49:458–70. doi: 10.1021/jf001097b. [DOI] [PubMed] [Google Scholar]
- 32.Ritchey JA, Davis BM, Pleban PA, Bayse CA. Experimental and theoretical evidence for cyclic selenurane formation during selenomethionine oxidation. Org Biomol Chem. 2005;3:4337–42. doi: 10.1039/b513238j. [DOI] [PubMed] [Google Scholar]
- 33.Isab AA. A 1H NMR Study of the Reaction of Gold(III) with DL-Seleno-Methionine in Aqueous Solution. Inorgan Chim Acta. 1983;80:L3–L4. [Google Scholar]
- 34.Davis FA, Reddy RT. Asymmetric Oxidation of Simple Selenides to Selenoxides in High Enantiopurity - Stereochemical Aspects of the Allyl Selenoxide / Allyl Selenenate Rearrangement. J Org Chem. 1992;57:2599–606. [Google Scholar]
- 35.Kurose N, Takahashi T, Koizumi T. First synthesis of optically pure selenuranes and stereoselective alkaline hydrolysis. their application to asymmetric [2,3] sigmatropic rearrangements of allylic selenoxides. Tetrahedron. 1997;53:12115–29. [Google Scholar]
- 36.Uden PC, Bird SM, Kotrebai M, Nolibos P, Tyson JF, Block E, Denoyer E. Analytical selenoamino acid studies by chromatography with interfaced atomic mass spectrometry and atomic emission spectral detection. Fresenius J Analyt Chem. 1998;362:447–56. [Google Scholar]
- 37.Gammelgaard B, Cornett C, Olsen J, Bendahl L, Hansen SH. Combination of LC-ICP-MS, LC-MS and NMR for investigation of the oxidative degradation of selenomethionine. Talanta. 2003;59:1165–71. doi: 10.1016/S0039-9140(03)00026-2. [DOI] [PubMed] [Google Scholar]
- 38.Cooper AJL, Meister A. Enzymatic oxidation of L-homocysteine. Arch Biochem Biophys. 1985;239:556–66. doi: 10.1016/0003-9861(85)90725-8. [DOI] [PubMed] [Google Scholar]
- 39.Zhou ZS, Smith AE, Matthews RG. L-Selenohomocysteine: one-step synthesis from L-selenomethionine and kinetic analysis as substrate for methionine synthases. Bioorg Med Chem Lett. 2000;10:2471–5. doi: 10.1016/s0960-894x(00)00498-4. [DOI] [PubMed] [Google Scholar]
- 40.Banica A, Culetu A, Banica FG. Electrochemical and EQCM investigation of L-selenomethionine in adsorbed state at gold electrodes. J Electroanalyt Chem. 2007;599:100–10. [Google Scholar]
- 41.Harman LS, Mottley C, Mason RP. Free radical metabolites of L-cysteine oxidation. J Biol Chem. 1984;259:5606–11. [PubMed] [Google Scholar]
- 42.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. 2008;12:746–54. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 43.Devarie-Baez NO, Zhang D, Li S, Whorton AR, Xian M. Direct methods for detection of protein S-nitrosylation. Methods. 2013;62:171–6. doi: 10.1016/j.ymeth.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasaki E, Zhang X, Sun HG, Lu MYJ, Liu Tl, Ou A, Li Jy, Chen Yh, Ealick SE, Liu Hw. Co-opting sulphur-carrier proteins from primary metabolic pathways for 2-thiosugar biosynthesis. Nature. 2014;509:427–31. doi: 10.1038/nature13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwaoka M, Arai K. From sulfur to selenium. A new research area in chemical biology and biological chemistry. Curr Chem Biol. 2013;7:2–24. [Google Scholar]
- 46.Zhou ZS, Flohr A, Hilvert D. An antibody-catalyzed allylic sulfoxide-sulfenate rearrangement. J Org Chem. 1999;64:8334–41. doi: 10.1021/jo991299a. [DOI] [PubMed] [Google Scholar]
- 47.Zhou ZS, Peariso K, Penner-Hahn JE, Matthews RG. Identification of the zinc ligands in cobalamin-independent methionine synthase (MetE) from Escherichia coli. Biochemistry. 1999;38:15915–26. doi: 10.1021/bi992062b. [DOI] [PubMed] [Google Scholar]
- 48.Matthews RG, Smith AE, Zhou ZS, Taurog RE, Bandarian V, Evans JC, Ludwig M. Cobalamin-dependent and cobalamin-independent methionine synthases: are there two solutions to the same chemical problem? Helv Chim Acta. 2003;86:3939–54. [Google Scholar]
- 49.Mosley SL, Bakke BA, Sadler JM, Sunkara NK, Dorgan KM, Zhou ZS, Seley-Radtke KL. Carbocyclic pyrimidine nucleosides as inhibitors of S-adenosylhomocysteine hydrolase. Bioorg Med Chem Lett. 2006;14:7967–71. doi: 10.1016/j.bmc.2006.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zang T, Dai S, Chen D, Lee BWK, Liu S, Karger BL, Zhou ZS. Chemical methods for the detection of protein N-homocysteinylation via selective reactions with aldehydes. Anal Chem. 2009;81:9065–71. doi: 10.1021/ac9017132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Rejtar T, Zhou ZS, Karger BL. Desulfurization of cysteine-containing peptides resulting from sample preparation for protein characterization by mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:267–75. doi: 10.1002/rcm.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou ZS, Jiang N, Hilvert D. An antibody-catalyzed selenoxide elimination. J Am Chem Soc. 1997;119:3623–4. [Google Scholar]
- 53.Alfaro JF, Gillies LA, Sun HG, Dai S, Zang T, Klaene JJ, Kim BJ, Lowenson JD, Clarke SG, Karger BL, Zhou ZS. Chemo-enzymatic detection of protein isoaspartate using protein isoaspartate methyltransferase and hydrazine trapping. Anal Chem. 2008;80:3882–9. doi: 10.1021/ac800251q. [DOI] [PubMed] [Google Scholar]
- 54.Chen T, Nayak N, Majee SM, Lowenson J, Schafermeyer KR, Eliopoulos AC, Lloyd TD, Dinkins R, Perry SE, Forsthoefel NR, Clarke SG, Vernon DM, Zhou ZS, Rejtar T, Downie AB. Substrates of the Arabidopsis thaliana protein isoaspartyl methyltransferase 1 identified using phage display and biopanning. J Biol Chem. 2010;285:37281–92. doi: 10.1074/jbc.M110.157008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu M, Cheetham J, Cauchon N, Ostovic J, Ni W, Ren D, Zhou ZS. Protein isoaspartate methyltransferase-mediated 18O-labeling of isoaspartic acid for mass spectrometry analysis. Anal Chem. 2012;84:1056–62. doi: 10.1021/ac202652z. [DOI] [PubMed] [Google Scholar]
- 56.Gui S, Wooderchak-Donahue WL, Zang T, Chen D, Daly MP, Zhou ZS, Hevel JM. Substrate-induced control of product formation by protein arginine methyltransferase 1. Biochemistry. 2013;52:199–209. doi: 10.1021/bi301283t. [DOI] [PubMed] [Google Scholar]
- 57.De Silva V, Woznichak MM, Burns KL, Grant KB, May SW. Selenium redox cycling in the protective effects of organoselenides against oxidant-induced DNA damage. J Am Chem Soc. 2004;126:2409–13. doi: 10.1021/ja037294j. [DOI] [PubMed] [Google Scholar]
- 58.Iwig DF, Booker SJ. Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-L-methionine. Biochemistry. 2004;43:13496–509. doi: 10.1021/bi048693+. [DOI] [PubMed] [Google Scholar]
- 59.Willnow S, Martin M, Luscher B, Weinhold E. A selenium-based click AdoMet analogue for versatile substrate labeling with wild-type protein methyltransferases. ChemBioChem. 2012;13:1167–73. doi: 10.1002/cbic.201100781. [DOI] [PubMed] [Google Scholar]
- 60.Bothwell IR, Luo M. Large-Scale, Protection-Free Synthesis of Se-Adenosyl-L-selenomethionine Analogues and Their Application as Cofactor Surrogates of Methyltransferases. Org Lett. 2014;16:3056–9. doi: 10.1021/ol501169y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scovill JP, Thigpen DL, II, Lemley PV. A convenient method for the synthesis and raney nickel desulfurization of 5′-deoxy-5′-methylthioadenosine. Phosphorus, Sulfur, and Silicon. 1993;85:149–52. [Google Scholar]
- 62.Orlov IG, Markin VS, Moiseev YV, Khurgin UI. Infrared spectra of amino acids and peptides. Chem Nat Cpds. 1967;3:163–6. [Google Scholar]
- 63.Paetzold R, Lindner U, Bochmann G, Reich P. Dimethyl- und diathylselenoxide sowie ihre oxoniumsalze. darstellung, eigenschaften und schwingungsspektren. Zeits Anorgan Allge Chemie. 1967;352:295–308. [Google Scholar]
- 64.Wallace TJ, Mahon JJ. Reactions of thiols with sulfoxides. III. Catalysis by acids and bases. J Org Chem. 1965;30:1502–6. [Google Scholar]
- 65.Firouzabadi H, Jamalian A. Reduction of oxygenated organosulfur compounds. JSulfur Chem. 2008;29:53–97. [Google Scholar]
- 66.Zhao G, Wan W, Mansouri S, Alfaro JF, Bassler BL, Cornell KA, Zhou ZS. Chemical synthesis of S-ribosyl-L-homocysteine and activity assay as a LuxS substrate. Bioorg Med Chem Lett. 2003;13:3897–900. doi: 10.1016/j.bmcl.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 67.Ramalingam K, Woodard RW. A convenient synthesis of S-(5′-deoxy-5′-adenosyl)-(±)-2-methylhomocysteine. Tetrahedron Lett. 1985;26:1135–6. [Google Scholar]
- 68.Robins MJ, Hansske F, Wnuk SF, Kanai T. Nucleic acid related compounds. 66. improved syntheses of 5′-chloro-5′-deoxy- and 5′-S-aryl(or alkyl)-5′-thionucleosides. Can J Chem. 1991;69:1468–74. [Google Scholar]
- 69.Casiot C, Vacchina V, Chassaigne H, Szpunar J, Potin-Gautier M, Lobinski R. An approach to the identification of selenium species in yeast extracts using pneumatically-assisted electrospray tandem mass spectrometry. Anal Commun. 1999;36:77–80. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.