

Abstract
Therapeutic proteins are indispensable in treating numerous human diseases. However, therapeutic proteins often suffer short serum half-life. In order to extend the serum half-life, a natural albumin ligand (a fatty acid) has been conjugated to small therapeutic peptides resulting in a prolonged serum half-life via binding to patients' serum albumin in vivo. However, fatty acid-conjugation has limited applicability due to lack of site-specificity resulting in the heterogeneity of conjugated proteins and a significant loss in pharmaceutical activity. In order to address these issues, we exploited the site-specific fatty acid-conjugation to a permissive site of a protein, using copper-catalyzed alkyne-azide cycloaddition, by linking a fatty acid derivative to p-ethynylphenylalanine incorporated into a protein using an engineered pair of yeast tRNA/aminoacyl tRNA synthetase. As a proof-of-concept, we show that single palmitic acid conjugated to superfolder green fluorescent protein (sfGFP) in a site-specific manner enhanced a protein's albumin-binding in vitro about 20 times and the serum half-life in vivo 5 times when compared to those of the unmodified sfGFP. Furthermore, the fatty acid conjugation did not cause a significant reduction in the fluorescence of sfGFP. Therefore, these results clearly indicate that the site-specific fatty acid-conjugation is a very promising strategy to prolong protein serum half-life in vivo without compromising its folded structure and activity.
Keywords: Noncanonical amino acid, Fatty acid, Copper-catalyzed alkyne-azide cycloaddition, Albumin, Half-life
1. Introduction
Recombinant proteins with therapeutic activity have become critical for treating numerous diseases, and cover a wide range of therapeutics including monoclonal antibodies, hormones, growth factors, cytokines, and enzymes [1]. However, utility of therapeutic proteins is often hampered by their short serum half-life requiring frequent re-administration resulting in patient discomfort and noncompliance. Therefore, extending the serum half-life of therapeutic proteins will significantly enhance the utility of existing therapeutic proteins and will also enable development of new therapeutic proteins [2,3]. In the quest to extend the serum half-life, binding/conjugation of serum albumin or Fc portion of immunoglobulin G to therapeutic proteins is a very promising emerging strategy [4–6].
Human serum albumin (HSA) has an inherently long serum half-life (19 days) [6] due to neonatal Fc receptor (FcRn)-mediated recycling as well as reduced renal filtration [7,8]. HSA-binding/conjugation is a very attractive strategy for extending the serum half-life of a therapeutic protein when compared to conventional poly(ethylene)glycol (PEG) conjugation which mainly relies on renal filtration evasion. Furthermore, although it has long been considered that PEG is non-immunogenic, antibodies raised against PEG were observed in patients administered PEGylated uricase [9]. Therefore, the binding of therapeutic proteins to HSAs in patients' blood is actively investigated to mitigate most immune response issues. Despite the many benefits of HSA as a binding/conjugation partner, developing a general strategy to bind/conjugate therapeutic proteins to HSA remains a big challenge.
In order to facilitate HSA binding of therapeutic proteins in patients' blood, genetic fusion of an albumin binding domain to N-term or C-term of therapeutic proteins is performed. But this methodology has a potential risk of immunogenicity [10,11]. Furthermore, the end-to-end fusion does not provide steric control or favorable topology that retains both therapeutic efficacy and conformational stability [12–14]. Alternatively, synthetic or natural albumin-binding moieties have been chemically attached to a peptide, preferably to cysteine or lysine residues [15,16]. In particular, the conjugation of a natural HSA ligand, a fatty acid, has been successfully used to extend the serum half-life in vivo of two therapeutic peptides, insulin and glucagon-like peptide-1 agonist (GLP-1) via acylation at lysine residues [17,18]. Seven binding sites in HSA have been identified to accommodate saturated fatty acids with 10–18 carbons [19,20]. Compared to direct fusion/chemical conjugation of HSA to therapeutic proteins, this approach is advantageous for deep penetration into tissues [16], higher activity to mass ratio [21], and greatly reduced immunogenicity [22,23]. However, fatty acid-conjugation to multiple lysine residues of therapeutic proteins likely leads to heterogeneous mixtures of the conjugated proteins, compromising pharmaceutical activity and downstream processing. For instance, fatty acid-conjugation to lysine residues of interferon-alpha led to an 80% reduction in its antiviral potency [24]. Therefore, fatty acid-conjugation has been limited to peptides with a small number of lysine residues. In order to overcome the heterogeneity of the conjugated proteins and the compromised pharmaceutical activity, a fatty acid should be attached to a permissive site of a protein necessitating site-specific fatty acid-conjugation technique. Although therapeutic proteins are dominant over therapeutic peptides in clinical applications, to our knowledge, fatty acid-conjugation to a protein in a site-specific manner has not been reported. In this report, we describe a novel strategy to achieve site-specific conjugation of the natural albumin ligand, a fatty acid, to a protein with the combined use of copper-catalyzed alkyne-azide cycloaddition (CuAAC) and site-specific incorporation of noncanonical amino acid (NAA) technique. CuAAC is a popular reaction in which a terminal alkynyl group (HC ≡ C − R1) and an azido group (−N = N+ = N − R2) are united to give a 1,4-di-substituted 1,2,3-triazole in the presence of catalytic copper [25,26]. Its uniqueness lies in the bio-orthogonality of both moieties, since they are absent in all natural amino acids and thus ensure a highly selective reaction [27–29]. To employ CuAAC in protein engineering, amino acids containing either an alkynyl or azido group should be introduced into a protein. Among several techniques for expanding the chemical diversity of proteins [30–34], the site-specific genetic incorporation of NAAs is capable of adding new chemistries at a desired site. An orthogonal pair of tRNA amber suppressor and aminoacyl-tRNA synthetase from foreign species needs to be engineered to be specific for each NAA and utilized to incorporate it in response to an amber codon in the target protein sequence [35,36]. In order to achieve site-specific fatty acid-conjugation to a protein, p-ethynylphenylalanine (pEthF) was introduced to a model protein, superfolder green fluorescent protein (sfGFP), using the bacterial cells outfitted with the orthogonal pair of engineered yeast phenylalanyl-tRNA/phenylalanyl-tRNA synthetase. The sfGFP was chosen as a model protein thanks to its favorable properties for our study. First, its fluorescence is directly correlated to its folding [37]. Therefore, perturbation of its folded structure upon fatty acid-conjugation can be estimated by measuring its fluorescence. Second, its spectral properties greatly facilitate quantitative analyses in vitro including HSA binding assay. Third, the family of green fluorescent protein variants is generally known to be non-toxic to animals facilitating pharmacokinetics testing in vivo [38,39]. The sfGFP variant containing pEthF was coupled to a fatty acid derivative containing an azido group via CuAAC. Finally, using the fatty acid-conjugated sfGFP, we show that the site-specific fatty acid-conjugation to a protein enhances its binding to HSA in vitro and prolongs protein retention in blood when administered in vivo without any significant loss in its intrinsic folded structure and fluorescence.
2. Material and methods
2.1. Materials
p-Ethynylphenylalanine (pEthF) was synthesized as described previously [40]. Ni-NTA agarose and pQE-16 plasmid were obtained from Qiagen (Valencia, CA). Sequencing grade modified trypsin was obtained from Promega Corporation (Madison, WI). Amicon ultra centrifugal filters and ZipTip® with C18 media were purchased from Millipore Corporation (Billerica, MA). NHS-activated agarose and BCA protein assay kit were purchased from Thermo Scientific (Rockford, IL). GFP ELISA kit was purchased from Cell Biolabs, Inc. (San Diego, CA). Coumarin-azide was obtained from Glen Research (Sterling, VA). Palmitic acid-azide was obtained from Invitrogen Corporation (Carlsbad, CA). All other chemicals were purchased from Sigma-Aldrich Corporation (St. Louis, MO).
2.2. Plasmid construction and strains
Preparation of the plasmids pQE16am-yPheRST415A and pREP4-ytRNAPheCUA_UG is described previously [41]. pQE16am-yPheRST415G encodes the engineered yeast aminoacyl-tRNA synthetase and the murine dihydrofolate reductase (mDHFR) with an amber codon at 38th position and a C-terminal hexahistidine tag. A Phe/Trp/Lys triple auxotrophic Escherichia coli strain, designated AFWK, was prepared as described previously [41]. AFWK harboring both plasmids was used as an expression host for incorporation of pEthF into murine dihyrofolate reductase (mDHFR). A gene encoding a sfGFP with a C-terminal hexahistidine tag was synthesized from Epoch Life Science, Inc. (Sugar Land, Texas). The expression cassette of the sfGFP was inserted into the AatII/NheI site in pQE16am-yPheRST415A replacing the coding sequence of the mDHFR and yielding pQE16-sfGFP-yPheRST415A. An amber codon was generated by PCR mutagenesis in a position between the 214th and the 215th amino acid of the sfGFP resulting in pQE16-sfGFP215Amb-yPheRST415A. The mutagenic primer sequences were as follows: 215Amb_F, 5′-CCCAACGAAAAGTAGCGTGA CCACATGG-3′; 215Amb_R, 5′-CCATGTGGTCACGCTACTTTTCGTTGGG-3′. AFWK was co-transformed with pQE16-sfGFP215Amb-yPheRST415A and pREP4-ytRNAPheCUA_UG and then used as an expression host for incorporation of pEthF into the sfGFP.
2.3. Expression and purification of wild-type and mutant proteins
The wild-type sfGFP (sfGFP-WT) was expressed from AFWK harboring pQE16-sfGFP-yPheRST415GA by 1 mM IPTG induction in LB media containing 100 μg/mL ampicillin at 37 °C. To express the sfGFP mutant containing pEthF at the 215th position (sfGFP-pEthF), AFWK harboring pQE16-sfGFP215Amb-yPheRST415A and pREP4-ytRNAPheCUA_UG was used. Saturated overnight cultures grown at 37°C in M9 minimal medium supplemented with 100 μg/mL ampicillin, 30 μg/mL kanamycin, 0.4% (w/v) glucose, 1 mM MgSO4, 0.1 mM CaCl2, 10 μg/mL thiamine, and 20 amino acids (25 μg/mL each) were diluted 20 fold in the same fresh medium, and grown at 37 °C until an OD600 of 0.9 was reached. After incubation on ice for 15 min, cells were sedimented by centrifugation at 4000 g for 12 min, and washed with cold 0.9% (w/v) NaCl by gentle resuspension. After repeating twice, cells were shifted to M9 medium supplemented with the same ingredients described above except for different amino acid compositions: 17 amino acids (35 μg/mL each), 150 μM Lys, 60 μM Phe, 10 μM Trp, and 3 mM pEthF. To maximize the incorporation efficiency in condensed culture, the total volume of M9 medium was 20-fold smaller than the original volume. Upon induction by 1 mM IPTG, cells were incubated with shaking at 30 °C for 15 h before harvest. Expression of the wild-type mDHFR (mDHFR-WT) or the mDHFR mutant with pEthF (mDHFR-pEthF) at the 38th position was performed similarly except that the plasmid pQE16 for mDHFR-WT or pQE16am-yPheRST415GA for mDHFR-pEthF was used instead of pQE16-sfGFP215Amb-yPheRST415A. Cells were pelleted by centrifugation, and the protein was purified by gravity-flow affinity chromatography using Ni-NTA agarose beads under native (sfGFP-WT and sfGFP-pEthF) or denaturing (mDHFR-WT and mDHFR-pEthF) condition according to the supplier's instructions (Qiagen). Purified proteins were directly used or buffer-exchanged using PD-10 desalting columns to appropriate buffers. If necessary, the protein solutions were concentrated using centrifugal filters.
2.4. CuAAC-mediated dye labeling and fatty acid-conjugation
Palmitic acid-azide was reacted with the mDHFR-pEthF under the following condition yielding the mDHFR-Pal: 30 μM mDHFR-pEthF in the denaturing elution buffer (8 M urea, 10 mM Tris, 100 mM NaH2PO4, pH = 4.5), 150 μM palmitic acid-azide, 1 mM CuSO4, 2 mM sodium ascorbate, and at room temperature for 2 h. sfGFP-pEthF was conjugated to palmitic acid-azide or fluorogenic coumarin-azide under the following condition generating sfGFP-Pal or sfGFP-CM: 30 μM sfGFP-pEthF in 20 mM potassium phosphate (pH = 8) and 35% (v/v) DMSO, 150 μM palmitic acid-azide or coumarin-azide, 1.0 mM TBTA, 1.5 mM CuSO4, 2.0 mM DTT, and at 25 °C for 10 h. Reactions were quenched by adding 200 mM imidazole and 5 mM EDTA. Upon completion of reaction, the reaction mixture was desalted and buffer-exchanged using PD-10 desalting columns to appropriate buffers for downstream uses. Protein concentrations were determined by BCA assay.
2.5. Verification of pEthF incorporation and fatty acid-conjugation by mass spectrometry
Tryptic digestion of the mDHFR-WT, the mDHFR-pEthF or the mDHFR-Pal in the denaturing elution buffer (8 M urea, 10 mM Tris, 100 mM NaH2PO4, pH = 4.5) was performed by diluting 10 μL of a protein with 90 μL of NH4HCO3 and then adding 0.5 μL of modified trypsin (0.1 μg). Following incubation at 37 °C for 2 h, the reaction mixture was mixed with 12 μL of 5% (v/v) trifluoroacetic acid (TFA) to quench the reaction and then desalted on a ZipTip® C18. The site-specific incorporation of pEthF into mDHFR and palmitic acid-conjugation was confirmed by MALDI-TOF mass spectrometry (MS) analysis of the tryptic digests of mDHFR. The MS analysis was performed using 20 mg/mL of 2,5-dihydroxybenzoic acid and 2 mg/mL of l-(−)-fucose dissolved in 10% ethanol as a matrix by Microflex™ MALDI-TOF MS (Bruker Corporation, Billerica, MA). LC–MS/MS analyses of tryptic digests of mDHFR were conducted on a thermo electron LTQ VelosOrbitrap mass spectrometer. The tryptic digests of mDHFR were separated on a reverse phase column (75 μm) with acetonitrile gradient. The column eluent was introduced to the microspray source, and amino acid sequence analysis was carried out by fragmentation of the precursor ion corresponding to the Peptide_Z38. The site-specific incorporation of pEthF into sfGFP and palmitic acid-conjugation were confirmed by LC–MS coupled with electron spray ionization (ESI). The chromatographic separation was performed using a BEH C4 (2.1 × 100 mm, 1.7 μm) column at a flow rate of 0.4 μL/min with mobile phase consisting of water and n-propanol. The eluent was introduced into the ion source of the LTQ-Orbitrap mass spectrometer operated in a positive mode at a spray voltage of 3.0 kV. The data were acquired by XCalibur (Thermo Scientific) and processed using ProMass deconvolution (Thermo Scientific).
2.6. In vitro albumin-binding assay
N-hydroxysuccinimide-activated agarose was coated with HSA according to the supplier's protocol or inactivated by adding an excess amount of glycine to generate HSA-coated and inactivated resin, respectively. The resins were mixed with the sfGFP-WT, the sfGFP-pEthF, or the sfGFP-Pal and incubated at room temperature for 1 h. After washing with PBS multiple times, the fluorescence images and intensities of the resins were obtained at λex = 480 nm and λem = 510 nm using the Biospectrum imaging system (UVP Inc., Upland, CA) and the Biotek fluorescence plate reader, respectively. For membrane-based binding assay, the HSA solution (10 mg/mL) was spotted on the nitrocellulose membrane. After extensive washing with PBS, the membrane was blocked with casein solution. Two microliters of protein solutions (2 mg/mL) was overlaid on the HSA spot, and the membrane was washed with PBS and analyzed by the imaging system.
2.7. In vivo studies of the sfGFP-WT and sfGFP-Pal
The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Virginia. Pharmacokinetic properties of sfGFP-WT and sfGFP-Pal were investigated by injecting 50 μg of each sfGFP sample in 200 μL PBS into the tail vein of young female C57BL/6 mice (n = 4). The blood was sampled at 0 (10 min), 3, and 6 h post-injection for the sfGFP-WT, and at 0 (10 min), 3, 6, 18, 24, and 30 h post-injection for the sfGFP-Pal.
3. Results
3.1. Site-specific incorporation of p-ethynylphenylalanine into the murine dihydrofolate reductase and the CuAAC-mediated dye labeling
In order to investigate site-specific fatty acid-conjugation to a protein via CuAAC (Fig. 1A), we introduced pEthF into murine dihydrofolate reductase (mDHFR) in a site-specific manner. Since the expression and purification of DHFRs in E. coli are well established [41,42], we routinely used mDHFR to study site-specific incorporation of a NAA. pEthF is a phenylalanine analog with an alkyne moiety at para-position of the phenyl ring (Fig. 1B) and expected to act as a molecular handle for CuAAC with azide-functionalized molecules. Previously the yeast-originated pair of phenylalanine-tRNA suppressor/phenylalanyl-tRNA synthetase variant (ytRNAPheCUA_UG/yPheRST415A) was designed to incorporate a Phe analog into mDHFR in E. coli expression system [41,43]. The relaxed substrate specificity of yPheRST415A also allows recognition of a panel of Phe and Trp analogs with a bulky functional group at para-position of the phenyl ring. Therefore, we hypothesize that the ytRNAPheCUA_UG/yPheRST415A pair will also allow efficient site-specific incorporation of pEthF in response to an amber codon at the 38th position of the mDHFR mutant (mDHFR-38Am). The mDHFR-38Am was expressed in the presence of 3 mM pEthF, purified under denaturing condition, and trypsin-digested for MALDI-TOF analysis as described previously with minor alterations [41,42]. The mDHFR mutant containing pEthF at the 38th position is designated as mDHFR-pEthF. For wild-type mDHFR (mDHFR-WT), pep-tide F38 (residues 26-39; NGDLPWPPLRNEAFK), one of the tryptic digests, was detected with a monoisotopic mass of 1682.7 Da, in accord with its theoretical mass (Fig. 2A). Peptide Z38 of the mDHFR-pEthF (residue 26–39; NGDLPWPPLRNEAmK where Am indicates an amber codon) was detected with a strong signal at a mass of 1706.8 Da, supporting the incorporation of pEthF in response to the amber codon. Furthermore, liquid chromatography–tandem mass spectrometry confirmed this assignment (Supplementary Fig. S1).
Fig. 1.
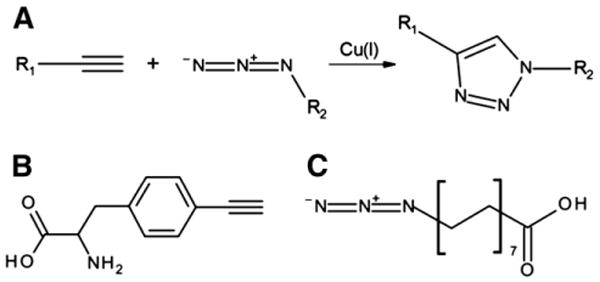
Reaction scheme and chemical structures. (A) Copper-catalyzed alkyne-azide cycloaddition. Structures of p-ethynylphenylalanine (B) and palmitic acid-azide (C).
Fig. 2.
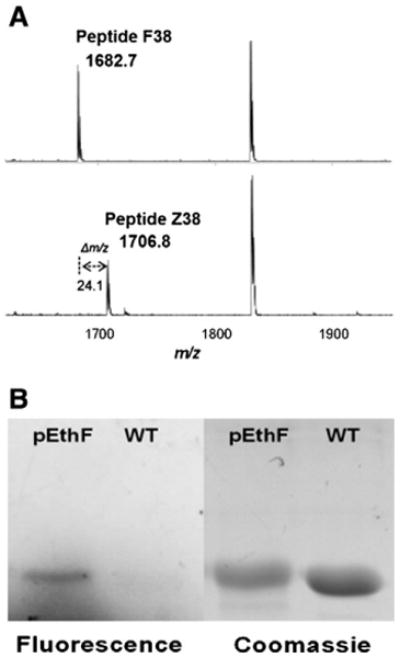
Site-specific incorporation of pEthF into the mDHFR-38Am and CuAAC-mediated coumarin-labeling. (A) MALDI-TOF analysis of trypsin-digested mDHFR-WT and mDHFR-pEthF. Peptide F38 (top) of the mDHFR-WT and Peptide Z38 of the mDHFR-pEthF (bottom). (B) Protein gel images of the fluorogenic dye-treated mDHFR-pEthF (pEthF) and mDHFR-WT (WT). The gel was subjected to UV (390 nm) irradiation to excite the fluorophore (fluorescence panel), and then stained with Coomassie brilliant blue (Coomassie panel) to visualize proteins.
To validate orthogonal reactivity of the alkyne end group of pEthF with an azide moiety via CuAAC, fluorogenic coumarin azide was reacted with the purified mDHFR-pEthF or mDHFR-WT in a CuSO4/ascorbate system. Since the reaction of the coumarin azide with an alkyne group produces a strongly fluorescent triazole-linked conjugate [44], the evolution of fluorescence is an indicator of pEthF reactivity for CuAAC. In SDS-PAGE analysis, the protein gel under UV exposure (λex = 390 nm) clearly exhibited the fluorescence confirming the formation of a triazole linkage between coumarin azide and the alkynyl group of the mDHFR-pEthF (Fig. 2B) as well as a strong protein band stained with Coomassie blue dye, whereas the mDHFR-WT did not exhibit any fluorescence despite a strong protein band stained with Coomassie blue dye. The combined results of the mass spectrometric analysis and the fluorogenic dye conjugation strongly support the idea that pEthF was site-specifically incorporated into a protein using the E. coli expression system containing the orthogonal pair of ytRNAPheCUA_UG/yPheRST415A, and the pEthF introduced into a protein is reactive for bio-orthogonal CuAAC.
3.2. Site-specific fatty acid-conjugation to the mDHFR-pEthF
Next, we tested if a fatty acid with an intrinsic affinity for HSA can be grafted to the mDHFR-pEthF through CuAAC. Palmitic acid-azide (15-azidopentadecanoic acid), a palmitic acid analog containing an azide moiety at the end of the carbon chain (Fig. 1C), was used for this purpose. The mDHFR-pEthF was reacted with palmitic acid-azide, and then subjected to tryptic digestion. The MALDI-TOF mass spectrum of the tryptic digests shows that anew signal with a monoisotopic mass of 1989.9 appears whereas Peptide Z38 signal is substantially reduced (Supplementary Fig. S2). Considering that palmitic acid-azide conjugation will add 283.4 Da to the mass of Peptide Z38 (the actual mass shift of 283.2 in the spectrum), the new peak is considered as palmitic acid-conjugated Peptide Z38 (Peptide Z38-PAL). This result clearly indicates that palmitic acid-azide has been conjugated to the mDHFR-pEthF in a site-specific manner.
3.3. Site-specific fatty acid-conjugation to superfolder green fluorescent protein without compromising its folded structure and intrinsic fluorescence
To investigate site-specific fatty acid-conjugation to a native protein without compromising intrinsic properties, we examined the site-specific fatty acid-conjugation to superfolder green fluorescent protein (sfGFP) [37]. Intrinsic fluorescence of sfGFP correlated to its folding facilitates the estimation of the extent of structure perturbation during CuAAC and the determination of sfGFP quantity in the following characterization steps. In order to allow efficient fatty acid-conjugation with least protein structure perturbation, we chose a position between the 214th and the 215th amino acid as pEthF incorporation site by using a server-based solvent accessibility calculation program (ASAView) [45] and examining the crystal structure of sfGFP. This position is located in a loop region with high solvent accessibility (0.72 score in the ASAView) and distal from the chromophore (Supplementary Fig. S3). Furthermore, it was reported that another NAA at this position can be used for CuAAC [28]. pEthF was introduced into the amber codon site using the E. coli expression host harboring ytRNAPheCUA_UG/yPheRST415A orthogonal pair. The sfGFP variant containing pEthF at position 215 (sfGFP-pEthF) was purified via metal-ion affinity chromatography using a six-histidine tag. Based on the expression medium volume, about 80 mg/L of purified sfGFP-pEthF was obtained. Site-specific incorporation of pEthF into wild-type sfGFP (sfGFP-WT) was confirmed by mass spectrometry coupled with electron spray ionization (ESI-MS) (Supplementary Fig. S4). The measured mass of the full length sfGFP variant (sfGFP-pEthF) is 27,755.2 Da, which is consistent with the calculated mass of 27,755.8 Da. The sfGFP-pEthF was then subjected to CuAAC-mediated palmitic acid-conjugation in a native condition using a CuSO4/dithiothreitol (DTT)/Tris[(1-benzyl-1H-1,2,3-triazol-4-yl) methyl]amine (TBTA) system where TBTA acts as an accelerating ligand as well as a radical scavenger [25]. The palmitic acid-conjugate (sfGFP-Pal) was analyzed by ESI-MS and found to have a molecular weight greater than that of the sfGFP-pEthF by 281.2 Da, strongly indicating site-specific conjugation at a stoichiometry of one palmitic acid per protein. As reported previously [46,47], the N-terminal methionine was cleaved in a portion of the sfGFP-pEthF (ΔM-sfGFP-pEthF) generating a peak with 27,624.1 m/z (Supplementary Fig. S4). However, an additional peak corresponding to the palmitic acid-conjugated ΔM-sfGFP-pEthF (27,905.7 m/z) (Supplementary Fig. S4) was also detected, indicating that both the intact and the methionine-cleaved sfGFPs are successfully conjugated to the palmitic acid.
Next, we investigated whether the fatty acid-conjugation perturbs the sfGFP folded structure. As an indicator of the portion of correctly folded sfGFPs, the fluorescence intensity of sfGFPs was monitored (Fig. 3). The fluorescence intensity of the sfGFP-pEthF is 25% higher than that of the sfGFP-WT. Even after the sfGFP-pEthF was conjugated to a fatty acid, the fluorescence intensity of the sfGFP-Pal remains unchanged. Similarly, the incubation of the sfGFP-WT under the same condition used for the fatty acid-conjugation of the sfGFP-pEthF did not significantly alter the fluorescence intensity (Fig. 3). These results clearly demonstrate that neither the site-specific incorporation of pEthF at position 215 of sfGFP nor the fatty acid-conjugation via CuAAC compromises the intrinsic fluorescence and thereby folded structure of sfGFP.
Fig. 3.
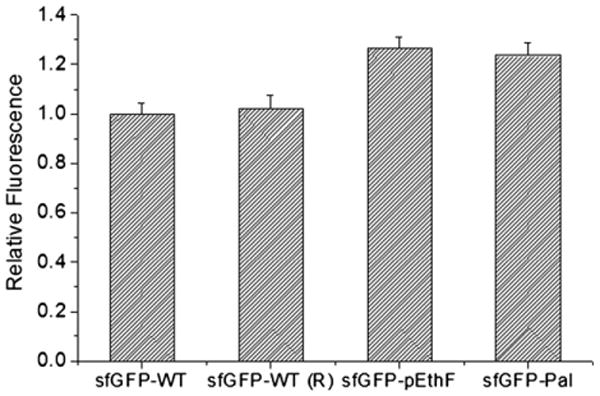
The relative fluorescence of the sfGFP-WT and sfGFP variants. Protein solutions (20 μg/mL) were loaded onto a 96-well microplate at 100 μL per well, and read on the plate reader at λex = 480 nm and λem = 510 nm. Values were averaged for each protein (n = 5), and normalized to the fluorescence of the sfGFP-WT. In order to investigate the effect of reagents used in CuAAC, the sfGFP-WT was treated in parallel with the sfGFP-pEthF subjected to the fatty acid-conjugation, and designated sfGFP-WT (R).
3.4. HSA-binding of the sfGFP-Pal in vitro
To investigate whether the fatty acid-conjugation to a protein generates albumin-binding affinity, the sfGFP-WT and the sfGFP-Pal were mixed with HSA-coupled agarose beads or, as a control, inactivated beads in which amine-reactive N-hydroxysuccinimide groups had been blocked by glycine. After washing the beads multiple times with PBS on a gravity-flow column, the fluorescence intensity of the beads were qualitatively or quantitatively analyzed by using a fluorescence imager and a fluorescence microplate reader, respectively (Fig. 4). The HSA-coupled beads mixed with the sfGFP-Pal exhibit significant fluorescence while the HSA-coupled beads mixed with the sfGFP-WT display negligible fluorescence (Fig. 4A). Furthermore, the inactivated beads mixed with the sfGFP-Pal exhibit negligible fluorescence. These results strongly support the idea that the sfGFP-Pal binds the HSA-coupled beads via HSA-specific interactions. In order to quantitatively compare the binding affinities of the sfGFP-WT and the sfGFP-Pal to HSA, the fluorescence intensities of the HSA-coupled beads mixed with the sfGFP-WT and the sfGFP-Pal were measured. The HSA-coupled beads mixed with the sfGFP-Pal exhibit about 20-fold greater fluorescence than those with the sfGFP-WT. The inactivated beads mixed with the sfGFP-WT and the sfGFP-Pal exhibit 1.7- and 1.3-fold greater fluorescence than that of the HSA-coupled beads mixed with the sfGFP-WT, likely because the sfGFP-WT and the sfGFP-Pal reacted with a small amount of residual amine-reactive N-hydroxysuccinimide groups in the inactivated beads (Fig. 4A). Without any significant structural perturbation, the fluorescence intensity is directly correlated to the amount of sfGFP. Therefore, these results suggest that the fatty acid-conjugation leads to a substantial increase in albumin-binding affinity. To eliminate the possibility that the fluorescence increase is caused by aggregation of the sfGFP-Pal leading to its retention in the column during the HSA-coupled bead binding assay, the binding was also examined using the nitrocellulose membrane blot which was subjected to extensive washing. In contrast to the sfGFP-WT and the sfGFP-pEthF that were washed away after being spotted on HSA-coated membrane, the sfGFP-Pal was tightly bound to HSA as confirmed using fluorescence image analysis (Fig. 4B). These results unambiguously indicate that the site-specific palmitic acid-conjugation remarkably enhances HSA-binding affinity of the sfGFP compared to the unmodified sfGFP.
Fig. 4.
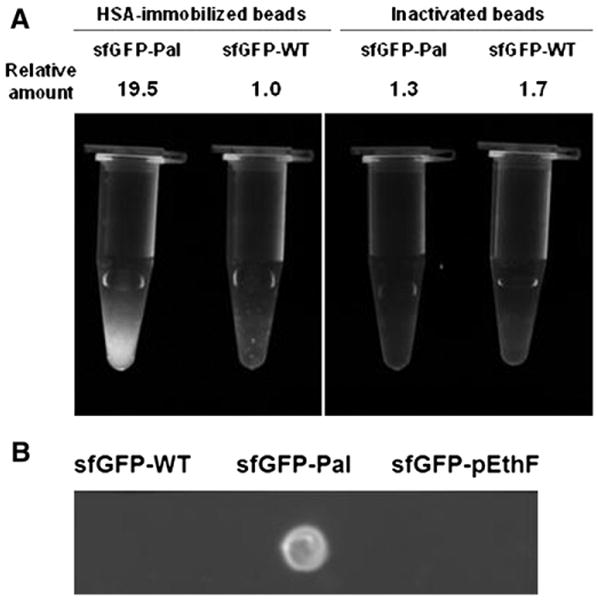
Relative albumin-binding affinities of sfGFP-variants. (A) Inactivated (amine-reactive functional groups blocked by glycine) or HSA-immobilized agarose beads were mixed with the sfGFP-WT and the sfGFP-Pal. After washing extensively with PBS, the fluorescence image was taken on the UV epi-illuminator at λex = 480 nm, and emitted light above 510 nm was captured. For a quantitative fluorescence measurement, the same amounts of agarose beads were loaded on a 96-well microplate and read on the plate reader at λex = 480 nm and λem = 510 nm. The relative amounts of sfGFP samples were calculated from the relative fluorescence intensities. (B) Four micrograms of each protein in 2 μL of PBS were dotted onto the HSA-coated nitrocellulose membrane and air-dried. After washing in PBS for 5 min and air-dry, the membrane was epi-illuminated at λex = 480 nm, and emitted light above 510 nm was captured.
3.5. Pharmacokinetic study of the sfGFP-Pal
In order to evaluate clinical benefits from albumin-binding capacity generated from the site-specific fatty acid-conjugation, a single dose of either the sfGFP-WT or the sfGFP-Pal was intravenously administered to mice (n = 4), and the sfGFP serum concentrations in the serum samples taken at different time points were measured by using GFP-specific ELISA kit. Assuming one-compartment distribution and first-order elimination of the sfGFP in the serum [48,49], its logarithmic residual serum concentrations versus time were plotted, and the data were fitted into a straight line to calculate the serum half-life (Fig. 5). The serum half-life of the sfGFP-Pal calculated (5.2 h) is approximately 5-fold longer than that of the sfGFP-WT (1.0 h).
Fig. 5.
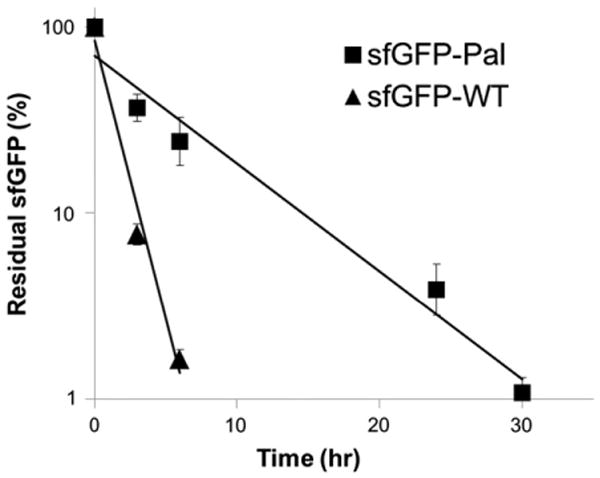
Pharmacokinetics of the sfGFP-WT and the sfGFP-Pal. Four mice were intravenously administered the sfGFP-Pal (square) or the sfGFP-WT (triangle), respectively, and serum concentrations were measured by ELISA at different time points (mean ± s.d.): 0 (10 min), 3, and 6 h for the sfGFP-WT; 0 (10 min), 3, 6, 24, and 30 h for the sfGFP-Pal. Data were normalized with regard to the initial value, plotted in a logarithmic scale versus time post-injection, and fitted into a straight line (R2 = 0.98 for the sfGFP-WT and 0.97 for the sfGFP-Pal).
4. Discussion
The fatty acid-conjugation is an attractive methodology for developing long-acting protein therapeutics. A natural occurrence of a fatty acid in the blood greatly reduces the risk of immunogenicity and toxicity when it is used as an albumin-binding tag. In addition, its small size relative to other albumin-binding motifs [50,51], including albumin-binding domain, is less likely to impair protein folded structure and function upon conjugation. The recently FDA-approved long-acting peptide analogs in which a fatty acid has been chemically linked to a lysine residue represent the potential of a fatty acid as a safe and reliable half-life extender in clinical settings. To render it broadly applicable to large-sized proteins as well as small peptides, new protein conjugation chemistry is required to prevent the production of positional isomers and detrimental loss of inherent activity arising from random coupling to multiple lysine residues.
Since its advent in 2002, CuAAC has found numerous applications in diverse fields, providing highly selective reactivity. To implement CuAAC for the site-specific attachment of a fatty acid to a protein, pEthF was introduced by using the engineered orthogonal pair of ytRNAPheCUA_UG/yPheRST415A with the yield of approximately 80 mg/L based on the volume of protein expression medium. Research efforts witnessed over the past couple of years have demonstrated near-optimal expression of a NAA-incorporated protein (up to 800 mg/L) comparable to that of its wild-type, thereby showing great promise for its expanded application to protein therapeutics [52–54]. Successful bioconjugation via CuAAC is critically dependent on stabilizing catalytically active Cu(I) oxidation state while simultaneously preventing generation of reactive byproducts leading to undesirable protein aggregation. We discovered that the CuSO4/DTT/TBTA system is suitable for the fatty acid-conjugation to a protein resulting in a high yield with minimal side products. The use of TBTA ligand was essential for a high yield and an optimal reaction rate, but its low solubility in water required the addition of a polar solvent, DMSO, in the CuAAC reaction. Newly-developed water-soluble ligands such as THPTA and BTTAA might be alternatives for bioconjugation of proteins intolerant to DMSO [55].
We report here the utility of a fatty acid as an albumin-binding tag attached to a large protein with absolute site selectivity. Site-specificity is the key advantage of our technique over other albumin-binding strategies relying on the genetic fusion of affinity motifs or random chemical attachment of synthetic binding molecules. Another key to exploiting this technology is imparting albumin-binding capability to a protein with minimal perturbation of its native activity and stability. As demonstrated in this paper, the site-specific fatty acid-conjugation via CuAAC does not cause any significant loss of the sfGFP fluorescence, strongly indicating that the native sfGFP structure was not perturbed. Based on examination of a crystal structure of a target protein and the solvent accessibility prediction, optimal sites for NAA incorporation and subsequent fatty acid-conjugation can be chosen, which has not been possible previously. Furthermore, the utility of this technology to modulate pharmacokinetics can be easily expanded by varying carbon chain lengths or by adding distinct chemical linkers between a fatty acid and a target protein. Tailoring the half-life of a therapeutic protein offers the advantage of being able to optimize the requirements of its intended clinical application [56,57].
The animal study has clearly revealed the significance of albumin-binding effect on in vivo half-life extension. Five-fold longer retention of the sfGFP-Pal in blood compared to the sfGFP-WT is most likely attributed to FcRn-mediated recycling of the sfGFP-HSA complex, which is supported by in vitro HSA-binding assay. Previously, a GFP variant C-terminally attached to a PEG-like polymer exhibited only 2 h of serum half-life when injected intravenously into mice [58]. Similarly, a single-chain diabody (scDb) C-terminally fused to an albumin-binding domain showed 2.6 h of serum half-life despite the 13-fold half-life extension compared to that of an unmodified one [59]. Therefore, the half-lives of the GFP and the scDb conjugates were found to be smaller than that of the sfGFP-Pal (5.2 h). Although differences in dose, concentration measurement, and data analysis complicate a direct comparison between half-life extension technologies, it is evident that the technique and approach described in this paper constitutes a significant impact on the optimization of therapeutic efficacy of a protein by virtue of unique features including immuno-safety and orthogonal chemistry unrestricted in site of modification, and thereby has broad applications to short-lived proteins.
5. Conclusions
In this paper we successfully demonstrated that a fatty acid can be conjugated to a pEthF site of a recombinant protein via CuAAC leading to an increase in HSA binding in vitro and in serum half-life in vivo by 20-fold and 5-fold, respectively. To our knowledge, this is the first time to show the site-specific conjugation of a biomolecule into a single pEthF site of a protein via CuAAC. Furthermore, a careful choice of the pEthF incorporation site in a protein allows the successful fatty acid-conjugation without any significant perturbation of the folded protein. In the future, development of an orthogonal tRNA/aaRS pair for pEthF incorporation into a protein expressed in yeasts and mammalian cells will extend the application of the technique described here to broader ranges of recombinant proteins.
Supplementary Material
Acknowledgments
We appreciate Dr. Nicholas E. Sherman at the University of Virginia for his assistance with mass spectrometry analysis. The W.M. Keck Biomedical Mass Spectrometry Laboratory is funded by a grant from the University of Virginia Pratt Fund through the School of Medicine.
Footnotes
Appendix A. Supplementary data: Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2013.05.023.
References
- 1.Aggarwal SR. What's fueling the biotech engine-2011 to 2012. Nat Biotechnol. 2012;30(12):1191–1197. doi: 10.1038/nbt.2437. [DOI] [PubMed] [Google Scholar]
- 2.Jevsevar S, Kunstelj M, Porekar VG. PEGylation of therapeutic proteins. Biotechnol J. 2010;5(1):113–128. doi: 10.1002/biot.200900218. [DOI] [PubMed] [Google Scholar]
- 3.Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868–876. doi: 10.1016/j.copbio.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO Mol Med. 2012;4(10):1015–1028. doi: 10.1002/emmm.201201379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kontos S, Hubbell JA. Drug development: longer-lived proteins. Chem Soc Rev. 2012;41(7):2686–2695. doi: 10.1039/c2cs15289d. [DOI] [PubMed] [Google Scholar]
- 6.Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012;161(2):429–445. doi: 10.1016/j.jconrel.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhury C, Brooks CL, Carter DC, Robinson JM, Anderson CL. Albumin binding to FcRn: distinct from the FcRn–IgG interaction. Biochemistry. 2006;45(15):4983–4990. doi: 10.1021/bi052628y. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197(3):315–322. doi: 10.1084/jem.20021829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherman MR, Saifer MG, Perez-Ruiz F. PEG-uricase in the management of treatment-resistant gout and hyperuricemia. Adv Drug Deliv Rev. 2008;60(1):59–68. doi: 10.1016/j.addr.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 10.Libon C, Corvaia N, Haeuw JF, Nguyen TN, Stahl S, Bonnefoy JY, Andreoni C. The serum albumin-binding region of streptococcal protein G (BB) potentiates the immunogenicity of the G130-230 RSV-A protein. Vaccine. 1999;17(5):406–414. doi: 10.1016/s0264-410x(98)00198-4. [DOI] [PubMed] [Google Scholar]
- 11.Stork R, Muller D, Kontermann RE. A novel tri-functional antibody fusion protein with improved pharmacokinetic properties generated by fusing a bispecific single-chain diabody with an albumin-binding domain from streptococcal protein G. Protein Eng Des Sel. 2007;20(11):569–576. doi: 10.1093/protein/gzm061. [DOI] [PubMed] [Google Scholar]
- 12.Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes. 2004;53(9):2492–2500. doi: 10.2337/diabetes.53.9.2492. [DOI] [PubMed] [Google Scholar]
- 13.Schulte S. Half-life extension through albumin fusion technologies. Thromb Res. 2009;124(Suppl. 2):S6–S8. doi: 10.1016/S0049-3848(09)70157-4. [DOI] [PubMed] [Google Scholar]
- 14.Zhao HL, Xue C, Wang Y, Li XY, Xiong XH, Yao XQ, Liu ZM. Circumventing the heterogeneity and instability of human serum albumin-interferon-alpha2b fusion protein by altering its orientation. J Biotechnol. 2007;131(3):245–252. doi: 10.1016/j.jbiotec.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Jonassen I, Havelund S, Hoeg-Jensen T, Steensgaard DB, Wahlund PO, Ribel U. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm Res. 2012;29(8):2104–2114. doi: 10.1007/s11095-012-0739-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trussel S, Dumelin C, Frey K, Villa A, Buller F, Neri D. New strategy for the extension of the serum half-life of antibody fragments. Bioconjug Chem. 2009;20(12):2286–2292. doi: 10.1021/bc9002772. [DOI] [PubMed] [Google Scholar]
- 17.Havelund S, Plum A, Ribel U, Jonassen I, Volund A, Markussen J, Kurtzhals P. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm Res. 2004;21(8):1498–1504. doi: 10.1023/b:pham.0000036926.54824.37. [DOI] [PubMed] [Google Scholar]
- 18.Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thogersen H, Wilken M, Agerso H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43(9):1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 19.Bhattacharya AA, Grune T, Curry S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J Mol Biol. 2000;303(5):721–732. doi: 10.1006/jmbi.2000.4158. [DOI] [PubMed] [Google Scholar]
- 20.Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16(3):165–179. [PubMed] [Google Scholar]
- 21.Pollaro L, Heinis C. Strategies to prolong the plasma residence time of peptide drugs. Medchemcomm. 2010;1(5):319–324. [Google Scholar]
- 22.Buse JB, Garber A, Rosenstock J, Schmidt WE, Brett JH, Videbaek N, Holst J, Nauck M. Liraglutide treatment is associated with a low frequency and magnitude of antibody formation with no apparent impact on glycemic response or increased frequency of adverse events: results from the liraglutide effect and action in diabetes (LEAD) trials. J Clin Endocrinol Metab. 2011;96(6):1695–1702. doi: 10.1210/jc.2010-2822. [DOI] [PubMed] [Google Scholar]
- 23.Zinman B, Philis-Tsimikas A, Cariou B, Handelsman Y, Rodbard HW, Johansen T, Endahl L, Mathieu C. Insulin degludec versus insulin glargine in insulin-naive patients with type 2 diabetes: a 1-year, randomized, treat-to-target trial (BEGIN Once Long) Diabetes Care. 2012;35(12):2464–2471. doi: 10.2337/dc12-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shechter Y, Sasson K, Lev-Goldman V, Rubinraut S, Rubinstein M, Fridkin M. Newly designed modifier prolongs the action of short-lived peptides and proteins by allowing their binding to serum albumin. Bioconjug Chem. 2012;23(8):1577–1586. doi: 10.1021/bc3000854. [DOI] [PubMed] [Google Scholar]
- 25.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org Lett. 2004;6(17):2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 26.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41(14):2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Lallana E, Riguera R, Fernandez-Megia E. Reliable and efficient procedures for the conjugation of biomolecules through Huisgen azide-alkyne cycloadditions. Angew Chem Int Ed Engl. 2011;50(38):8794–8804. doi: 10.1002/anie.201101019. [DOI] [PubMed] [Google Scholar]
- 28.Bundy BC, Swartz JR. Site-specific incorporation of p-propargyloxyphenylalanine in a cell-free environment for direct protein–protein click conjugation. Bioconjug Chem. 2010;21(2):255–263. doi: 10.1021/bc9002844. [DOI] [PubMed] [Google Scholar]
- 29.Patel KG, Swartz JR. Surface functionalization of virus-like particles by direct conjugation using azide-alkyne click chemistry. Bioconjug Chem. 2011;22(3):376–387. doi: 10.1021/bc100367u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Budisa N. Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire. Angew Chem Int Ed Engl. 2004;43(47):6426–6463. doi: 10.1002/anie.200300646. [DOI] [PubMed] [Google Scholar]
- 31.Hohsaka T, Ashizuka Y, Taira H, Murakami H, Sisido M. Incorporation of nonnatural amino acids into proteins by using various four-base codons in an Escherichia coli in vitro translation system. Biochemistry (Mosc) 2001;40(37):11060–11064. doi: 10.1021/bi0108204. [DOI] [PubMed] [Google Scholar]
- 32.Jester BC, Levengood JD, Roy H, Ibba M, Devine KM. Nonorthologous replacement of lysyl-tRNA synthetase prevents addition of lysine analogues to the genetic code. Proc Natl Acad Sci U S A. 2003;100(24):14351–14356. doi: 10.1073/pnas.2036253100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Link AJ, Tirrell DA. Reassignment of sense codons in vivo. Methods. 2005;36(3):291–298. doi: 10.1016/j.ymeth.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292(5516):498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 36.Zheng S, Kwon I. Manipulation of enzyme properties by noncanonical amino acid incorporation. Biotechnol J. 2012;7(1):47–60. doi: 10.1002/biot.201100267. [DOI] [PubMed] [Google Scholar]
- 37.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 38.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28(1):41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 39.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407(3):313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 40.Takasu A, Kondo S, Ito A, Furukawa Y, Higuchi M, Kinoshita T, Kwon I. Artificial extracellular matrix proteins containing phenylalanine analogues biosynthesized in bacteria using T7 expression system and the PEGylation. Biomacromolecules. 2011;12(10):3444–3452. doi: 10.1021/bm200522d. [DOI] [PubMed] [Google Scholar]
- 41.Kwon I, Wang P, Tirrell DA. Design of a bacterial host for site-specific incorporation of p-bromophenylalanine into recombinant proteins. J Am Chem Soc. 2006;128(36):11778–11783. doi: 10.1021/ja0626281. [DOI] [PubMed] [Google Scholar]
- 42.Kwon I, Tirrell DA. Site-specific incorporation of tryptophan analogues into recombinant proteins in bacterial cells. J Am Chem Soc. 2007;129(34):10431–10437. doi: 10.1021/ja071773r. [DOI] [PubMed] [Google Scholar]
- 43.Kwon I, Kirshenbaum K, Tirrell DA. Breaking the degeneracy of the genetic code. J Am Chem Soc. 2003;125(25):7512–7513. doi: 10.1021/ja0350076. [DOI] [PubMed] [Google Scholar]
- 44.Beatty KE, Xie F, Wang Q, Tirrell DA. Selective dye-labeling of newly synthesized proteins in bacterial cells. J Am Chem Soc. 2005;127(41):14150–14151. doi: 10.1021/ja054643w. [DOI] [PubMed] [Google Scholar]
- 45.Ahmad S, Gromiha M, Fawareh H, Sarai A. ASAView: database and tool for solvent accessibility representation in proteins. BMC Bioinforma. 2004;5:51. doi: 10.1186/1471-2105-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirel PH, Schmitter MJ, Dessen P, Fayat G, Blanquet S. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc Natl Acad Sci U S A. 1989;86(21):8247–8251. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang F, Niu W, Guo J, Schultz PG. Unnatural amino acid mutagenesis of fluorescent proteins. Angew Chem Int Ed Engl. 2012;51(40):10132–10135. doi: 10.1002/anie.201204668. [DOI] [PubMed] [Google Scholar]
- 48.DiPiro JT. Concepts in Clinical Pharmacokinetics. American Society of Health-System Pharmacists; Bethesda: 2010. [Google Scholar]
- 49.Yang Z, Wang J, Lu Q, Xu J, Kobayashi Y, Takakura T, Takimoto A, Yoshioka T, Lian C, Chen C, Zhang D, Zhang Y, Li S, Sun X, Tan Y, Yagi S, Frenkel EP, Hoffman RM. PEGylation confers greatly extended half-life and attenuated immunogenicity to recombinant methioninase in primates. Cancer Res. 2004;64(18):6673–6678. doi: 10.1158/0008-5472.CAN-04-1822. [DOI] [PubMed] [Google Scholar]
- 50.Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem. 2002;277(38):35035–35043. doi: 10.1074/jbc.M205854200. [DOI] [PubMed] [Google Scholar]
- 51.Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR, van Dongen GA. Improved tumor targeting of anti-epidermal growth factor receptor nanobodies through albumin binding: taking advantage of modular nanobody technology. Mol Cancer Ther. 2008;7(8):2288–2297. doi: 10.1158/1535-7163.MCT-07-2384. [DOI] [PubMed] [Google Scholar]
- 52.Cho H, Daniel T, Buechler YJ, Litzinger DC, Maio Z, Putnam AM, Kraynov VS, Sim BC, Bussell S, Javahishvili T, Kaphle S, Viramontes G, Ong M, Chu S, Becky GC, Lieu R, Knudsen N, Castiglioni P, Norman TC, Axelrod DW, Hoffman AR, Schultz PG, DiMarchi RD, Kimmel BE. Optimized clinical performance of growth hormone with an expanded genetic code. Proc Natl Acad Sci U S A. 2011;108(22):9060–9065. doi: 10.1073/pnas.1100387108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson DB, Xu J, Shen Z, Takimoto JK, Schultz MD, Schmitz RJ, Xiang Z, Ecker JR, Briggs SP, Wang L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat Chem Biol. 2011;7(11):779–786. doi: 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395(2):361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 55.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Increasing the efficacy of bioorthogonal click reactions for bioconjugation: a comparative study. Angew Chem Int Ed Engl. 2011;50(35):8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schellenberger V, Wang CW, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, Cleland JL, Silverman J, Stemmer WP. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat Biotechnol. 2009;27(12):1186–1190. doi: 10.1038/nbt.1588. [DOI] [PubMed] [Google Scholar]
- 57.Nguyen A, Reyes AE, II, Zhang M, Mc Donald P, Wong WL, Damico LA, Dennis MS. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel. 2006;19(7):291–297. doi: 10.1093/protein/gzl011. [DOI] [PubMed] [Google Scholar]
- 58.Gao W, Liu W, Christensen T, Zalutsky MR, Chilkoti A. In situ growth of a PEG-like polymer from the C terminus of an intein fusion protein improves pharmacokinetics and tumor accumulation. Proc Natl Acad Sci U S A. 2010;107(38):16432–16437. doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hopp J, Hornig N, Zettlitz KA, Schwarz A, Fuss N, Muller D, Kontermann RE. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng Des Sel. 2010;23(11):827–834. doi: 10.1093/protein/gzq058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.