

Summary
All three members of the endothelin (ET) family of peptides, ET-1, ET-2, and ET-3, are expressed in the human kidney, with ET-1 being the predominant isoform. ET-1 and ET-2 bind to two G-protein–coupled receptors, ETA and ETB, whereas at physiological concentrations ET-3 has little affinity for the ETA receptor. The human kidney is unusual among the peripheral organs in expressing a high density of ETB. The renal vascular endothelium only expresses the ETB subtype and ET-1 acts in an autocrine or paracrine manner to release vasodilators. Endothelial ETB in kidney, as well as liver and lungs, also has a critical role in scavenging ET-1 from the plasma. The third major function is ET-1 activation of ETB in in the nephron to reduce salt and water re-absorption. In contrast, ETA predominate on smooth muscle, causing vasoconstriction and mediating many of the pathophysiological actions of ET-1. The role of the two receptors has been delineated using highly selective ETA (BQ123, TAK-044) and ETB (BQ788) peptide antagonists. Nonpeptide antagonists, bosentan, macitentan, and ambrisentan, that are either mixed ETA/ETB antagonists or display ETA selectivity, have been approved for clinical use but to date are limited to pulmonary hypertension. Ambrisentan is in clinical trials in patients with type 2 diabetic nephropathy. This review summarizes ET-receptor antagonism in the human kidney, and considers the relative merits of selective versus nonselective antagonism in renal disease.
Keywords: Ambrisentan, antagonist, bosentan, endothelin-1, macitentan, sitaxentan
All three members of the endothelin (ET) family of peptides, ET-1, ET-2, and ET-3, are expressed in the human kidney, although ET-1 is the predominant isoform. ET-1 and ET-2 bind to two G-protein–coupled receptors, ETA and ETB, whereas at physiological concentrations ET-3 has little affinity for the ETA receptor. The endothelin receptors are members of the Family A G-protein–coupled receptors, a class of proteins that has been exploited very successfully as targets for the development of drugs.
The human kidney is unusual among the peripheral organs in expressing a high density of ETB. The renal vascular endothelium only expresses the ETB subtype and ET-1 acts in an autocrine or paracrine manner to release vasodilators. Endothelial ETB in kidney, as well as liver and lungs, has a critical role in scavenging ET-1 from the plasma. The third major function is for ET-1 activation of ETB in medullary epithelial cells to reduce salt and water reabsorption. ETA predominate on the vasculature to cause vasoconstriction. The pathophysiological actions of ET-1 are mediated mainly via the ETA subtype. The role of the two subtypes has been delineated in preclinical and acute experimental studies using highly selective ETA (including BQ123, TAK-044) and ETB (BQ788) peptide antagonists. Three nonpeptide antagonists, bosentan, macitentan, and ambrisentan, that are either mixed ETA/ETB antagonists or display ETA selectivity, have been approved for clinical use, primarily in pulmonary arterial hypertension.
In renal pathophysiological conditions ET-1 contributes to vascular remodeling, proliferation of mesangial cells, and extracellular matrix production, mainly through binding to ETA. Beneficial actions of ET-1 on sodium and water regulation mainly are ETB-mediated. These findings suggest an ETA-selective antagonist would have a therapeutic advantage over a mixed antagonist in renal disease. Acute studies directly comparing mixed and selective peptide antagonists suggest selective ETA blockade, however, sparing ETB may be beneficial. However, this was balanced by a greater prevalence of side effects for small-molecule, orally active ETA antagonists compared with mixed antagonists, although the latter also have their limitations. The ET signaling pathway in the kidney remains a promising clinical target for receptor antagonism, which may be realized by the next generation of antagonists.
ET Receptors
The ET family comprises three isoforms, ET-1, ET-2, and ET-3.1,2 Although messenger RNA encoding all three has been detected in human kidney, ET-1 is the predominant intrarenal isoform.3 ETs interact with two distinct G-protein–coupled receptors, ETA4 and ETB5 (Fig. 1), which were identified 2 years after the discovery of the endogenous peptides in 1988. They are both class A, G-protein–coupled receptors; this class is the target of nearly half of currently available medicines. This has resulted from well-developed medicinal chemistry strategies and high-throughput screening programs to identify small-molecule drugs, stimulating considerable effort to discover ET-receptor antagonists. The initial clue to the existence of two subtypes and the key to classifying the receptors was that ET-1 and ET-2 are equipotent at the ETA subtype whereas ET-3 shows at least 100-fold lower potency and at physiological concentration ET-3 is unlikely to activate this subtype (Table 1). All three ETs bind to ETB with similar affinity.6,7 This review focuses on the role of ET receptors in the human kidney and considers the clinical pharmacology of ET antagonists that have been used to block these receptors. The effects of ET-1 on the kidney are complex and more detailed information can be found in reviews on renal endothelin physiology8 and pathology,9 and the pharmacology of the endothelin signaling pathway.10,11
Figure 1.

Model of the ET-1 signaling pathway in the renal vasculature. The primary source of ET-1 production is the vascular endothelium, although it also is produced by other cell types in the kidney including epithelial cells. ET-1 is synthesized within the secretory vesicles of the constitutive pathway. Pro–ET-1 is processed to big ET-1 by the action of a furin convertase. Big ET-1 then is transformed to the mature, biologically active peptide ET-1, mainly through the action of ECE-1, although a second related enzyme, ECE-2, also may play a role, particularly under acidic pathophysiological conditions. ET-1 also is released from Weibel-Palade bodies of the regulated pathway in response to external stimuli. ET-1 released abluminally causes the underlying smooth muscle to contract, mainly via ETA. The ligand-receptor complex then undergoes internalization to the endosome before recycling of the receptor to the cell surface. Some smooth muscle cells from specific vascular beds express a low density of ETB, but ETA antagonists are able to fully reverse an established ET-1 constrictor response, implying a minimal contribution. Binding to endothelial ETB elicits an opposing vasodilatation via the release of relaxing factors as well as removal of ET-1 from the circulation by internalization to the lysosome and degradation.
Table 1.
Key ET Agonists and Antagonists Used in Research and Clinical Studies (Including Only Those Approved for Clinical Use in Clinical Studies), and the Functional Role of Renal ET Receptors
| ETA | ETA/ETA | ETB | |
|---|---|---|---|
| Potency | ET-1 = ET-2 > ET-3 | ET-1 = ET-2 = ET-3 | |
| Peptide agonists | Sarafotoxin S6C | ||
| BQ3020 | |||
| IRL1620 | |||
| Peptide antagonists | BQ123 | BQ788 | |
| FR139317 | |||
| Tak-044 | |||
| Clinically approved antagonists | |||
| Selective | Sitaxentan | ||
| Ambrisentan | |||
| Mixed | Bosentan | ||
| Macitentan | |||
| Renal Function | ETA | ETB |
|---|---|---|
| Vasculature | Cortical vasoconstriction | Medullary vasodilatation |
| Conduit vessel vasoconstriction | Conduit vessel vasodilatation | |
| Afferent arteriolar constriction | Afferent arteriolar dilatation | |
| Efferent arteriolar constriction | Efferent arteriolar dilatation | |
| Glomerulus | Mesangial cell contraction | |
| Mesangial cell proliferation | ||
| Podocyte injury | ||
| Epithelium | Endothelial function not known | |
| Inner medullary | Extracellular matrix accumulation | Natriuresis |
| collecting duct | Interstitial fibrosis | |
| Possible natriuresis |
No Evidence for Further ET-Receptor Subtypes
Further receptor subclassifications have been proposed including suggestions that ETB could be subdivided into ETB1, present on endothelial cells, and ETB2 on smooth muscle cells, but there currently is no evidence that the receptors expressed by these two cell types can be distinguished pharmacologically.10,11 ET-receptor antagonists have not been successful in certain conditions such as heart failure,12 perhaps implying that ETs may mediate their actions via previously unsuspected receptors; however, this is unlikely. Following the sequencing of the human genome, it is accepted that all genes that potentially encode a G-protein–coupled receptor have been identified and currently are classified as ‘orphan’ to indicate that their endogenous ligand is not yet known.13,14 These remaining orphan receptors (approximately 80) have been screened against more than 20 ET peptides (including all three endogenous isoforms and their corresponding big ET precursors, C-terminal metabolites, the ETA antagonist BQ123, and the ETB agonist BQ3020) without detectable binding. The screen also included two of the most closely related orphan receptors to ETA and ETB, GPR37 (also known as endothelin-receptor type B-like receptor or Parkin-associated endothelin receptor-like receptor) and its related receptor GPR37L1. Two neuropeptides, prosaptide and prosaposin, that are structurally distinct from the ETs have been suggested to be the endogenous ligands for GPR37 and GPR37L1.15
Peptide Agonists
Experimental medicine studies in volunteers mainly use ET-1 that is equipotent for ETA and ETB (Table 1). ET-3, which is modestly selective for ETB,7 also has been used but greater ETB selectivity is shown by sarafotoxin S6c, one of the isoforms originally identified from snake venom.16 IRL1620 (Suc-[Glu9,Ala11,15]-endothelin-18-21)17 is a truncated linear analogue in which the N-terminus has an N-succinyl modification, reducing metabolism by nonspecific peptidases. It was developed as an ETB agonist but now is used in clinical trials as a potential vasodilator in the delivery of anticancer agents and in neuroprotection where it is known as SPI-1620 (licensed by Spectrum Pharmaceuticals, Henderson, NV). The second widely used ETB agonist is BQ3020 ([Ala11,15]Ac-ET-l6-21),18 however, this compound has not been used clinically.
Peptide Antagonists
The first endothelin-receptor antagonists to be discovered were from natural product screening, compound libraries, or drug design based on the structure of the endogenous ET peptides (Table 1). The most widely used, according to the number of published articles, is the cyclic pentapeptide BQ-123 (D-Asp-L-Pro-D-Val-L-Leu-D-Trp-) (Ihara et al19), based on peptides isolated from Streptomyces misakiensis, a highly selective competitive ETA antagonist with low nanomolar affinity for the receptor. The second most widely used is FR 139317 (N-[(hexahydro-1-azepinyl)carbonyl]L-Leu[1-Me]D-Trp-3 [2-pyridyl]-D-Ala),20 a linear tripeptide. These are both highly ETA selective for human (as well as rodent) ET receptors and at concentrations used in experimental medicine or in vivo animal experiments are likely to block only the ETA receptor; data from these studies can be interpreted with confidence. TAK-044 is a cyclic hexapeptide also isolated from S misakiensis with a more modest degree of ETA selectivity.21 BQ788 (N-[([2R,6S]-2,6-dimethyl-1-piperidinyl)carbonyl]-4-methyl-L-leucyl-N-[(1R)-1-carboxylatopentyl]-1-[methoxycarbonyl]-D-tryptophanamide) is a modified tripeptide developed by structure-activity analysis22 and is a selective competitive ETB antagonist (usually showing one to two orders of magnitude selectivity for ETB over ETA) in human beings and across species. Because these compounds are all peptides, they have little or no oral bioavailability, require intra-arterial administration, and are metabolized or excreted over comparatively short periods of time. An advantage in their use is that they are soluble and do not bind plasma proteins. Therefore, they are used for short-term, acute investigations in both animal models and in experimental medicine studies.
ETA Receptors Predominate on Smooth Muscle of Renal Vessels and Mediate Vasoconstriction
A major physiological action of ET-1 is to function as one of the most powerful vasoconstrictors of human blood vessels. As such, ET-1 plays a major role in regulating vascular function in all organ systems, including the kidney (Fig. 1). As in other vessels, ET-1 is thought to be released from endothelial cells lining intrarenal vessels throughout the cortex and medulla. In the human vasculature, including that of the kidney, under normal physiological conditions release of ET-1 from endothelial cells causes sustained vasoconstriction via ETA that predominate on the underlying smooth muscle. Under pathophysiological conditions in which ET-1 is overproduced, vascular cells also may undergo proliferation and contribute to vascular remodeling and the development of renal fibrosis. Figure 1 shows the ratio of the densities of the two receptor subtypes measured by radioligand binding assays with the ETA subtype representing greater than 90% of ET receptors in the smooth muscle layer of all renal vessels studied. This includes the large conduit vessels, the arcuate arteries, and veins at the corticomedullary junction, as well as small intrarenal vessels such as the afferent and efferent vessels of the glomerulus.23–27
In a detailed study using human isolated main stem renal arteries and veins in organ baths,28 ET-1 was, as expected, a potent vasoconstrictor, with the concentration producing half-maximal response (EC50) values of 4 and 1 nmol/L, respectively. In renal artery, ET-3 and the ETB agonist sarafotoxin 6c showed little or no activity up to 300 nmol/L. In veins, some but not all samples responded to ET-3, but this peptide was much less potent than ET-1, consistent with an ETA- mediated action. Interestingly, S6c concentration-related contractions were found in some individuals and, although more potent than ET-1, the maximum response was 30% to 60% of that obtained with ET-1. Crucially, however, the ETA antagonist BQ123 fully reversed the ET-1 contractions in both arteries and veins without reducing the maximum agonist response, consistent with a competitive antagonist. Therefore, in renal vessels the endogenous peptides ET-1 and ET-3 appear to mediate vasoconstriction via the ETA, indicating that ETB-mediated responses in human renal vessels are of little importance. The pharmacology of isolated renal arteries and veins is similar to vessels obtained from other human vascular beds, with ETA antagonists fully reversing an ET-1 response.29 This is critical to understanding the importance of selectivity for the two subtypes. Sarafotoxin S6c–induced constrictor responses have been used previously as evidence of significant ETB constrictor responses in human vessels. However, it is not an endogenous ligand and ET-1 responses are fully reversed using ET antagonists. Bohm et al30 performed key experimental medicine studies that showed in volunteers in vivo that BQ123 inhibited the ET-1–mediated increase in renal vascular resistance whereas BQ788 (ETB antagonist) potentiated the ET-1 effect, implying a constrictor role for ETA and that ETB clears ET-1 from the plasma. Kaasjager et al31 also concluded that the systemic and renal vasoconstrictor effects of ET-1 in human beings are mediated by the ETA.
A further unusual feature of ET-1 compared with other vasoconstrictors is that the constrictor response is sustained over a considerable period of time, lasting for several hours or in some cases several days.32 Contractions compared with many other vasoconstrictors are slow to wash out, which is consistent with a slow dissociation rate for ET-1 and may contribute to sustained hypertension and/or ET-induced vasospasm associated with pathophysiological conditions such as chronic kidney disease. Importantly, ET antagonists are able to relax ETA- mediated vasoconstriction in vessels preconstricted with ET-133 and this may reflect rapid internalization of the ligand receptor complex for recycling to the membrane (Fig. 1). In contrast, binding of ET-1 to ETB in vivo often is not displaced by ETB antagonists,34 which is in agreement with ETB being internalized by a different pathway and degraded in the lysosome.
How Important is the Small Population of ETB Receptors Expressed by Vascular Smooth Muscle?
In some, but not all, human vessels, a small population of ETB (usually <15%) can be measured by ligand binding. Although in some human beings isolated renal vessel responses to high concentrations of ET-3 were detected,28 comparison of equipotent concentrations of ET-3 and ET-1 in healthy volunteers found that ET-3 had no effect on blood pressure or renal hemodynamics,31 which might have been expected if ETB contributed significantly to a contractile response. Whether the proportion of vascular ETB changes with disease remains controversial and has not been studied in detail in pathophysiological renal tissue. However, detailed studies in vitro in human coronary arteries with atherosclerotic lesions did not show any increase.35 In agreement, in experimental medicine studies in both heart failure patients and volunteer controls, selective ETA antagonism (BQ123) caused the expected potent vasodilatation in the peripheral circulation. However, BQ788 caused vasoconstriction in both groups, consistent with blocking endothelial cell ETB-mediated vasodilatation, with no evidence of contractile ETB.36,37
Other Cell Types Expressing ETA Receptors
ETA have been shown to be present on human and rat podocytes (glomerular epithelial cells) that wrap around the capillaries of the glomerulus within Bowman׳s capsule.38,39 Ortmann et al40 also detected messenger RNA encoding ETB as well as ETA on human podocytes. However, ETA contribute to podocyte injury through cytoskeleton disruption and apoptosis and only ETA antagonists are effective in preventing podocyte injury. In renal disease, proliferation in mesangial cells, extracellular matrix production, and inflammation41,42 are mediated mainly by ETA.
ETB Receptors Predominate in the Kidney and Mediate Beneficial Vasodilatation, Clearing of ET-1 from Plasma, and Natriuresis
In peripheral tissues such as the heart (Fig. 1), ETA are more abundant (>60%) than ETB (Fig. 2). In marked contrast, in the kidney, lungs, and liver this ratio is reversed. Although measurements of receptors within smooth muscle throughout the renal vasculature show a predominance of ETA, 70% of the ET receptors in both cortex and medulla in human kidney are ETB. ETB predominate, reflecting, at least in part, that these are endothelial cell–rich tissues similar to liver and lungs.24–26 Endothelial cells line every vessel wall and have a mass comparable with other endocrine organs. Although ETB also are expressed by other cell types, selective deletion of the endothelial cell ETB, leaving ETB on other cells intact, shows that in many organs, including the kidney, liver, and lungs, endothelial cells represent the majority of the receptors.43
Figure 2.

Ratio of the density of human ETA and ETB measured using radioligand binding in the whole organ (brain, kidney, lung, liver, and heart) and measured in the medial smooth muscle layer of the vasculature within each organ. In human beings, kidney, lung, and liver are ETB-rich, reflecting the expression of ETB receptors on endothelium of the vasculature and other cell types such as the epithelium. In contrast, in the heart, ETA are the principal subtype reflecting expression on myocytes. In the smooth muscle layer of all human vessels, ETA are more abundant than ETB.
A consensus has emerged that ETB mediates vasodilatation by the release of endothelium-derived relaxing factors (nitric oxide, prostacyclin, and/or endothelium-derived hyperpolarizing factor), acting as a feedback mechanism to limit the vasoconstrictor action of ET-1. Infusions of ET-1 into the brachial artery of volunteers produces a biphasic response: low doses of ET-1 cause ETB-mediated vasodilatation, however, as the concentration increases to higher pathophysiological concentrations, vasodilatation is overwhelmed by ETA-mediated constrictor responses. When endothelial dysregulation occurs in renal disease there is a loss of opposing vasodilators, leading to increased vasoconstriction and vasospasm.
Endothelial cell ETB function as scavenging or clearing receptors to remove ET-1 from the circulation,23,43,44 particularly by the ETB-rich tissues: kidney, lungs, and liver34 (Fig. 1). Selectively blocking ETA, but not ETB, with a low dose of the peptide antagonist TAK-044 infused into volunteers caused no change in measured plasma ET-1 levels. However, a higher dose that blocked both subtypes increased ET-1 levels by more than three-fold as a result of reducing clearing by ETB.45
In renal circulation, in agreement with other vascular beds in human beings, systemic infusion of ET-1, which activates both receptors, into volunteers increased blood pressure (6 mm Hg), and decreased renal plasma flow, glomerular filtration rate, and sodium excretion rate.31 Although BQ123 infused alone did not affect basal arterial blood pressure or renal or splanchnic vascular resistance, the antagonist inhibited the increase in vascular resistance induced by co-infusion of ET-1. In contrast, BQ788 alone caused the opposite effect: increased renal or splanchnic vascular resistance, consistent with blocking endothelial cell–receptor vasodilatation. Second, BQ788 potentiated the ET-1–induced increase in vascular resistance mediated by ETA, suggesting that blocking the scavenging receptors modulated plasma ET-1 levels.30 Inhibition of tonic nitric oxide production by inhibition of nitric oxide synthase elicits vasoconstriction with an increase in mean arterial pressure and vascular resistance in many organs, including the kidney. Renal and systemic vasoconstriction in volunteers caused by the nitric oxide synthase inhibitor N-nitro-L-arginine methyl ester were attenuated by BQ123, supporting the concept that the balance between endogenous nitric oxide production and ET-1/ETA activity contributes to renal and systemic tone in human beings.46,47
The role of ETB clearing receptors has been studied in detail in endothelial cell–specific ETB knock-out mice. In these animals, clearance of an intravenous bolus of labeled ET-1 was reduced significantly compared with wild-type controls. Importantly, functioning ETB were retained on all other cell types such as epithelial cells.43,48 Dynamic imaging of rats using positron emission tomography showed that after infusion of 18F ET-1, there was remarkably fast clearance of the radioligand from the circulation (plasma-half life (t1/2) = 0.43 min), with high levels of radioligand accumulated in the kidney, liver, and lung, which rapidly reached equilibrium, and this was maintained for at least 20 minutes. Infusion of BQ788 before injecting 18F ET-1 reduced the amount of radioligand visualized in the lung and kidney by 85% and 55%, respectively, consistent with blockade of ETB. However, infusion of BQ788 after 18F ET-1 did not displace the bound ligand.34 This finding is consistent with the internalization of the ligand-receptor complex to the lysosome where ET-1 is thought to be degraded, similar to other peptides, by cathepsin A. In support, cathepsin A knock-out mice showed reduced ET-1 degradation and significantly increased arterial blood pressure.49 Inactivation of ET-1 by kidney, liver, and lungs may be particularly important for ET-1 because it is structurally unusual compared with other vasoactive peptides, possessing two disulfide bridges that confer resistance to degradation by nonspecific peptidases.
ET-1 promotes diuresis and natriuresis via ETB located on epithelial cells throughout the tubular epithelium, particularly the inner medullary collecting duct cells.50 Deletion of ETB, but not ETA, leads to salt-sensitive hypertension.51 In agreement, the effects of three doses of BQ-123 (0.1, 0.2, and 0.3 mg/kg) on renal hemodynamics, tubular function, and vasoactive hormones were measured in volunteers in a randomized, placebo-controlled, double-blind, dose-response study. The main effect was a dose-dependent increase in renal sodium excretion despite stimulation of the renin-angiotensin system as evidenced by an increase in angiotensin II levels, whereas there was little effect on atrial and brain natriuretic peptides or vasopressin.52
Goddard et al53 elegantly showed that ETA antagonism by BQ123 and angiotensin-converting enzyme (ACE) inhibition using enalapril were synergistic in reducing mean arterial pressure in volunteers. However, BQ-123 increased renal blood flow, increased urinary sodium excretion, and reduced renal vascular resistance only during ACE inhibition. These effects were abolished by ETB blockade using BQ788 and nitric oxide synthase inhibition, whereas cyclooxygenase inhibition had no effect. These results showed that synergism between ETA antagonism and ACE inhibition occurs via an ETB-mediated, nitric oxide–dependent, cyclooxygenase-independent mechanism. In patients with chronic kidney disease, TAK-044 beneficially reduced the mean arterial and systemic vascular resistance index and tended to increase renal plasma flow. TAK-044 had no effect on sodium or lithium clearance, or on the fractional excretion of sodium and lithium.54
Combining the results from a number of different studies has led to the proposal that antagonism of ETB may be undesirable in conditions such as chronic renal failure, and therefore ETA-selective antagonists might be superior to mixed ETA/ETB antagonists. This hypothesis was tested experimentally by comparing the action of BQ123 or BQ788 alone or in combination in hypertensive patients with chronic renal failure.55 Blocking the ETA alone significantly reduced blood pressure in these patients. The magnitude of change was significantly higher than when ETB also was blocked by BQ788. BQ788 alone caused the expected systemic and renal vasoconstriction, supporting the concept that ETB maintain tonic renal vasodilatation in patients. BQ-123 infused alone increased renal blood flow and renal vascular resistance and reduced proteinuria, consistent with a renoprotective action. This effect was lost when ETB were blocked by infusing both BQ788 and BQ123. There was no change in sodium excretion but this may have been the result of a comparatively small number of subjects.55
In a larger study of 22 patients with nondiabetic proteinuric chronic kidney disease, BQ-123 produced significant natriuresis, resulting from increased renal blood flow. In addition, ETA antagonism reduced blood pressure and proteinuria, and, a new finding, decreased arterial stiffness.56 However, in diabetic patients with chronic kidney disease, avosentan (ETA-selective nonpeptide antagonist) was reported to be detrimental as a result of fluid overload.57
ET-Receptor Blockade in Chronic Kidney Disease
Receptor antagonists have emerged as the only strategy in the clinic for blocking the unwanted actions of ET-1. To date, no alternative strategies, such as inhibitors of ET converting enzymes or combined endothelin-converting enzyme (ECE)/neutral endopeptidase (NEP) inhibitors, have been approved. Four compounds, bosentan, ambrisentan, sitaxentan, and macitentan, originally were approved for clinical use in pulmonary arterial hypertension (PAH) (Table 2).11 Sitaxentan, however, was withdrawn from clinical use in 201058 after idiosyncratic hepatitis occurred resulting from acute liver failure, leading to death. PAH affects approximately 100,000 patients in the United States and Europe and currently there is no cure. The disease is characterized by constriction and remodeling of pulmonary vessels, with high blood pressure in the lungs. This leads to right heart failure, which is the ultimate cause of death. Interestingly, although ETA are increased significantly in the failing right ventricle of patients with PAH59 and the failing left ventricle of patients with heart failure,60 clinical trails have failed to show a benefit in patients from the latter group.12 The reasons for this are unclear, but the action of ET antagonists on the vasculature may be more important in restoring the imbalance between ET-induced constriction and opposing vasodilatation of blood vessels.
Table 2.
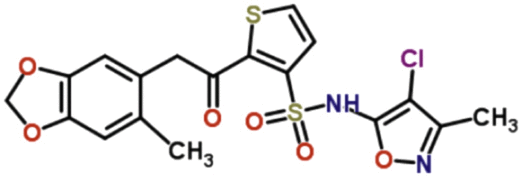
Structure and Pharmacokinetic Properties of ET-Receptor Antagonists in Clinical Use
| Bosentan | Macitentan | Active metabolite of macitentan | Ambrisentan | Sitaxentan (withdrawn from clinical use in 2010) | |
|---|---|---|---|---|---|
| Trade name | Tracleer (Actelion, Allschwil, Switzerland) | Opsumit (Actelion) | Letairis, Volibris (Gilead, Foster City, California) | Thelin (Pfizer, Groton, Connecticut) | |
| Other names | Ro47-0203 | ACT-064992 | ACT-132577 | LU-208075 | TBC-11251 |
| Chemical name | Benzenesulfonamide | Sulfamide | Sulfamide | Benzenepropanoic acid | 3-Thiophenesulfonamide |
| Structure | 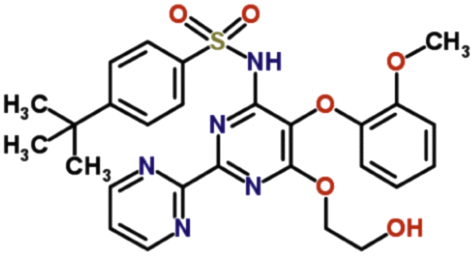 |
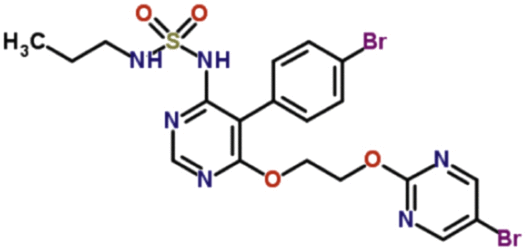 |
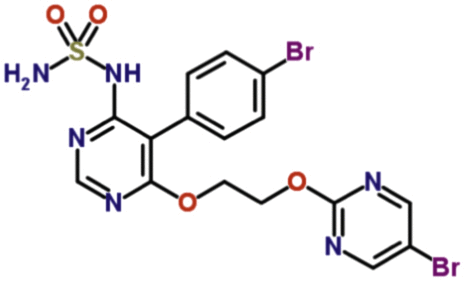 |
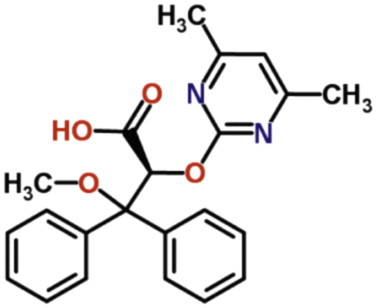 |
 |
| ET plasma levels after administration | ↑↑ | ↑↑ | - | ↑ | ↓ |
| Bioavailability | ~50% | Not reported | Not reported | High | 70%-100% |
| Time to maximum plasma concentration | 3-5 | 4-12 | 30 | 1.7-3.3 | 1-4 |
| Terminal half life (hours) | 5.4 | 16 | 40.2-65.6 | 15 | 10 |
| Excretion in urine (%) | <3 | Not detected | Not detected | Low | 50-60 |
In theory, the selectivity of antagonists should have pharmacologic and pathophysiological consequences. Selectively blocking smooth muscle ETA would be expected to lead to vasodilatation and attenuate proliferation, migration, fibrosis, and hypertrophy. Endothelial ETB, particularly in kidney, lung, and liver, should continue to bind and remove ET-1 where it is overexpressed in pathophysiological conditions, as well as releasing vasodilators to mediate their antiproliferative and antithrombotic actions.
How do we Define Antagonist-Receptor Selectivity?
These four antagonists represent a spectrum of selectivity ranging from bosentan, which is classified by the pharmaceutical company Actelion (Allschwil, Switzerland) as a mixed or balanced ETA/ETB antagonist, to sitaxentan, the most ETA selective. No consensus has emerged about the relative merits of mixed versus ETA-selective compounds in PAH.61,62 Some animal studies have suggested that selective ETA antagonism that leaves ETB unopposed and unblocked is beneficial,63 whereas other studies have shown mixed and ETA-selective antagonists have similar outcomes. The advantage of animal studies are that compounds can be compared head to head, but given differences in cell expression of subtypes these may not necessarily be informative of clinical studies in human beings. No clear-cut advantage of one over another has been reported for selective versus nonselective antagonism in PAH. However, in chronic kidney disease, the function and distribution of receptors suggests an ETA antagonist would be preferable in blocking ETA-mediated constriction and proliferation but sparing endothelial cell vasodilatation, clearing ET from the plasma, and natriuresis.64
The selectivity of a ligand for two receptors usually is calculated by measuring the equilibrium dissociation constant (KD) for the two subtypes, in this case ETA and ETB, to provide a ratio of selectivity65,66 in ligand-binding assays. There is no standardized method or general agreement among pharmaceutical companies to determine which compound should be classified as ETA selective versus a mixed antagonist.62 Accurate information is essential in interpreting results from experiments in animal models and clinical trials as to whether the doses used are likely to result in a compound occupying only ETA or both subtypes. We have proposed that ETA-selective compounds should have at least a 100-fold selectivity for the ETA subtype whereas mixed antagonists should have less than 100-fold ETA selectivity.65 The reason for this is that the degree of receptor occupancy achieved when an antagonist is administered in vivo or in vitro is proportional to the concentration and can be calculated from the affinity using the following formula: L*/(KD + L*), where L* is the free ligand concentration and KD is the affinity constant. For example, a compound that has an affinity measured in a ligand-binding assay of 1 nmol/L for ETA but 100 nmol/L for ETB would have 100-fold selectivity for ETA. By using this equation, at a concentration of 10 nmol/L, 90% of ETA are calculated to be blocked but less than 10% of the ETB. Although this concentration can be achieved accurately under controlled in vitro conditions, 100-fold selectivity is likely to represent the minimum that can be used in vivo to achieve selective ETA blockade. If the plasma concentration of this antagonist was increased to 100 nmol/L, 50% of ETB then would be occupied. Compounds of greater than 1,000-fold selectivity are likely to be needed for clinical or in vivo studies to ensure ETA selectivity is maintained.
As proof of principle, the effect of selective blockade was measured using TAK-044, a peptide antagonist with approximately 250-fold selectivity for the ETA subtype over ETB as measured by ligand binding in the human heart. A 30-mg infusion over 15 minutes of TAK-044 (providing a serum concentration of 2 nmol/L, calculated to block >95% of ETA but <5% ETB) had no effect on the immunoreactive plasma concentrations of ET-1. However, after a higher dose of 750 mg TAK-044 (providing a serum concentration of 80 nmol/L, calculated to block >99% of ETA and >75% ETB), the immunoreactive plasma ET-1 concentrations were increased more than three-fold over basal levels. Importantly, the concentrations of the ET-1 precursor or C-terminal fragment of big endothelin-1 were unchanged, indicating that the increase in ET-1 in the plasma was unlikely to be the result of increased synthesis or release. The most likely sources of endothelin contributing to the observed increase were displacement of receptor-bound peptide and a reduction in plasma clearance mediated by ETB.45
Does Selectivity Matter in Chronic Kidney Disease?
Blocking ETB clearly results in a significant increase in circulating plasma ET-1 levels. However, with a mixed antagonist, this increase is unlikely to be important because the vasoconstrictor ETA also is blocked. Side effects including headache, nausea, and nasal congestion have, to a certain extent, been reported for ETA/ETB mixed antagonists and with ETA-selective compounds. For ETA-selective compound such as ambrisentan, nasal congestion and peripheral edema are more prevalent but they have less of the hepatic effects such as an increase in liver enzyme levels that require liver function tests and drug–drug interactions that are associated with mixed antagonists such as bosentan.67 Studies in mice selectively knocking out ETA from the nephron or collecting duct did not show ETA antagonist-induced fluid retention and this was attenuated where ETA smooth muscle had been deleted, suggesting the mechanism is a direct action on collecting duct receptors and partially within the vasculature.68
Liver toxicity has been a significant problem with bosentan but its mechanism of action has been proposed to be independent of ET receptors and is thought to occur by inhibiting the bile salt export pump leading to accumulation of cytotoxic bile salts, resulting in hepatocellular damage. In contrast, macitentan is thought to enter the liver via passive diffusion and not by active uptake.69 As a result, macitentan has been reported to have a better safety profile compared with bosentan for hepatic toxicity. This is an important consideration in patients with renal or hepatic disease.70 Bosentan is a competitive antagonist of ETA and ETB of the sulfonamide class,71 with a comparatively short half-life and good bioavailability. Bosentan, as with other dual antagonists, tends to have lower rates of fluid retention and edema when used clinically. Although bosentan has been shown to be effective in animal models of renal disease, the compound has not been evaluated in detailed clinical trials involving renal patients. Ambrisentan represents the second chemical class,71 is less ETA selective than sitaxentan, but has good bioavailability and a long half-life. However, again clinical studies have not been reported in chronic kidney disease. Key clinical studies have been performed using sitaxentan,71 the most ETA-selective antagonist that largely supports the hypothesis of selective ET blockade. A randomized, double-blind, three-way, cross-over study of patients with proteinuric chronic kidney disease compared sitaxentan and nifedipine with placebo for proteinuria, blood pressure, and arterial stiffness. As expected, plasma levels of ET-1 were unchanged during sitaxentan treatment, indicating that ET-1 continued to be cleared from the circulation but urinary ET-1 levels were decreased. Blood pressure, arterial stiffness, and proteinuria also were reduced significantly over 6 weeks. Intriguingly, asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthases that is considered an independent marker of disease progression, was increased in these patients, supporting the concept that sparing ETB receptors from blockade with sitaxentan treatment would modulate the nitric oxide pathway. Importantly, these effects were seen in patients already receiving optimal treatment with ACE inhibitors and angiotensin blockers. A related study also found an increase in nocturnal dipping in blood pressure with sitaxentan.72 The results suggested that ETA antagonism had additional longer-term renoprotective effects in patients with chronic kidney disease.73
ET Antagonist for the Future: Macitentan and Atresentan
Macitentan is an insurmountable antagonist, resulting from structure-activity studies to improve the efficacy and tolerability of bosentan, and gained approval in the United States in 2013 for the treatment of PAH. Actelion describes the compound as a dual antagonist, but on the basis of their own data measuring inhibition of [125I]–ET-1 binding to human-expressed receptors it displays approximately 800-fold selectivity for the ETA subtype.74 However, plasma ET-1 concentrations were increased significantly (two-fold at the highest dose tested), suggesting blocking of ETB occurs at the dose used.75 A metabolite of macitentan, ACT-132577, is active pharmacologically, albeit with a lower potency, but reaches higher plasma concentrations and has a longer half-life than macitentan.76–79 Key pharmacologic parameters suggest macitentan will have the potential for greater efficacy and safety than bosentan. Macitentan has a much longer receptor occupancy (17 minutes compared with 70 seconds for bosentan), probably as a result of interaction with different amino acid residues in the ET receptors and is an order of magnitude more potent than bosentan, measured by in vitro assays. Pharmacokinetic benefits include fewer interactions with other drugs, with no requirement to alter doses in patients with renal (or hepatic) impairment. Crucially, the compound has improved hepatic safety and reduced edema/fluid retention compared with bosentan.79,80 A number of clinical trials are actively recruiting, however, these do not yet include chronic kidney disease patients.81
Clinical trials also recently were reported on an investigational ETA-selective antagonist: atrasentan (ABT 627).82 The aim of the double-blind study performed in parallel at two centers was to determine whether albuminuria was reduced further when atrasentan was administered at two different doses, with inhibitors of the renin-angiotensin system, to patients with type 2 diabetic nephropathy. Atrasentan reduced albuminuria at both doses tested, and reduced blood pressure, cholesterol, and triglyceride levels, with unwanted side effects being more manageable at the lower dose. These promising results lead to the initiation of a phase 3 multicenter trial (Study Of Diabetic Nephropathy With Atrasentan83) with 4,000 patients.
Perspectives
After more than 25 years since the discovery of ET, the peptide remains the most powerful and long-lasting constrictor of the human vasculature including the kidney described to date. In pathophysiological conditions, ET-1 contributes to vascular remodeling, proliferation of mesangial cells, and extracellular matrix production mainly through binding to ETA. Beneficial actions of ET-1 on sodium and water regulation are mainly ETB-mediated. These findings suggest an ETA-selective antagonist would have a therapeutic advantage over a mixed antagonist in renal disease, and indeed the small number of acute studies directly comparing the peptide antagonists BQ123 versus BQ788 suggest ETA blockade, sparing ETB, may be beneficial. However, this is balanced by the possible greater prevalence of side effects such as edema reported for small-molecule, orally active ETA antagonists compared with mixed antagonists, although the latter also have their limitations because of liver toxicity. In addition, head-to-head studies in patients comparing orally active ETA antagonists with mixed antagonists have not been performed. The renal ET system remains a compelling target: will new therapies be clinically relevant in the future? This question may be answered by the next generation of ET antagonists.
Footnotes
Financial support: Supported by the British Heart Foundation (PS/02/001, PG/05/127/19872, FS/12/64/130001) Wellcome Trust Programme in Metabolic and Cardiovascular Disease 096822/Z/11/Z, National Institute for Health Research Cambridge Biomedical Research Centre, and the Pulmonary Hypertension Association United Kingdom.
Conflict of interest statement: none.
References
- 1.Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 2.Inoue A., Yanagisawa M., Kimura S., Kasuya Y., Miyauchi T., Goto K. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc Natl Acad Sci U S A. 1989;86:2863–2867. doi: 10.1073/pnas.86.8.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karet F.E., Davenport A.P. Localization of endothelin peptides in human kidney. Kidney Int. 1996;49:382–387. doi: 10.1038/ki.1996.56. [DOI] [PubMed] [Google Scholar]
- 4.Arai H., Hori S., Aramori I., Ohkubo H., Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–732. doi: 10.1038/348730a0. [DOI] [PubMed] [Google Scholar]
- 5.Sakurai T., Yanagisawa M., Takuwa Y., Miyazaki H., Kimura S., Goto K. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature. 1990;348:732–735. doi: 10.1038/348732a0. [DOI] [PubMed] [Google Scholar]
- 6.Davenport A.P. International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharmacol Rev. 2002;54:219–226. doi: 10.1124/pr.54.2.219. [DOI] [PubMed] [Google Scholar]
- 7.Davenport A.P., Maguire J.J. Endothelin. Handbk Exp Pharmacol. 2006;152:295–329. doi: 10.1007/3-540-32967-6_9. [DOI] [PubMed] [Google Scholar]
- 8.Kohan D.E., Inscho E.W., Wesson D., Pollock D.M. Physiology of endothelin and the kidney. Compr Physiol. 2011;1:883–919. doi: 10.1002/cphy.c100039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohan D.E., Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014;86:896–904. doi: 10.1038/ki.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport A.P., Maguire J.J. Pharmacology of renal endothelin receptors. Contrib Nephrol. 2011;172:1–17. doi: 10.1159/000328678. [DOI] [PubMed] [Google Scholar]
- 11.Magure J.J., Davenport A.P. Endothelin@25–new agonists, antagonists, inhibitors and emerging research frontiers: IUPHAR review. Br J Pharmacol. 2014;17:5555–5572. doi: 10.1111/bph.12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohan D.E., Cleland J.G., Rubin L.J., Theodorescu D., Barton M. Clinical trials with endothelin receptor antagonists: what went wrong and where can we improve? Life Sci. 2012;91:528–539. doi: 10.1016/j.lfs.2012.07.034. [DOI] [PubMed] [Google Scholar]
- 13.Davenport A.P., Alexander S.P., Sharman J.L., Pawson A.J., Benson H.E., Monaghan A.E. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013;65:967–986. doi: 10.1124/pr.112.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Southern C., Cook J.M., Neetoo-Isseljee Z., Taylor D.L., Kettleborough C.A., Merritt A. Screening beta-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J Biomol Screen. 2013;18:599–609. doi: 10.1177/1087057113475480. [DOI] [PubMed] [Google Scholar]
- 15.Meyer R.C., Giddens M.M., Schaefer S.A., Hall R.A. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc Natl Acad Sci U S A. 2013;110:9529–9534. doi: 10.1073/pnas.1219004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.William D.L., Jones K.L., Pettibone D.J., Lis E.V., Clineschmidt B.V. Sarafotoxin S6c, an agonist which distinguishes between endothelin receptor subtypes. Biochem Biophys Res Commun. 1991;175:556–561. doi: 10.1016/0006-291x(91)91601-8. [DOI] [PubMed] [Google Scholar]
- 17.Watakabe T., Urade Y., Takai M., Umemura I., Okada T. A reversible radioligand specific for the ETB receptor: [125I]Tyr13-Suc-[Glu9,Ala11,15]-endothelin-1(8-21), [125I]IRL 1620. Biochem Biophys Res Commun. 1992;185:867–873. doi: 10.1016/0006-291x(92)91707-w. [DOI] [PubMed] [Google Scholar]
- 18.Ihara M., Saeki T., Fukuroda T., Kimura S., Ozaki S., Patel A.C. A novel radioligand [125I]BQ-3020 selective for endothelin (ETB) receptors. Life Sci. 1992;51:PL47–PL52. doi: 10.1016/0024-3205(92)90418-o. [DOI] [PubMed] [Google Scholar]
- 19.Ihara M., Noguchi K., Saeki T., Fukuroda T., Tsuchida S., Kimura S. Biological profiles of highly potent novel endothelin antagonists selective for the ET(A) receptor. Life Sci. 1992;50:247–255. doi: 10.1016/0024-3205(92)90331-i. [DOI] [PubMed] [Google Scholar]
- 20.Aramori I., Nirei H., Shoubo M., Sogabe K., Nakamura K., Kojo H. Subtype selectivity of a novel endothelin antagonist, FR139317, for the two endothelin receptors in transfected Chinese-hamster ovary cells. Mol Pharmacol. 1993;43:127–131. [PubMed] [Google Scholar]
- 21.Masuda Y., Sugo T., Kikuchi T., Kawata A., Satoh M., Fujisawa Y. Receptor binding and antagonist properties of a novel endothelin receptor antagonist, TAK-044 [cyclo[D-alpha-aspartyl-3-[(4-phenylpiperazin-1-yl) carbonyl]-L-alanyl-L-alpha-aspartyl-D-2-(2-thienyl) glycyl-L-leucyl-D-tryptophyl]disodium salt], in human endothelinA and endothelinB receptors. J Pharmacol Exp Ther. 1996;279:675–685. [PubMed] [Google Scholar]
- 22.Ishikawa K., Ihara M., Noguchi K., Mase T., Mino N., Saeki T. Biochemical and pharmacological profile of a potent and selective endothelinB-receptor antagonist, BQ-788. Proc Natl Acad Sci U S A. 1994;91:4892–4896. doi: 10.1073/pnas.91.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karet F.E., Davenport A.P. Human kidney: endothelin isoforms detected by HPLC with radioimmunoassay and receptor subtypes detected using ligands BQ123 and BQ3020. J Cardiovasc Pharmacol. 1993;22(Suppl 8):S29–S33. doi: 10.1097/00005344-199322008-00010. [DOI] [PubMed] [Google Scholar]
- 24.Kuc R., Davenport A.P. Comparison of endothelin-A and endothelin-B receptor distribution visualized by radioligand binding versus immunocytochemical localization using subtype selective antisera. J Cardiovasc Pharmacol. 2004;44(Suppl 1):S224–S226. doi: 10.1097/01.fjc.0000166260.35099.d5. [DOI] [PubMed] [Google Scholar]
- 25.Kuc R.E., Karet F.E., Davenport A.P. Characterization of peptide and nonpeptide antagonists in human kidney. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S373–S375. [PubMed] [Google Scholar]
- 26.Karet F.E., Kuc R.E., Davenport A.P. Novel ligands BQ123 and BQ3020 characterize endothelin receptor subtypes ETA and ETB in human kidney. Kidney Int. 1993;44:36–42. doi: 10.1038/ki.1993.210. [DOI] [PubMed] [Google Scholar]
- 27.Davenport A.P., Kuc R.E., Hoskins S.L., Karet F.E., Fitzgerald F. [125I]-PD151242: a selective ligand for endothelin ETA receptors in human kidney which localizes to renal vasculature. Br J Pharmacol. 1994;113:1303–1310. doi: 10.1111/j.1476-5381.1994.tb17140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maguire J.J., Kuc R.E., O׳Reilly G., Davenport A.P. Vasoconstrictor endothelin receptors characterized in human renal artery and vein in vitro. Br J Pharmacol. 1994;113:49–54. doi: 10.1111/j.1476-5381.1994.tb16172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maguire J.J., Davenport A.P. ETA receptor-mediated constrictor responses to endothelin peptides in human blood vessels in vitro. Br J Pharmacol. 1995;115:191–197. doi: 10.1111/j.1476-5381.1995.tb16338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohm F., Pernow J., Lindstrom J., Ahlborg G. ETA receptors mediate vasoconstriction, whereas ETB receptors clear endothelin-1 in the splanchnic and renal circulation of healthy men. Clin Sci (Lond) 2003;104:143–151. doi: 10.1042/CS20020192. [DOI] [PubMed] [Google Scholar]
- 31.Kaasjager K.A., Shaw S., Koomans H.A., Rabelink T.J. Role of endothelin receptor subtypes in the systemic and renal responses to endothelin-1 in humans. J Am Soc Nephrol. 1997;8:32–39. doi: 10.1681/ASN.V8132. [DOI] [PubMed] [Google Scholar]
- 32.Asano T.1, Ikegaki I., Suzuki Y., Satoh S., Shibuya M. Endothelin and the production of cerebral vasospasm in dogs. Biochem Biophys Res Commun. 1989;159:1345–1351. doi: 10.1016/0006-291x(89)92258-4. [DOI] [PubMed] [Google Scholar]
- 33.Pierre L.N., Davenport A.P. Blockade and reversal of endothelin-induced constriction in pial arteries from human brain. Stroke. 1999;30:638–643. doi: 10.1161/01.str.30.3.638. [DOI] [PubMed] [Google Scholar]
- 34.Johnstrom P., Fryer T.D., Richards H.K., Harris N.G., Barret O., Clark J.C. Positron emission tomography using 18F-labelled endothelin-1 reveals prevention of binding to cardiac receptors owing to tissue-specific clearance by ET B receptors in vivo. Br J Pharmacol. 2005;144:115–122. doi: 10.1038/sj.bjp.0706064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maguire J.J., Davenport A.P. No alteration in vasoconstrictor endothelin-B-receptor density or function in human coronary artery disease. J Cardiovasc Pharmacol. 2000;36(Suppl 1):S380–S381. doi: 10.1097/00005344-200036051-00110. [DOI] [PubMed] [Google Scholar]
- 36.Love M.P., Haynes W.G., Webb D.J., McMurray J.J. Venous endothelin receptor function in patients with chronic heart failure. Clin Sci (Lond) 2000;98:65–70. [PubMed] [Google Scholar]
- 37.Love M.P., Ferro C.J., Haynes W.G., Plumpton C., Davenport A.P., Webb D.J. Endothelin receptor antagonism in patients with chronic heart failure. Cardiovasc Res. 2000;47:166–172. doi: 10.1016/s0008-6363(00)00081-x. [DOI] [PubMed] [Google Scholar]
- 38.Gauquelin G., Thibault G., Garcia R. Characterization of renal glomerular endothelin receptors in the rat. Biochem Biophys Res Commun. 1989;164:54–57. doi: 10.1016/0006-291x(89)91681-1. [DOI] [PubMed] [Google Scholar]
- 39.Rebibou J.M., He C.J., Delarue F., Peraldi M.N., Adida C., Rondeau E. Functional endothelin 1 receptors on human glomerular podocytes and mesangial cells. Nephrol Dial Transplant. 1992;7:288–292. doi: 10.1093/oxfordjournals.ndt.a092130. [DOI] [PubMed] [Google Scholar]
- 40.Ortmann J., Amann K., Brandes R.P., Kretzler M., Munter K., Parekh N. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension. 2004;44:9749–9781. doi: 10.1161/01.HYP.0000149249.09147.b4. [DOI] [PubMed] [Google Scholar]
- 41.Barton M. Therapeutic potential of endothelin receptor antagonists for chronic proteinuric renal disease in humans. Biochim Biophys Acta. 2010;1802:1203–1213. doi: 10.1016/j.bbadis.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 42.Neuhofer W., Pittrow D. Endothelin receptor selectivity in chronic kidney disease: rationale and review of recent evidence. Eur J Clin Invest. 2009;39(Suppl 2):50–67. doi: 10.1111/j.1365-2362.2009.02121.x. [DOI] [PubMed] [Google Scholar]
- 43.Kelland N.F., Kuc R.E., McLean D.L., Azfer A., Bagnall A.J., Gray G.A. Endothelial cell-specific ETB receptor knockout: autoradiographic and histological characterisation and crucial role in the clearance of endothelin-1. Can J Physiol Pharmacol. 2010;88:644–651. doi: 10.1139/Y10-041. [DOI] [PubMed] [Google Scholar]
- 44.Fukuroda T., Fujikawa T., Ozaki S., Ishikawa K., Yano M., Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199:1461–1465. doi: 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 45.Plumpton C., Ferro C.J., Haynes W.G., Webb D.J., Davenport A.P. The increase in human plasma immunoreactive endothelin but not big endothelin-1 or its C-terminal fragment induced by systemic administration of the endothelin antagonist TAK-044. Br J Pharmacol. 1996;119:311–314. doi: 10.1111/j.1476-5381.1996.tb15987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montanari A., Carra N., Perinotto P., Iori V., Fasoli E., Biggi A. Renal hemodynamic control by endothelin and nitric oxide under angiotensin II blockade in man. Hypertension. 2002;39:715–720. doi: 10.1161/hy0202.104399. [DOI] [PubMed] [Google Scholar]
- 47.Montanari A., Biggi A., Carra N., Fasoli E., Calzolari M., Corsini F. Endothelin-A blockade attenuates systemic and renal hemodynamic effects of L-NAME in humans. Hypertension. 2000;35:518–523. doi: 10.1161/01.hyp.35.1.518. [DOI] [PubMed] [Google Scholar]
- 48.Bagnall A.J., Kelland N.F., Gulliver-Sloan F., Davenport A.P., Gray G.A., Yanagisawa M. Deletion of endothelial cell endothelin B receptors does not affect blood pressure or sensitivity to salt. Hypertension. 2006;48:286–293. doi: 10.1161/01.HYP.0000229907.58470.4c. [DOI] [PubMed] [Google Scholar]
- 49.Seyrantepe V., Hinek A., Peng J., Fedjaev M., Ernest S., Kadota Y. Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation. 2008;117:1973–1981. doi: 10.1161/CIRCULATIONAHA.107.733212. [DOI] [PubMed] [Google Scholar]
- 50.Gariepy C.E., Ohuchi T., Williams S.C., Richardson J.A., Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest. 2000;105:925–933. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ge Y., Bagnall A., Stricklett P.K., Strait K., Webb D.J., Kotelevtsev Y. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2006;291:F1274–F1280. doi: 10.1152/ajprenal.00190.2006. [DOI] [PubMed] [Google Scholar]
- 52.Pedersen E.B., Thomsen I.M., Fjordside L.S. Effect of BQ-123, an endothelin antagonist, on renal hemodynamics, tubular function, vasoactive hormones, and blood pressure in healthy humans: a dose response study. Am J Hypertens. 2005;18:1578–1585. doi: 10.1016/j.amjhyper.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 53.Goddard J., Eckhart C., Johnston N.R., Cumming A.D., Rankin A.J., Webb D.J. Endothelin A receptor antagonism and angiotensin-converting enzyme inhibition are synergistic via an endothelin B receptor-mediated and nitric oxide-dependent mechanism. J Am Soc Nephrol. 2004;15:2601–2610. doi: 10.1097/01.ASN.0000141313.84470.4B. [DOI] [PubMed] [Google Scholar]
- 54.Dhaun N., Ferro C.J., Davenport A.P., Haynes W.G., Goddard J., Webb D.J. Haemodynamic and renal effects of endothelin receptor antagonism in patients with chronic kidney disease. Nephrol Dial Transplant. 2007;22:3228–3234. doi: 10.1093/ndt/gfm364. [DOI] [PubMed] [Google Scholar]
- 55.Goddard J., Johnston N.R., Hand M.F., Cumming A.D., Rabelink T.J., Rankin A.J. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation. 2004;109:1186–1193. doi: 10.1161/01.CIR.0000118499.69469.51. [DOI] [PubMed] [Google Scholar]
- 56.Dhaun N., Macintyre I.M., Melville V., Lilitkarntakul P., Johnston N.R., Goddard J. Blood pressure-independent reduction in proteinuria and arterial stiffness after acute endothelin-a receptor antagonism in chronic kidney disease. Hypertension. 2009;54:113–119. doi: 10.1161/HYPERTENSIONAHA.109.132670. [DOI] [PubMed] [Google Scholar]
- 57.Mann J.F., Green D., Jamerson K., Ruilope L.M., Kuranoff S.J., Littke T., ASCEND Study Group Avosentan for overt diabetic nephropathy. J Am Soc Nephrol. 2010;21 doi: 10.1681/ASN.2009060593. 527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Don G.W., Joseph F., Celermajer D.S., Corte T.J. Ironic case of hepatic dysfunction following the global withdrawal of sitaxentan. Intern Med J. 2012;42:1351–1354. doi: 10.1111/imj.12007. [DOI] [PubMed] [Google Scholar]
- 59.Kuc R.E., Carlebur M., Maguire J.J., Yang P., Long L., Toshner M. Modulation of endothelin receptors in the failing right ventricle of the heart and vasculature of the lung in human pulmonary arterial hypertension. Life Sci. 2014;118:391–396. doi: 10.1016/j.lfs.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zolk O., Quattek J., Sitzler G., Schrader T., Nickenig G., Schnabel P. Expression of endothelin-1, endothelin-converting enzyme, and endothelin receptors in chronic heart failure. Circulation. 1999;99:2118–2123. doi: 10.1161/01.cir.99.16.2118. [DOI] [PubMed] [Google Scholar]
- 61.Opitz C.F., Ewert R., Kirch W., Pittrow D. Inhibition of endothelin receptors in the treatment of pulmonary arterial hypertension: does selectivity matter? Eur Heart J. 2008;29:1936–1948. doi: 10.1093/eurheartj/ehn234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vachiéry J.L., Davenport A. The endothelin system in pulmonary and renal vasculopathy: les liaisons dangereuses. Eur Respir Rev. 2009;18:260–271. doi: 10.1183/09059180.00005709. [DOI] [PubMed] [Google Scholar]
- 63.Allcock G.H., Warner T.D. Inhibition of ETB receptors limits the efficacy of nonselective endothelin antagonists in vivo. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S177–S179. [PubMed] [Google Scholar]
- 64.Ohkita M., Takaoka M., Matsumura Y. Drug discovery for overcoming chronic kidney disease (CKD): the endothelin ET B receptor/nitric oxide system functions as a protective factor in CKD. J Pharmacol Sci. 2009;109:7–13. doi: 10.1254/jphs.08r10fm. [DOI] [PubMed] [Google Scholar]
- 65.Maguire J.J., Davenport A.P. Importance of sub-type selectivity for endothelin receptor antagonists in the human vasculature. In: Abraham D., Handler C., Dashwood M., Coghlan G., editors. Translational vascular medicine: pathogenesis, diagnosis, and treatment. Springer; London: 2012. 151-72. [Google Scholar]
- 66.Maguire J.J., Kuc R.E., Davenport A.P. Defining the affinity and receptor sub-type selectivity of four classes of endothelin antagonists in clinically relevant human cardiovascular tissues. Life Sci. 2012;91:681–686. doi: 10.1016/j.lfs.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 67.Vizza C.D., Fedele F., Pezzuto B., Rubin L.J. Safety and efficacy evaluation of ambrisentan in pulmonary hypertension. Expert Opin Drug Saf. 2012;11:1003–1011. doi: 10.1517/14740338.2012.714770. [DOI] [PubMed] [Google Scholar]
- 68.Stuart D., Chapman M., Rees S., Woodward S., Kohan D.E. Myocardial, smooth muscle, nephron, and collecting duct gene targeting reveals the organ sites of endothelin A receptor antagonist fluid retention. J Pharmacol Exp Ther. 2013;346:182–189. doi: 10.1124/jpet.113.205286. [DOI] [PubMed] [Google Scholar]
- 69.Treiber A., Aanismaa P., de Kanter R., Delahaye S., Treher M., Hess P. Macitentan does not interfere with hepatic bile salt transport. J Pharmacol Exp Ther. 2014;350:130–143. doi: 10.1124/jpet.114.214106. [DOI] [PubMed] [Google Scholar]
- 70.Dingemanse J., Sidharta P.N., Maddrey W.C., Rubin L.J., Mickail H. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf. 2014;13:391–405. doi: 10.1517/14740338.2014.859674. [DOI] [PubMed] [Google Scholar]
- 71.Palmer M.J. Endothelin receptor antagonists: status and learning 20 years on. Prog Med Chem. 2009;47:203–237. doi: 10.1016/S0079-6468(08)00205-1. [DOI] [PubMed] [Google Scholar]
- 72.Dhaun N., Moorhouse R., MacIntyre I.M., Melville V., Oosthuyzen W., Kimmitt R.A. Diurnal variation in blood pressure and arterial stiffness in chronic kidney disease: the role of endothelin-1. Hypertension. 2014;64:296–304. doi: 10.1161/HYPERTENSIONAHA.114.03533. [DOI] [PubMed] [Google Scholar]
- 73.Dhaun N., Melville V., Blackwell S., Talwar D.K., Johnston N.R., Goddard J. Endothelin-A receptor antagonism modifies cardiovascular risk factors in CKD. J Am Soc Nephrol. 2013;24:31–36. doi: 10.1681/ASN.2012040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pulido T., Adzerikho I., Channick R.N., Delcroix M., Galie N., Ghofrani H.A. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 75.Sidharta P.N., van Giersbergen P.L., Halabi A., Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67:977–984. doi: 10.1007/s00228-011-1043-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iglarz M., Binkert C., Morrison K., Fischli W., Gatfield J., Treiber A. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736–745. doi: 10.1124/jpet.108.142976. [DOI] [PubMed] [Google Scholar]
- 77.Sidharta P.N., Lindegger N., Ulc I., Dingemanse J. Pharmacokinetics of the novel dual endothelin receptor antagonist macitentan in subjects with hepatic or renal impairment. J Clin Pharmacol. 2014;54:291–300. doi: 10.1002/jcph.193. [DOI] [PubMed] [Google Scholar]
- 78.Sidharta P.N., van Giersbergen P.L., Safety Dingemanse J. tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol. 2013;53:1131–1138. doi: 10.1002/jcph.152. [DOI] [PubMed] [Google Scholar]
- 79.Bruderer S., Hopfgartner G., Seiberling M., Wank J., Sidharta P.N. Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans. Xenobiotica. 2012;42:901–910. doi: 10.3109/00498254.2012.664665. [DOI] [PubMed] [Google Scholar]
- 80.Gatfield J., Mueller Grandjean C., Sasse T., Clozel M., Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One. 2012;7:e47662. doi: 10.1371/journal.pone.0047662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patel T.1, McKeage K. Macitentan: first global approval. Drugs. 2014;74:127–133. doi: 10.1007/s40265-013-0156-6. [DOI] [PubMed] [Google Scholar]
- 82.de Zeeuw D., Coll B., Andress D., Brennan J.J., Tang H., Houser M. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J Am Soc Nephrol. 2014;25:1083–1093. doi: 10.1681/ASN.2013080830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Study Of Diabetic Nephropathy With Atrasentan (SONAR). Available from: http://clinicaltrials.gov/show/NCT01858532. Accessed: August 10, 2014.