

Abstract
Psoriasis is a common inflammatory skin disease with an incompletely understood etiology. The disease is characterized by red, scaly and well-demarcated skin lesions formed by the hyperproliferation of epidermal keratinocytes. This hyperproliferation is driven by cytokines secreted by activated resident immune cells, an infiltrate of T cells, dendritic cells and cells of the innate immune system, as well as the keratinocytes themselves. Psoriasis has a strong hereditary character and has a complex genetic background. Genome-wide association studies have identified polymorphisms within or near a number of genes encoding cytokines, cytokine receptors or elements of their signal transduction pathways, further implicating these cytokines in the psoriasis pathomechanism. A considerable number of inflammatory cytokines have been shown to be elevated in lesional psoriasis skin, and the serum concentrations of a subset of these also correlate with psoriasis disease severity. The combined effects of the cytokines found in psoriasis lesions likely explain most of the clinical features of psoriasis, such as the hyperproliferation of keratinocytes, increased neovascularization and skin inflammation. Thus, understanding which cytokines play a pivotal role in the disease process can suggest potential therapeutic targets. A number of cytokines have been therapeutically targeted with success, revolutionizing treatment of this disease. Here we review a number of key cytokines implicated in the pathogenesis of psoriasis.
Keywords: Psoriasis, Skin, Inflammation, Cytokine, Interleukin
1. Psoriasis, an immune-mediated skin disease
Psoriasis is a common immune-mediated inflammatory skin disease affecting all major human populations with the greatest prevalence of 2–3% in those of northern European ancestry [1]. The most common form of the disease is chronic plaque psoriasis (psoriasis vulgaris), which manifests as plaques of red, scaly and well-demarcated regions of inflamed skin. These plaques are the result of increased keratinocyte proliferation, where up to an eight-fold increase in epidermal cell turnover has been demonstrated [2], leading to a thickening of the epidermis (acanthosis) and altered keratinocyte differentiation. This marked hastening of the transit of keratinocytes to the upper layers of the epidermis results in perturbation of their normal maturation program, resulting in altered protein expression, loss of a mature granular layer and retention of keratinocyte nuclei (parakeratosis). These changes are accompanied by dermal angiogenesis leading to an increasingly complex underlying vascular system, giving the plaques their deep red coloration (Fig. 1). This increased vascularity allows for a greater influx of inflammatory cells into the skin, further driving the inflammation. T cells and a myriad of cells from the adaptive and innate arms of the immune system are present early in lesions, forming characteristic nests of activated leukocytes in the reticular dermis, with a mixed CD4/CD8+ T cell infiltrate in the papillary dermis and an exclusively CD8+ T cell population in the epidermis (Fig. 1). As such, psoriasis is now generally regarded as a T cell-mediated immune disease with a mixed Th1/Th17 cytokine environment [3–5].
Fig. 1.
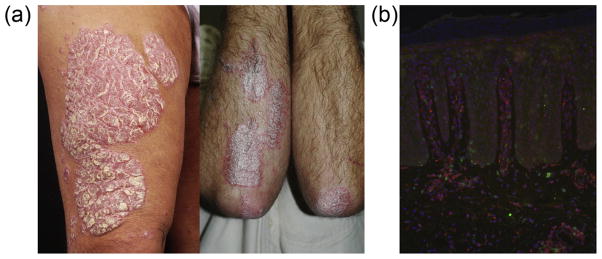
Plaque psoriasis lesions. Characteristic well-demarcated skin lesions, common on the elbows, knees, trunk and extensor surfaces of the limbs (a). Immunofluorescent staining of a biopsy of lesional psoriasis skin showing infiltration of CD4+ T cells (red) in the dermis and CD8+ T cells (green) in the dermal and epidermal compartments. Cell nuclei counterstained with DAPI (blue). 100× original magnification. Photo courtesy of Dr. Johann E. Gudjonsson, University of Michigan Medical School. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2. Psoriasis, the product of a cytokine storm
A decade ago we [6] and others [7] suggested that the interplay between cytokines expressed in psoriasis skin (Fig. 2) could explain most of the clinical features of psoriasis, such as the hyperproliferation of keratinocytes, increased neovascularization and inflammation, and that by determining which cytokines played a central role in the disease process, interesting therapeutic targets could be identified [6]. In the time since, drugs targeting tumor necrosis factor (TNF)-α, interleukin (IL)-12/23, IL-17, IL-22, IL-23, granulocyte monocyte-colony stimulating factor (GM-CSF), as well as inhibitors of the Janus kinases (JAK1/2/3) downstream of a number of cytokine receptors, have reached the clinic or are currently in clinical trials [8]. Recently several technologies have been developed that allow the quantification of multiple cytokines and growth factors in tissues both at the level of protein [9] and mRNA using cDNA microarrays [10] and high-throughput complementary DNA sequencing (RNA-seq) [11] giving an increasingly more sensitive and global view of the psoriasis transcriptome (Fig. 2). Here we present a short survey of key cytokines implicated in the pathogenesis of psoriasis.
Fig. 2.

Psoriasis lesions contain multiple up-regulated cytokines. RNA-seq data [11] queried for transcripts of interleukins 1 through 38. Values calculated for the fold change in reads per kilobase of transcript per million mapped reads (RKPM) of involved skin vs. uninvolved skin.
3. The IL-1 family in psoriasis: masters of inflammation
IL-1 is viewed as the archetypal pro-inflammatory cytokine, studied for its fever-inducing and inflammatory properties since the 1940s and as such IL-1 was the first cytokine detected in skin [12]. The IL-1 family of cytokines now contains 11 ligands and 9 receptors, many of which are altered in their expression in both non-lesional and lesional psoriasis skin compared with healthy control skin [13]. The canonical IL-1 family members IL-1α and IL-1β are both present in normal healthy epidermis [14,15] and act by recruiting IL-1R1 and the accessory protein IL-1RAcP, a process moderated by the decoy receptor IL-1R2 and the receptor antagonist IL-1Ra. IL-1α and IL-1β are both expressed as immature pro-proteins but have differential requirements for activation. Keratinocytes constitute a major reservoir of IL-1α [16] and although pro-IL-1α is active as a cell membrane-associated cytokine, under conditions of cell stress, particularly stimuli that trigger cytoplasmic NOD-like receptor activation, cytoplasmic pro-IL-1α is rapidly processed by calpain-like proteases and secreted. Depending on the stimulus, this process may be either independent of caspase-1 or require the presence, but not catalytic activity of, caspase-1 [17]. On the other hand, the activity of IL-1β, along with IL-1 family member IL-18, always requires post-translational cleavage of the pro-protein by caspase-1 for activity. Cell stress, infection or local danger signals trigger the assembly of inflammasome complexes [15,18,19], which activate caspase-1, which in turn cleaves pro-IL-1β to its active form. The IL-1 family conspicuously lacks signal sequences for conventional cytokine secretion via the endoplasmic reticulum – Golgi pathways and the mechanism of secretion may vary with cell type and inducing stimulus, with proposed mechanisms involving lysosome exocytosis, multivesicular body formation and exosome release or pyroptosis [20]. Interestingly, pro-IL-1α also contains a nuclear localization motif in its N-terminal domain [21] which permits its translocation to the nucleus and activation of NF-κB and AP-1, a mechanism that may lower the signaling threshold for an inflammatory response by IL-1α-expressing cells.
Once secreted, the two IL-1 isoforms have similar functions, acting in an autocrine and paracrine fashion on keratinocytes and also on local fibroblasts, vascular endothelium and lymphocytes. IL-1 has rapid and profound effects on keratinocytes, inducing a swath of gene transcripts involved with inflammation and antimicrobial responses, a transcriptional signature which closely resembles differences seen in lesional versus non-lesional psoriatic skin [22,23], suggesting that IL-1 could be an important mediator in psoriasis pathogenesis. IL-1 also drives the expression of ICAM and VCAM-1 by dermal endothelial cells, as well as the secretion of platelet aggregating factor, nitric oxide and prostaglandin I2, leading to the increased recruitment of immune cells to the skin. Interestingly, IL-1α but not IL-1β appears to be critical for the formation of T cell-APC dermal clusters, which provide an extra-lymphoid environment for intimate contact between DCs, T cells and macrophages for the elicitation of immune reposes [24]. APCs are exquisitely sensitive to IL-1, upregulating a host of maturation markers, priming for antigen presentation to T cells. Moreover, IL-1 appears to be a key cytokine in the development of skin Th17 responses in psoriasis, with IL-1 and IL-23 cooperating in the induction of IL-17 production by T cells [25]. The use of synthetic IL-1R antagonists, such as anakinra, are helpful in the treatment of rheumatoid arthritis [26] but have failed to show efficacy for psoriasis [27], possibly because IL-1Ra is already abundant in psoriatic lesions [28]. A number of case reports suggest that IL-1 antagonism may be a useful approach for treating pustular variants of psoriasis [29], however, there is currently a lack of adequately controlled trials to support this.
4. The IL-36 sub-family: specialists in epithelial inflammation
Analogous to IL-1α, -1β, -1Ra, and their receptor IL-1RI, the sub-family of IL-36 cytokines includes three receptor agonists: IL-36α (formerly known as IL-1F6) [30], IL-36β (IL-1F8) [31,32], and IL-36γ (IL-1F9) [32,33], and a receptor antagonist, IL-36Ra (IL-1F5). These cytokines bind their cognate receptor IL-36R (IL-1Rrp2)/IL-1RAcP (shared with IL-1RI) to signal via NF-κB and MAP kinases [32]. In humans, the IL-36 receptor is widely expressed by epithelia [13,34,35] and antigen-presenting cells, but not human T cells or neutrophils [36].
The role of the IL-36 cytokine system in skin inflammation has been extensively demonstrated [13,30,32,33]. All of the IL-36 family members have been shown to be upregulated in human psoriasis lesions [13,30,33] with IL-36γ correlating particularly well with psoriasis disease severity [37]. In addition, the IL-36 family is active in a number of mouse models of skin inflammation [13,30,36,38]. Expression of murine IL-36α induces leukocyte infiltration and skin inflammation [30,36] and blockade of the IL-36 system could ameliorate imiquimod-Toll-like receptor (TLR)-7/8 induced skin inflammation [38], indicating that IL-36 cytokines are critical members of the cytokine milieu that drives skin inflammation in this model. Thus far, all IL-36 ligands appear to be functionally equivalent with respect to their activity on human cells. IL-36 induces the expression of antimicrobial peptides, cytokines and chemokines by keratinocytes [13,36], and drives the activation of APCs [36,39]. These observations are consistent with a role for IL-36 in driving psoriatic skin inflammation by attracting neutrophils, myeloid cells and T cells into developing psoriasis lesions and altering APC function to potentiate the inflammatory cycle. In early experiments, microgram quantities of recombinant IL-36 ligands were required for inducing keratinocyte responses [13,40], however removal of 5, 4 or 19 amino acids N terminal to a A-X-Asp motif of IL-36α, β, or γ, respectively, results in up to a 10,000-fold increase in activity [41]; however, these peptides do not contain a caspase-1 cleavage motif and the enzyme(s) responsible for cleaving the peptides have yet to be identified. Moreover, the IL-36 cytokines do not have a signal peptide to direct their secretion, thus like IL-1α, both their processing and release from the cell are currently unclear.
The potential importance of the IL-36 family in psoriasis is high-lighted by the discovery that loss-of-function mutations in the IL-36 receptor antagonist gene IL36RN underlie a rare but debilitating form of psoriasis, generalized pustular psoriasis (GPP) [42,43]. Such mutations leave IL-36 agonist activity unchecked, driving a neutrophilic skin inflammation. Although inhibition of the canonical IL-1 system has not proved to be an effective therapeutic approach in psoriasis [27], targeting the IL-36 system holds promise, particularly in the debilitating conditions GPP and the closely-related disease palmar-plantar pustulosis (PPP), particularly where mutations in IL36RN have been identified. The involvement of IL-36 may not be restricted entirely to GPP and PPP, as psoriasis tends to occur across a spectrum of phenotypes, and the IL-36 system may play a greater role in the more neutrophilic forms of the disease [44]. Expression of IL-36R has been shown to be restricted to epithelial cells in direct contact with the environment, including the skin [13,34,35], which when taken together with the observations above highlight IL-36 as an attractive new therapeutic target for psoriasis.
5. IL-37: a pacifist amongst war-mongers?
Unlike the closely related IL-1β and the IL-36 family members, IL-37 and protein expression is strongly decreased in lesional psoriatic skin compared with non-lesional skin [11], coinciding with altered keratinocyte maturation and loss of the granular cell layer. This cytokine, formerly known as IL-1F7, is encoded as a 6-exon gene in the IL-1 locus on chromosome 2 and is transcribed as 5 spliced isoforms IL-37a–e [45]. Being the most abundant, IL-37b is the best-characterized isoform, expressed in a range of tissues including lymphatic tissue, myeloid cells and epithelia [35]. Post-translational regulation of cytokine expression is a common feature of the IL-1 family and in this respect IL-37 is no different, in that exon 1 encodes a caspase 1 cleavage site [46], exon 2 a putative second cleavage site [47], and exon 3 an elastase cleavage site [48]. Exogenous synthetic IL-37b has been shown to act as an antagonist of IL-18 [46,47,49]. However, physiologic release of IL-37 has yet to be demonstrated. Transfection of a human macrophage cell line with IL-37b lead to a dramatic reduction in secretion of the pro-inflammatory cytokines IL-1α, IL-6, TNF-α and MIP2 (CXCL2), but not IL-10, on stimulation with lipopolysaccharide [50]. Similarly when pro-monocyte and alveolar epithelial cell lines were transfected to over-express IL-37b, both cell lines showed significantly restrained inflammatory responses [51]. This potent anti-inflammatory activity of IL-37b was critically dependent on nuclear translocation [50,51], a property IL-37 may share with IL-1α [21] and IL-33 [52]. These findings have been extended in vivo, by overexpression of human IL-37b in mice, as a murine ortholog of IL-37 has not been identified. Transgenic expression of IL-37b resulted in no obvious changes in mouse phenotype; however, it protected the mice in two models of inflammation [51]. The expression pattern of IL-37 in the upper epidermal layers of healthy skin and its loss in lesional psoriasis [11] make it tempting to speculate that IL-37 may have a role in modulating keratinocyte responses to external stimuli.
6. IL-13: misrepresented as a Th2 cytokine?
IL-13 has been functionally categorized with IL-4 and IL-5 as a Th2 cytokine, and while true in central immunity, driving T cells towards a Th2 response, in the periphery IL-13 has effects divergent from a typical Th2 role. IL-13 can signal either by the IL-4R/ IL-13Ra1 receptor heterodimer or via the single IL-13Ra2 chain [53–55]. IL-13 is upregulated in the skin lesions of psoriasis while IL-4 and IL-5 are decreased (Fig. 2); both receptors for IL-13 are upregulated in psoriasis skin (J.E. Gudjonsson, personal communication) with a detectable IL-13 cytokine signature present in lesions [56]. IL-13 has been shown to be co-secreted with IFN-γ, IL-17A and IL-22 by T cells [5,57], and the major source of IL-13 in psoriasis has been proposed to be CD4+CD161+ T cells [58], and blood-derived CD4+CD161+ T cells isolated from psoriatic patients have been shown to co-secrete IFN-γ with IL-13. Contrary to its role as a Th2 cytokine, IL-13 synergizes with the classic Th1, Th17 and Th22 cytokines IFN-γ, IL-17A and IL-22. In psoriasis, IL-13 and IFN-γ synergize, driving the expression and secretion of chemokines such as CCL2 and CCL5, which can promote the influx of monocytes and dendritic cells into the skin [59]. A non-synonymous single nucleotide polymorphismin the IL13 gene, leading to a change in the amino acid sequence of the IL-13 cytokine, has been identified as a risk allele for psoriasis [60]; However, the functional consequences of this polymorphism on skin immunity and psoriatic inflammation remain unclear.
7. IL-17: The key to a durable remission in inflammatory diseases?
Since the breaking of the Th1/Th2 paradigm, IL-17 has stolen much of the limelight in the pathogenesis of many inflammatory diseases and their animal models. The IL-17 family consists of six ligands (IL-17A through IL-17E) and five receptors (IL-17RA through IL-17RE), with IL-17A and IL-17F sharing the greatest homology and binding to the same IL-17RA and IL-17RC receptor complex [61]. IL-17 plays a key role in host defense against certain pathogens including Candida species, through stimulating the release of antimicrobial peptides and pro-inflammatory cytokines and chemokines. The increased expression of IL-17A at sites of inflammation in psoriasis [25,62] as well as other autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, and Crohn’s disease strongly suggests a role in promoting autoimmune pathology [63,64]. A recent study indicated that IL-17C is more strongly expressed than IL-17A in psoriasis lesions and targeted over-expression of IL-17C in mouse keratinocytes leads to development of a psoriasiform skin phenotype [65]. IL-17C is mainly expressed by keratinocytes in lesional skin, whereas IL-17A emanates from innate immune cells including NK cells, gamma-delta T cells, mast cells, neutrophils (Lin et al., 2011), NKp44+ CD3-negative innate lymphoid cells (ILCs) [66] and mucosa-associated invariant T cells (MAIT) [67] as well as the Th17 (CD4+ IL-17+) and Tc17 (CD8+ IL-17+) cells of the acquired immune system [25,68,69]. Given how IL-17A has been implicated in several inflammatory diseases, three biologic drugs that target this cytokine or its receptor have been developed, including two which neutralize IL-17A (secukinumab and ixekinumab) and one that blocks the IL-17 receptor (brodalumab). These are currently undergoing phase III clinical trials for the treatment of psoriasis (http://www.clinicaltrials.gov: NCT01961609, NCT02267135, NCT01777191, NCT01646177, NCT01597245, NCT01624233) having shown promising results in preliminary phase II studies [70–73]. These join the ranks of the biologic agents targeting psoriasis that have proven extremely useful in limiting the impact of the disease (Fig. 3). Given the attractiveness of IL-17 as a therapeutic target in psoriasis, several more avenues are being explored, including inhibiting IL-17 signal transduction pathways [74] such as RORγt inverse agonists for inhibiting Th17-specific gene expression [75].
Fig. 3.

Psoriasis is now regarded as a mixed Th1/Th17 disease as products of these cells dominate the cytokine network in psoriasis and drive the keratinocyte hyperproliferation and skin inflammation characteristic of this disease. Treatments targeting TNF-α (etanercept, adalimumab, infliximab), IL-12/23p40 (ustekinumab), and more recently IL-17A (secukinumab, ixekinumab), and the IL-17 receptor (brodalumab), are proving effective in decreasing psoriasis disease severity (indicated in green, for details see main text). On the other hand, inhibition of IL-1α/β, IL-6, IFN-γ have thus far proven ineffective (red). Other members of the cytokine network such as IL-19, 20, 22 (fezakinumab), IL-20R1 and IL-20R2, and IL-23p19 (CNTO1959), as well as the IL-36 cytokines and their receptor are now being explored as potential targets for new interventions (blue). The efficacy of any anti-psoriatic treatment likely depends on its ability to disassemble the inflammatory cytokine network, by targeting a critical component and also the remarkable synergies between component cytokines. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
8. The IL-10 family: sheep in wolves’ clothing?
The IL-10 family of cytokines is comprised of IL-10, IL-28, IL-29 and the IL-20 subfamily (IL-19, IL-20, IL-22, IL-24 and IL-26) [76]. IL-19, IL-20, and IL-24 signal via the “type I” IL-20R made up of IL-20R1 and IL-20R2 (Table 2), while IL-20 and IL-24 can additionally signal through the “type II” IL-20R, an IL-20R2 and IL-22R1 heterodimer. Both of these receptor complexes are primarily expressed on epithelial cells and activate the transcription factor STAT3 driving tissue restoration by promoting tissue remodeling, wound healing, and antimicrobial peptide expression [77]. IL-22 is of interest in psoriasis as it is elevated in the skin and plasma of psoriasis patients [78] where it is produced by lesional T cells [79] and ILCs [80]. IL-22 induces epidermal hyperplasia [77] but not keratinocyte proliferation [81]; it inhibits epidermal differentiation and, either alone or in synergy with IL-1 and IL-17, drives the induction of pro-inflammatory gene expression [77,81]. Interestingly, a genetic variant that increases activity at the IL22 promoter has been associated with childhood-onset psoriasis [82]. Thus both genetic and functional data support the view that IL-22 is another critical cytokine in the pathogenesis of psoriasis and as such is a target of drug development [8]. Unlike IL-22, which is primarily secreted by lymphocytes, IL-19, IL-20, and IL-24 are expressed primarily by myeloid and epithelial cells [76]. Although IL-19, IL-20, IL-22 and IL-24 are all elevated in lesional psoriasis skin, IL-19 is the most upregulated mRNA transcript in lesional versus non-lesional skin biopsies [10,83,84] (Fig. 2) and thus has been proposed as a biomarker for psoriasis disease activity [85]. IL-19 appears to have similar but much weaker effects on keratinocytes compared with IL-22 [77]. However, given its more abundant expression, IL-19 may be a significant part of the cytokine storm in that it can amplify the effects of IL-17A and promote the IL-23/IL-17 inflammatory axis [85].
Table 2.
IL-10 cytokine family share receptor subunits.
| Ligand | Receptors | |
|---|---|---|
| IL-10 | IL-10r1 | IL-10r2 |
| IL-22 | IL-22r1 | IL-10r2 |
| IL-26 | IL-20r1 | IL-10r2 |
| IL-19 | IL-20r1 or IL-22r1 | IL-20r2 |
| IL-20 | ||
| IL-24 |
The parental member of this cytokine family, IL-10 has been characterized as an anti-inflammatory cytokine, produced by regulatory T cells, shown to blunt both pro-inflammatory T cell responses and keratinocyte inflammatory markers, and has shown promise in a phase II clinical trial [86]. However, the long-term administration of a large recombinant protein limited enthusiasm for this therapy. Given that the IL-20 cytokine sub-family acts directly on the epidermis, they present a number of other attractive targets for development of anti-psoriatic therapies. Thus far results have not been favorable: an anti-IL-22 biologic (fezakinumab, ILV-094) has been investigated in a phase I clinical trial (NCT00563524) but to date no results have been released, and a phase I/II trial of anti-IL-20 biologic (NCT01261767) was terminated due to an apparent lack of psoriasis response. Given the apparent redundancy of function between these cytokines [77], a more advantageous approach may be to target the IL-20R1 or IL-22R1 subunits that bind IL-19, 20, 24 and 26 or IL-22 and IL-24, respectively (Table 2). However, one recent observation suggests a possible caveat to targeting the IL-20 family: IL-19 (and IL-20, IL-24) may, like their parental cytokine IL-10, have regulatory roles, serving in a feedback inhibitory loop and dampening responses to inflammatory stimuli [87]. Thus despite that IL-19 has been shown to potentiate the effects of IL-17A on keratinocytes in vitro, the regulatory role of these cytokines within the complexities of skin tissue may be worthy of consideration.
9. The IL-12 family: guardians of the Th1/Th17 balance?
Members of the IL-12 family (IL-12, IL-23, IL-27, IL-35; see Table 1) are of interest to those studying psoriasis for a number of reasons: IL12B (IL12p40), IL23A (IL-23p19) and EBI (p35) subunits are overexpressed in psoriasis lesions, several psoriasis-associated genetic polymorphisms have been identified in both cytokines (IL12B) and receptors (IL23R) [88,89], and these cytokines are intimately involved in balancing the Th1/Th17 immune response, which may be particularly important in the epithelia of the gut, lung and skin. Compared with non-lesional or healthy control skin, psoriasis lesions are rich in IL-12 (particularly the IL-12p40 subunit) and IL-23p19 (Fig. 2). Skin macrophages were found to be the main source of IL-12 in psoriasis [90]. For some time, psoriasis has been viewed as amixed Th1/Th17 disease and as such, IL-12 is a major inducer of IFN-γ-producing (Th1) T cells, as well as having the ability to induce skin-homing characteristics (cutaneous lymphocyte-associated antigen [CLA] expression) on memory T cells [91]. Recently we showed that a psoriasis-associated polymorphism in IL12B disposes to increased expression of the IL-12p40 subunit by APC [92], the primary cellular source of IL-12 and IL-23 [93], and this was enhanced by IFN-γ pre-treatment. Individuals carrying this mutation have been shown to have increased IL-12 and decreased IL-23 levels in their serum and a more pronounced Th1/IFN-γ inflammatory signature in their skin lesions [92]. Interestingly, several reports point to involvement of the nervous system in the development of psoriasis skin lesions [94], and as such IL-12 can also serve as a potential link between the skin immune system and the nervous system as it is produced by peripheral nerve cells [95]. On the other hand, IL-23 is produced from both keratinocytes [96] and dendritic cells [93,97]. The importance of IL-23 in T cell immunology is underscored by the identification of several polymorphisms in IL23R associated with Crohn’s disease, psoriasis and psoriatic arthritis [60,88,89,98]. One of the key functions of IL-23 appears to be its ability to amplify and sustain Th17 T cells [97] and as such, is targeted by the anti-IL12p40 biologic ustekinumab (Fig. 3) as well as the more specific approach of targeting the IL-23p19 subunit (CNTO1959 currently in phase 2 trials for palmar plantar pustulosis) and the IL-23 receptor itself [99,100] as a means of targeting IL-17 activity upstream of Th17 cells.
Table 1.
IL-12 family cytokine subunits.
| Ligand | Subunits | |
|---|---|---|
| IL-12 | p35 | p40 |
| IL-23 | p19 | p40 |
| IL-27 | Ebi3 | p28 |
| IL-35 | Ebi3 | p35 |
10. TNF-α: inflammatory synergy
TNF-α is now regarded as a central cytokine in the development of several autoimmune diseases. As such, its role in the pathogenesis of arthritis and its targeting in animal models of arthritis [101] opened up a new approach to targeting cytokines in inflammatory diseases. TNF-α is a somewhat enigmatic cytokine with respect to psoriasis pathogenesis; although it is produced by most activated T cells and APC, TNF-α alone does not evoke significant responses from cultured keratinocytes; however, in combination with IL-17A [102], IL-17C [65] and other cytokines it forms strong synergies, amplifying responses and thus is a significant element of the cytokine storm in psoriasis. Underlying the powerful synergism between TNF-α and IL-17A is the stabilization of IL-17A mRNA [103] by TNF-α, potentiating the effects of IL-17A, in addition to the ability of TNF-α to increase the expression of IL-17R by keratinocytes [9] and IL-17A to induce TNFR expression. Following the success of targeting TNF-α in arthritis, this approach was taken in psoriasis where several biologics are particularly effective (Fig. 3). Not all psoriasis patients show a significant response to the anti-TNF biologics [104], suggesting that there may be differences in the inflammatory networks in the skin lesions of patients [56], perhaps driven by genetic background heterogeneity, with a different balance of protective and disease-associated alleles across several loci [83]. Some loss of efficacy of the anti-TNF biologics has been reported over time, even with fully humanized antibodies, suggesting the production of both anti-idiotype antibodies and shift of the set-points in the cytokine network [56]. Whether or not the anti-IL-17 biologics will eventually suffer from the same drawbacks as the anti-TNF agents remains to be seen.
11. The interferons: antiviral responses gone astray?
Type I IFNs (including IFN-α and IFN-β) are key cytokines in antiviral host defense, stemming from their ability to inhibit viral replication and to provoke immune activation. Type I IFNs are preferentially expressed by plasmacytoid dendritic cells (pDCs) which have been shown to produce large amounts of type I IFNs following TLR7-and TLR9-mediated recognition of viral RNA and DNA [105,106]. Since their first association with psoriasis [107] type I IFNs have been waiting to have their roles in the disease defined. Type I IFNs are not expressed in healthy skin, but are induced in virally infected skin where pDCs are present, as well as in skin wounds where mechanical injury induces rapid infiltration of pDCs, and in psoriasis lesions where sustained type I IFN production by pDCs has been demonstrated [108]. Type I IFNs are most likely to be involved in the triggering of psoriasis skin lesions, given that pDCs have been shown to infiltrate early developing lesions, but are notably absent in chronic lesions [109]. In this respect, targeting of type I IFNs could prevent the transformation of non-lesional to lesional skin in a xenograft model of psoriasis [108]. The importance of type I IFNs in the triggering of psoriasis is strengthened by the well-documented induction of new-onset psoriasis or exacerbation of pre-existing psoriasis during treatment with IFN-α2 [110]. However, during phase I clinical trial for treating chronic plaque psoriasis, an anti-IFN-α antibody (MEDI-545) failed to show efficacy [111]. A likely explanation is that IFN-α may only be a key member of the cytokine network in early lesions, or perhaps in the established skin lesions in a small subset of patients [56].
Psoriasis lesions have long been known to contain elevated levels of IFN-γ [112], mostly secreted by skin T cells [113,114] and intradermal injection of IFN-γ has been shown to induce a psoriatic skin phenotype [115,116]. In line with these observations, serum levels [117] and the frequency of circulating CD8+IFN-γ+ T cells in the blood of patients [118] have been shown to correlate with psoriasis disease severity. Although IFN-γ has been shown to drive inflammation in skin [115,116], anti-proliferative effects were seen on monolayer keratinocyte cultures [119], highlighting the need to examine cytokines in their correct tissue context. A central or critical role for IFN-γ in psoriasis was recently cast into doubt with the failure of an IFN-γ-targeted therapy [116]. However, IFN-γ likely contributes to the cytokine storm in psoriasis [7] by, at the very least, aiding and abetting other cytokines, in particular, IL-17A [25,120], as well as playing a role in priming APCs [25], which drives IL-1/IL-23 production to augment Th17 responses.
12. Conclusions and outlook
This is an exciting era where a better appreciation of the cellular interactions and complexities of the cytokine network in psoriasis is beginning to emerge. As we transition from data based on the effects of cytokines on monolayer keratinocytes and isolated immune cells, to judging their effects on three-dimensional or organotypic cultures which better recapitulate the state of cellular differentiation and spatial relationships, we will build a more complete picture of cytokine pathways in psoriasis. These data can then be used to test specific pathways in suitable animal models [121] and suggest how to target pathways to bring about durable disease remission. New techniques for examining the effects of cytokines on tissues, such as using cytokine and inflammatory gene expression signatures [56], add an extra layer of detail complementing proteomic [122] and transcriptomic analyses [10]. Susceptibility loci within or near genes encoding cytokines (e.g., IL13/IL4, IL12B, IL23A), cytokine receptors (e.g., IL23R, IL28RA) and elements of their signal transduction pathways (e.g., TYK2, TNIP1, TRAF3IP2, NFKBIA) and transcription factors (e.g., REL, STAT2, ETS1) [83,123] may all contribute to altered cytokine signaling in psoriasis which could drive an otherwise balanced inflammatory response into a self-sustaining cycle of disease-causing inflammation. The influence of these genetic susceptibility loci can be further analyzed by predicting the particular cell types in the skin that might be most affected by changes in the regulation or function of particular genes [123]. The use of systems biology approaches [56,124] is likely to be a key development in understanding the pathogenesis of psoriasis. These techniques have the potential to integrate information from all of these resources, thereby allowing the construction of more sophisticated models of cell and cytokine interactions in psoriasis, which should yield a better appreciation of the disease mechanism and help identify novel targets for therapeutic interventions.
Acknowledgments
This work was supported in part by a Babcock Foundation Endowment and NIH K01 AR064765, R01 AR065183, R01 AR062546.
Abbreviations
- NHK
normal human keratinocyte
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- pDC
plasmacytoid dendritic cells
- mDC
myeloid dendritic cell
- MO-DC
monocyte-derived dendritic cell
- APC
antigenpresenting cell
- TLR
Toll-like receptor
- GPP
generalized pustular psoriasis
- PPP
palmar-plantar pustulosis
References
- 1.Parisi R, Symmons DP, Griffiths CE, Ashcroft DM. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. 2012 doi: 10.1038/jid.2012.339. [DOI] [PubMed] [Google Scholar]
- 2.Bata-Csorgo Z, Hammerberg C, Voorhees JJ, Cooper KD. Flow cytometric identification of proliferative subpopulations within normal human epidermis and the localization of the primary hyperproliferative population in psoriasis. J Exp Med. 1993;178:1271–81. doi: 10.1084/jem.178.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–50. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 4.Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-10 e1–395. doi: 10.1016/j.jaci.2009.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS, et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol. 2013;133:973–9. doi: 10.1038/jid.2012.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudjonsson JE, Johnston A, Sigmundsdottir H, Valdimarsson H. Immunopathogenic mechanisms in psoriasis. Clin Exp Immunol. 2004;135:1–8. doi: 10.1111/j.1365-2249.2004.02310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nickoloff BJ, Nestle FO. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest. 2004;113:1664–75. doi: 10.1172/JCI22147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudjonsson JE, Johnston A, Ellis CN. Novel systemic drugs under investigation for the treatment of psoriasis. J Am Acad Dermatol. 2012;67:139–47. doi: 10.1016/j.jaad.2011.06.037. [DOI] [PubMed] [Google Scholar]
- 9.Johnston A, Guzman AM, Swindell WR, Wang F, Kang S, Gudjonsson JE. Early tissue responses in psoriasis to the antitumour necrosis factor-alpha biologic etanercept suggest reduced interleukin-17 receptor expression and signalling. Br J Dermatol. 2014;171:97–107. doi: 10.1111/bjd.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gudjonsson JE, Ding J, Johnston A, Tejasvi T, Guzman AM, Nair RP, et al. Assessment of the psoriatic transcriptome in a large sample: additional regulated genes and comparisons with in vitro models. J Invest Dermatol. 2010;130:1829–40. doi: 10.1038/jid.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, et al. Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. J Invest Dermatol. 2014;134:1828–38. doi: 10.1038/jid.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luger TA, Stadler BM, Luger BM, Mathieson BJ, Mage M, Schmidt JA, et al. Murine epidermal cell-derived thymocyte-activating factor resembles murine interleukin 1. J Immunol. 1982;128:2147–52. [PubMed] [Google Scholar]
- 13.Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol. 2011;186:2613–22. doi: 10.4049/jimmunol.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kupper TS, Ballard DW, Chua AO, McGuire JS, Flood PM, Horowitz MC, et al. Human keratinocytes contain mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA. Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J Exp Med. 1986;164:2095–100. doi: 10.1084/jem.164.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol: CB. 2007;17:1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 16.Murphy JE, Robert C, Kupper TS. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J Invest Dermatol. 2000;114:602–8. doi: 10.1046/j.1523-1747.2000.00917.x. [DOI] [PubMed] [Google Scholar]
- 17.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 19.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. 2011;22:189–95. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werman A, Werman-Venkert R, White R, Lee JK, Werman B, Krelin Y, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA. 2004;101:2434–9. doi: 10.1073/pnas.0308705101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mee JB, Johnson CM, Morar N, Burslem F, Groves RW. The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: dominance of innate immune responses in psoriasis. Am J Pathol. 2007;171:32–42. doi: 10.2353/ajpath.2007.061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yano S, Banno T, Walsh R, Blumenberg M. Transcriptional responses of human epidermal keratinocytes to cytokine interleukin-1. J Cell Physiol. 2008;214:1–13. doi: 10.1002/jcp.21300. [DOI] [PubMed] [Google Scholar]
- 24.Natsuaki Y, Egawa G, Nakamizo S, Ono S, Hanakawa S, Okada T, et al. Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat Immunol. 2014;15:1064–9. doi: 10.1038/ni.2992. [DOI] [PubMed] [Google Scholar]
- 25.Kryczek I, Bruce A, Gudjonsson J, Johnston A, Aphale A, Vatan L, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181:4733–41. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campion GV, Lebsack ME, Lookabaugh J, Gordon G, Catalano M. Dose-range and dose-frequency study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis. The IL-1Ra Arthritis Study Group. Arthritis Rheum. 1996;39:1092–101. doi: 10.1002/art.1780390704. [DOI] [PubMed] [Google Scholar]
- 27.Jung N, Hellmann M, Hoheisel R, Lehmann C, Haase I, Perniok A, et al. An open-label pilot study of the efficacy and safety of anakinra in patients with psoriatic arthritis refractory to or intolerant of methotrexate (MTX) Clin Rheumatol. 2010;29:1169–73. doi: 10.1007/s10067-010-1504-5. [DOI] [PubMed] [Google Scholar]
- 28.Hammerberg C, Arend WP, Fisher GJ, Chan LS, Berger AE, Haskill JS, et al. Interleukin-1 receptor antagonist in normal and psoriatic epidermis. J Clin Invest. 1992;90:571–83. doi: 10.1172/JCI115896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tauber M, Viguier M, Le Gall C, Smahi A, Bachelez H. Is it relevant to use an interleukin-1-inhibiting strategy for the treatment of patients with deficiency of interleukin-36 receptor antagonist? Br J Dermatol. 2014;170:1198–9. doi: 10.1111/bjd.12805. [DOI] [PubMed] [Google Scholar]
- 30.Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204:2603–14. doi: 10.1084/jem.20070157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magne D, Palmer G, Barton JL, Mezin F, Talabot-Ayer D, Bas S, et al. The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res Ther. 2006;8:R80. doi: 10.1186/ar1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279:13677–88. doi: 10.1074/jbc.M400117200. [DOI] [PubMed] [Google Scholar]
- 33.Debets R, Timans JC, Homey B, Zurawski S, Sana TR, Lo S, et al. Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J Immunol. 2001;167:1440–6. doi: 10.4049/jimmunol.167.3.1440. [DOI] [PubMed] [Google Scholar]
- 34.Kumar S, McDonnell PC, Lehr R, Tierney L, Tzimas MN, Griswold DE, et al. Identification and initial characterization of four novel members of the interleukin-1 family. J Biol Chem. 2000;275:10308–14. doi: 10.1074/jbc.275.14.10308. [DOI] [PubMed] [Google Scholar]
- 35.Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE. Four new members expand the interleukin-1 superfamily. J Biol Chem. 2000;275:1169–75. doi: 10.1074/jbc.275.2.1169. [DOI] [PubMed] [Google Scholar]
- 36.Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol. 2014;192:6053–61. doi: 10.4049/jimmunol.1301481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’Erme A, Wilsmann-Theis D, Wagenpfeil J, Holzel M, Ferring-Schmitt S, Sternberg S, et al. IL-36γ (IL-1F9) is a valuable biomarker for psoriasis skin lesions. J Invest Dermatol. 2014 doi: 10.1038/jid.2014.532. [in press] [DOI] [PubMed] [Google Scholar]
- 38.Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, et al. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest. 2012;122:3965–76. doi: 10.1172/JCI63451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mutamba S, Allison A, Mahida Y, Barrow P, Foster N. Expression of IL-1Rrp2 by human myelomonocytic cells is unique to DCs and facilitates DC maturation by IL-1F8 and IL-1F9. Eur J Immunol. 2012;42:607–17. doi: 10.1002/eji.201142035. [DOI] [PubMed] [Google Scholar]
- 40.Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428–37. doi: 10.1038/jid.2011.234. [DOI] [PubMed] [Google Scholar]
- 41.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36{alpha}, IL-36{beta} and IL-36{gamma}) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286:42594–602. doi: 10.1074/jbc.M111.267922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89:432–7. doi: 10.1016/j.ajhg.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365:620–8. doi: 10.1056/NEJMoa1013068. [DOI] [PubMed] [Google Scholar]
- 44.Setta-Kaffetzi N, Navarini AA, Patel VM, Pullabhatla V, Pink AE, Choon SE, et al. Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol. 2013;133:1366–9. doi: 10.1038/jid.2012.490. [DOI] [PubMed] [Google Scholar]
- 45.Taylor SL, Renshaw BR, Garka KE, Smith DE, Sims JE. Genomic organization of the interleukin-1 locus. Genomics. 2002;79:726–33. doi: 10.1006/geno.2002.6752. [DOI] [PubMed] [Google Scholar]
- 46.Kumar S, Hanning CR, Brigham-Burke MR, Rieman DJ, Lehr R, Khandekar S, et al. Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine. 2002;18:61–71. doi: 10.1006/cyto.2002.0873. [DOI] [PubMed] [Google Scholar]
- 47.Pan G, Risser P, Mao W, Baldwin DT, Zhong AW, Filvaroff E, et al. IL-1H, an interleukin 1-related protein that binds IL-18 receptor/IL-1Rrp. Cytokine. 2001;13:1–7. doi: 10.1006/cyto.2000.0799. [DOI] [PubMed] [Google Scholar]
- 48.Boraschi D, Lucchesi D, Hainzl S, Leitner M, Maier E, Mangelberger D, et al. IL-37: a new anti-inflammatory cytokine of the IL-1 family. Eur Cytokine Netw. 2011;22:127–47. doi: 10.1684/ecn.2011.0288. [DOI] [PubMed] [Google Scholar]
- 49.Bufler P, Azam T, Gamboni-Robertson F, Reznikov LL, Kumar S, Dinarello CA, et al. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci USA. 2002;99:13723–8. doi: 10.1073/pnas.212519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma S, Kulk N, Nold MF, Graf R, Kim SH, Reinhardt D, et al. The IL-1 family member 7b translocates to the nucleus and down-regulates proinflammatory cytokines. J Immunol. 2008;180:5477–82. doi: 10.4049/jimmunol.180.8.5477. [DOI] [PubMed] [Google Scholar]
- 51.Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11:1014–22. doi: 10.1038/ni.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282–7. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wills-Karp M, Finkelman FD. Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci Signal. 2008;1:pe55. doi: 10.1126/scisignal.1.51.pe55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 55.Chen W, Sivaprasad U, Tabata Y, Gibson AM, Stier MT, Finkelman FD, et al. IL-13R alpha 2 membrane and soluble isoforms differ in humans and mice. J Immunol. 2009;183:7870–6. doi: 10.4049/jimmunol.0901028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swindell WR, Xing X, Stuart PE, Chen CS, Aphale A, Nair RP, et al. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PLoS ONE. 2012;7:e34594. doi: 10.1371/journal.pone.0034594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. 2008;128:2625–30. doi: 10.1038/jid.2008.111. [DOI] [PubMed] [Google Scholar]
- 58.Nickoloff BJ, Bonish B, Huang BB, Porcelli SA. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J Dermatol Sci. 2000;24:212–25. doi: 10.1016/s0923-1811(00)00120-1. [DOI] [PubMed] [Google Scholar]
- 59.Purwar R, Werfel T, Wittmann M. IL-13-stimulated human keratinocytes preferentially attract CD4+CCR4+ T cells: possible role in atopic dermatitis. J Invest Dermatol. 2006;126:1043–51. doi: 10.1038/sj.jid.5700085. [DOI] [PubMed] [Google Scholar]
- 60.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–67. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–11. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 63.Zhu S, Qian Y. IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci. 2012;122:487–511. doi: 10.1042/CS20110496. [DOI] [PubMed] [Google Scholar]
- 64.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 65.Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. 2013;190:2252–62. doi: 10.4049/jimmunol.1201505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol. 2014;134:984–91. doi: 10.1038/jid.2013.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teunissen MB, Yeremenko NG, Baeten DL, Chielie S, Spuls PI, de Rie MA, et al. The IL-17A-producing CD8 T cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol. 2014 doi: 10.1038/jid.2014.261. [DOI] [PubMed] [Google Scholar]
- 68.Ortega C, Fernandez AS, Carrillo JM, Romero P, Molina IJ, Moreno JC, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. 2009;86:435–43. doi: 10.1189/JLB.0109046. [DOI] [PubMed] [Google Scholar]
- 69.Res PC, Piskin G, de Boer OJ, van der Loos CM, Teeling P, Bos JD, et al. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS ONE. 2010;5:e14108. doi: 10.1371/journal.pone.0014108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papp KA, Langley RG, Sigurgeirsson B, Abe M, Baker DR, Konno P, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol. 2013;168:412–21. doi: 10.1111/bjd.12110. [DOI] [PubMed] [Google Scholar]
- 71.Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–9. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 72.Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190–9. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 73.Rich P, Sigurgeirsson B, Thaci DP, Ortonne JP, Paul C, Schopf RE, et al. Secukinumab induction and maintenance therapy in moderate-to-severe plaque psoriasis: a randomised, double-blind, placebo-controlled, phase II regimen-finding study. Br J Dermatol. 2012 doi: 10.1111/bjd.12112. [DOI] [PubMed] [Google Scholar]
- 74.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, et al. Pharmacologic inhibition of RORgammat regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol. 2014;192:2564–75. doi: 10.4049/jimmunol.1302190. [DOI] [PubMed] [Google Scholar]
- 76.Commins S, Steinke JW, Borish L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J Allergy Clin Immunol. 2008;121:1108–11. doi: 10.1016/j.jaci.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 77.Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. 2007;178:2229–40. doi: 10.4049/jimmunol.178.4.2229. [DOI] [PubMed] [Google Scholar]
- 78.Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–23. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 79.Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. 2014;13:21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 80.Ward NL, Umetsu DT. A new player on the psoriasis block: IL-17A- and IL-22-producing innate lymphoid cells. J Invest Dermatol. 2014;134:2305–7. doi: 10.1038/jid.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- 82.Nikamo P, Cheuk S, Lysell J, Enerback C, Bergh K, Xu Landen N, et al. Genetic variants of the IL22 promoter associate to onset of psoriasis before puberty and increased IL-22 production in T cells. J Invest Dermatol. 2014;134:1535–41. doi: 10.1038/jid.2014.5. [DOI] [PubMed] [Google Scholar]
- 83.Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44:1341–8. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suarez-Farinas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Invest Dermatol. 2012;132:2552–64. doi: 10.1038/jid.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol. 2014;134:2757–67. doi: 10.1038/jid.2014.308. [DOI] [PubMed] [Google Scholar]
- 86.Asadullah K, Sterry W, Stephanek K, Jasulaitis D, Leupold M, Audring H, et al. IL-10 is a key cytokine in psoriasis. Proof of principle by IL-10 therapy: a new therapeutic approach. J Clin Invest. 1998;101:783–94. doi: 10.1172/JCI1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Myles IA, Fontecilla NM, Valdez PA, Vithayathil PJ, Naik S, Belkaid Y, et al. Signaling via the IL-20 receptor inhibits cutaneous production of IL-1beta and IL-17A to promote infection with methicillin-resistant Staphylococcus aureus. Nat Immunol. 2013;14:804–11. doi: 10.1038/ni.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–390. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nair RP, Ruether A, Stuart PE, Jenisch S, Tejasvi T, Hiremagalore R, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol. 2008;128:1653–61. doi: 10.1038/sj.jid.5701255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yawalkar N, Karlen S, Hunger R, Brand CU, Braathen LR. Expression of interleukin-12 is increased in psoriatic skin. J Invest Dermatol. 1998;111:1053–7. doi: 10.1046/j.1523-1747.1998.00446.x. [DOI] [PubMed] [Google Scholar]
- 91.Sigmundsdottir H, Johnston A, Gudjonsson JE, Valdimarsson H. Differential effects of interleukin 12 and interleukin 10 on superantigen-induced expression of cutaneous lymphocyte-associated antigen (CLA) and alphaEbeta7 integrin (CD103) by CD8+ T cells. Clin Immunol. 2004;111:119–25. doi: 10.1016/j.clim.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 92.Johnston A, Xing X, Swindell WR, Kochkodan J, Riblett M, Nair RP, et al. Susceptibility-associated genetic variation at IL12B enhances Th1 polarization in psoriasis. Hum Mol Genet. 2013;22:1807–15. doi: 10.1093/hmg/ddt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–30. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saraceno R, Kleyn CE, Terenghi G, Griffiths CE. The role of neuropeptides in psoriasis. Br J Dermatol. 2006;155:876–82. doi: 10.1111/j.1365-2133.2006.07518.x. [DOI] [PubMed] [Google Scholar]
- 95.Turka LA, Goodman RE, Rutkowski JL, Sima AA, Merry A, Mitra RS, et al. Interleukin 12: a potential link between nerve cells and the immune response in inflammatory disorders. Mol Med. 1995;1:690–9. [PMC free article] [PubMed] [Google Scholar]
- 96.Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. 2006;176:1908–15. doi: 10.4049/jimmunol.176.3.1908. [DOI] [PubMed] [Google Scholar]
- 97.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 98.Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122:201–6. doi: 10.1007/s00439-007-0397-0. [DOI] [PubMed] [Google Scholar]
- 99.Desmet J, Verstraete K, Bloch Y, Lorent E, Wen Y, Devreese B, et al. Structural basis of IL-23 antagonism by an Alphabody protein scaffold. Nat Commun. 2014;5:5237. doi: 10.1038/ncomms6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Quiniou C, Dominguez-Punaro MC, Cloutier F, Erfani A, Ennaciri J, Sivanesan D, et al. Specific targeting of the Il-23 receptor using a novel small peptide non-competitive antagonist, decreases the inflammatory response. Am J Physiol Regul Integr Comparative Physiol. 2014 doi: 10.1152/ajpregu.00540.2013. [ajpregu 00540 2013] [DOI] [PubMed] [Google Scholar]
- 101.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. 2011;131:677–87. doi: 10.1038/jid.2010.340. [DOI] [PubMed] [Google Scholar]
- 103.Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–41. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 104.Papp KA, Poulin Y, Bissonnette R, Bourcier M, Toth D, Rosoph L, et al. Assessment of the long-term safety and effectiveness of etanercept for the treatment of psoriasis in an adult population. J Am Acad Dermatol. 2012;66:e33–45. doi: 10.1016/j.jaad.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 105.Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983–94. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–9. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 107.Kariniemi AL. Effect of human leucocyte interferon on DNA synthesis in human psoriatic skin cultured in diffusion chambers. Acta Pathol et Microbiol Scand Sect A, Pathol. 1977;85A:270–2. doi: 10.1111/j.1699-0463.1977.tb00427.x. [DOI] [PubMed] [Google Scholar]
- 108.Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202:135–43. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Albanesi C, Scarponi C, Pallotta S, Daniele R, Bosisio D, Madonna S, et al. Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J Exp Med. 2009;206:249–58. doi: 10.1084/jem.20080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Afshar M, Martinez AD, Gallo RL, Hata TR. Induction and exacerbation of psoriasis with interferon-alpha therapy for hepatitis C: a review and analysis of 36 cases. J Eur Acad Dermatol Venereol. 2013;27:771–8. doi: 10.1111/j.1468-3083.2012.04582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bissonnette R, Papp K, Maari C, Yao Y, Robbie G, White WI, et al. A randomized, double-blind, placebo-controlled, phase I study of MEDI-545, an anti-interferon-alfa monoclonal antibody, in subjects with chronic psoriasis. J Am Acad Dermatol. 2010;62:427–36. doi: 10.1016/j.jaad.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 112.Gearing AJ, Fincham NJ, Bird CR, Wadhwa M, Meager A, Cartwright JE, et al. Cytokines in skin lesions of psoriasis. Cytokine. 1990;2:68–75. doi: 10.1016/1043-4666(90)90045-u. [DOI] [PubMed] [Google Scholar]
- 113.Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG. The majority of epidermal T cells in Psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752–9. doi: 10.1046/j.1523-1747.1999.00749.x. [DOI] [PubMed] [Google Scholar]
- 114.Vollmer S, Menssen A, Trommler P, Schendel D, Prinz JC. T lymphocytes derived from skin lesions of patients with psoriasis vulgaris express a novel cytokine pattern that is distinct fromthat of T helper type 1 and T helper type 2 cells. Eur J Immunol. 1994;24:2377–82. doi: 10.1002/eji.1830241018. [DOI] [PubMed] [Google Scholar]
- 115.Fierlbeck G, Rassner G, Muller C. Psoriasis induced at the injection site of recombinant interferon gamma. Results of immunohistologic investigations. Arch Dermatol. 1990;126:351–5. [PubMed] [Google Scholar]
- 116.Johnson-Huang LM, Suarez-Farinas M, Pierson KC, Fuentes-Duculan J, Cueto I, Lentini T, et al. A single intradermal injection of IFN-gamma induces an inflammatory state in both non-lesional psoriatic and healthy skin. J Invest Dermatol. 2012;132:1177–87. doi: 10.1038/jid.2011.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arican O, Aral M, Sasmaz S, Ciragil P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005:273–9. doi: 10.1155/MI.2005.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sigmundsdottir H, Gudjonsson J, Jonsdottir I, Ludviksson B, Valdimarsson H. The frequency of CLA+CD8+ T cells in the blood of psoriasis patients correlates closely with the severity of their disease. Clin Exp Immunol. 2001;126:365–9. doi: 10.1046/j.1365-2249.2001.01688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nickoloff BJ, Mitra RS, Elder JT, Fisher GJ, Voorhees JJ. Decreased growth inhibition by recombinant gamma interferon is associated with increased transforming growth factor-alpha production in keratinocytes cultured from psoriatic lesions. Br J Dermatol. 1989;121:161–74. doi: 10.1111/j.1365-2133.1989.tb01795.x. [DOI] [PubMed] [Google Scholar]
- 120.Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–9. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 121.Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, Elder JT. Mouse models of psoriasis. J Invest Dermatol. 2007;127:1292–308. doi: 10.1038/sj.jid.5700807. [DOI] [PubMed] [Google Scholar]
- 122.Piruzian E, Bruskin S, Ishkin A, Abdeev R, Moshkovskii S, Melnik S, et al. Integrated network analysis of transcriptomic and proteomic data in psoriasis. BMC Syst Biol. 2010;4:41. doi: 10.1186/1752-0509-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Swindell WR, Stuart PE, Sarkar MK, Voorhees JJ, Elder JT, Johnston A, et al. Cellular dissection of psoriasis for transcriptome analyses and the post-GWAS era. BMC Med Genomics. 2014;7:27. doi: 10.1186/1755-8794-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Valeyev NV, Hundhausen C, Umezawa Y, Kotov NV, Williams G, Clop A, et al. A systems model for immune cell interactions unravels the mechanism of inflammation in human skin. PLoS Comput Biol. 2010;6:e1001024. doi: 10.1371/journal.pcbi.1001024. [DOI] [PMC free article] [PubMed] [Google Scholar]