

Abstract
In neurons, enhanced PKA signaling elevates synaptic plasticity, promotes neuronal development, and increases dopamine synthesis. On the other hand, a decline in PKA signaling contributes to the etiology of several brain degenerative diseases including Alzheimer’s disease and Parkinson’s disease suggesting that PKA predominantly plays a neuroprotective role. A-kinase anchoring proteins (AKAP) are large multi-domain scaffold proteins that target PKA and other signaling molecules to distinct subcellular sites to strategically localize PKA signaling at dendrites, dendritic spines, cytosol, and axons. PKA can be recruited to outer mitochondrial membrane by associating with three different AKAPs to regulate mitochondrial dynamics, structure, mitochondrial respiration, trafficking, dendrite morphology, and neuronal survival. In this review, we survey the myriad of essential neuronal functions modulated by PKA but place a special emphasis on mitochondrially-localized PKA. Finally, we offer an updated overview of how loss of PKA signaling contributes to the etiology of several brain degenerative diseases.
Keywords: A-kinase anchoring proteins, protein kinase A, mitochondrial dynamics, mitochondrial trafficking, neurodegeneration, dendrites
Introduction
Protein Kinase A (PKA) is a master regulator of most cyclic-AMP dependent physiological processes in eukaryotic cells. PKA is a tetrameric holoenzyme composed of two regulatory subunits (classified as type I or type II regulatory subunits) bound to two catalytic subunits. The association of cAMP to a specific site in the molecular surface of the PKA holoenzyme (interface between regulatory and catalytic subunits) leads to the destabilization of the regulatory (R) subunits and induces release of the catalytic (C) subunits. Depending on the subcellular localization of PKA, the liberated C subunits phosphorylate downstream substrates to regulate critical physiological functions in eukaryotic cells (Feliciello, et al. 2001).
PKA can be targeted to the outer mitochondrial membrane via three different A-kinase anchoring proteins (AKAPs), large multi-domain PKA anchoring proteins. Mitochondrial AKAPs contain small unique positively charged mitochondrial leader sequences that bind the lipid membrane of mitochondria. A fourth AKAP, termed sphingosine kinase interacting protein (SKIP), targets PKA to the inner mitochondrial membrane to phosphorylate the coiled-coil helix protein ChChd3 (Means, et al. 2011). At the OMM, protein kinase A (PKA) has emerged as an essential regulator of different aspects of mitochondrial biology including mitochondrial morphology, dynamics, and turnover by phosphorylating OMM-localized dynamin-related protein 1 (Drp1) to inhibit mitochondrial fission, the proapoptotic protein BAD to promote survival, and mitochondrial-anchored cytoskeletal proteins to remodel dendrites (Affaitati, et al. 2003, Cardone, et al. 2002, Merrill, et al. 2011). At the matrix, PKA has been shown to phosphorylate a subunit of complex I (NDUFS4) in vitro and in vivo to elevate oxidative/phosphorylation (Papa. 2002).
The outer mitochondrial membrane (OMM) has emerged as a crucial platform by which a myriad of protein signaling pathways converge to regulate essential neuronal functions. The reversible phosphorylation of OMM-localized protein substrates by serine/threonine (ser/thr) kinases and phosphatases is a crucial posttranslational event that governs mitochondrial structure, metabolism, and mitochondrial oxidative phosphorylation (Pagliarini, et al. 2006).
Mitochondria are multi-faceted organelles that not only serve as the main energy hubs to power most eukaryotic physiological reactions, but act as calcium sinks, heat generators, regulate lipid/steroid metabolism and cell survival. Unlike proliferating cells, neurons predominantly rely on oxidative phosphorylation to power essential neuronal functions and survival. The high reliance of neurons on oxidative phosphorylation is evident as mitochondrial dysfunction underlies the etiology of several neurodegenerative disorders including Parkinson’s disease (PD) (Gusdon, et al. 2010). In addition to powering the soma, mitochondria are critical for the maintenance of complex and vast neurite networks. Indeed, in order to achieve this tall order, mitochondria have the challenging task of providing the necessary energy and calcium buffering capacity for the maintenance of dendrites and axons from long projecting neurons. Hence, a continuous turnover of effete mitochondria and a highly coordinated trafficking of de novo mitochondria towards sites of highest energy demands within dendritic arbors and axons are necessary to prevent premature loss of neurites (Cherra, et al. 2010, Schwarz. 2013).
In this review, we purvey the critical role that PKA plays on the maintenance of dendritic arbors, mitochondrial trafficking, mitochondrial function, and survival. Secondly, we discuss how dysregulation of the activity and localization of specific pools of PKA contributes to the molecular pathogenesis of various brain degenerative disorders including Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Andorfer, et al. 2000, Howells, et al. 2000, Pugazhenthi, et al. 2011, Vitolo, et al. 2002, Yamamoto, et al. 2000) and offer insight as to how ameliorating alterations in PKA signaling may reverse molecular pathogenesis in these diseases.
2. Role of PKA/AKAP in regulating mitochondrial function and dynamics in neurons
AKAPs, large multi-modular protein scaffolds that bind to PKA holoenzymes, have emerged as critical regulators of neuronal development, survival, and metabolism. AKAPs have the ability to relocate endogenous PKA -and other associated signaling players- to distinct subcellular compartments to strategically enhance cAMP-regulated signaling pathways (Feliciello, et al. 2001). For instance, D-AKAP79 is localized at postsynaptic sites to regulate dendritic spine development, morphology, and synaptic plasticity (Gomez, et al. 2002).In addition, D-AKAP150, also known as AKAP79 (human ortholog), targets PKA to dendrites to regulate the activity of acid-sensing ion channels in rat cortical neurons (Chai, et al. 2007). At the OMM, up to three AKAPs have been identified to interact with PKA including the dual-specificity A-kinase anchoring protein 1 (D-AKAP1), D-AKAP2, and rab32.
D-AKAP1 (also known as AKAP140/149 and other splice variants AKAP121, sAKAP84) is a dual specificity multi-domain mitochondrial protein scaffold that not only associates with endogenous PKA, but recruits a host of other signaling molecules to the OMM including PKC, src tyrosine kinase, protein phosphatase 2A (PP2A), protein tyrosine phosphatase-D1 (PTPD1) (Carnegie, et al. 2009, Feliciello, et al. 2001) (Cardone, et al. 2004), and various mRNA molecules (Ginsberg, et al. 2003). Based on Northern blot data alone, D-AKAP1 is widely expressed with high expression levels in the heart and testes while low protein levels have been observed in skeletal muscle and brain tissue (Huang, et al. 1997).
Splice variants of D-AKAP1 contain short divergent N-terminal and C-terminal regions that are spliced to a common core region that contains KH and Tudor domains. To date, six splice variants of D-AKAP1 have been identified (Feliciello, et al. 2001). Although numerous studies suggest that D-AKAP1 plays a critical role in sperm maturation and oocyte development (Carnegie, et al. 2009), the biological in vivo significance of various D-AKAP1 splice variants in neurons is, however, not clear given that very low levels of expression have been detected in the brain (Huang, et al. 1997). In vivo, knocking out endogenous D-AKAP1 is not embryonically lethal suggesting that other mitochondrial AKAPs (ie., D-AKAP2) or serine/threonine (ser/thr) kinases can compensate for loss of D-AKAP1 function (Newhall, et al. 2006).
D-AKAP1 plays a prominent neuroprotective role and is critical for mitochondrial structure/function in cell culture models. Indeed, decreased mitochondrial localization of endogenous D-AKAP1, accomplished by treating cells with anti-AKAP1 specific peptides or by transfecting cells with D-AKAP siRNA, leads to robust mitochondrial fragmentation, increased oxidative stress, and elevated basal cell death (Affaitati, et al. 2003, Livigni, et al. 2006, Merrill, et al. 2011, Perrino, et al. 2010). Conversely, overexpression of D-AKAP1 blocks serum withdrawal-induced apoptosis in neuronal PC12 cells by phosphorylating the BH3 only containing proapoptotic protein BAD. Mechanistically, D-AKAP1-induced neuroprotection requires PKA-mediated phosphorylation of BAD at serine 155 which leads to its cytosolic sequestration by 14-3-3 proteins and blocks the release of cytochrome C (Affaitati, et al. 2003). In addition to PKA-mediated phosphorylation of BAD, mitochondrial fusion through PKA-mediated phosphorylation of Drp1 has been recently identified as a second molecular mechanism by which D-AKAP1 confers neuroprotection. In hippocampal neurons, transient expression of a constitutively active form of the catalytic subunit of PKA targeted to the mitochondrion or of wild-type D-AKAP1, but not a PKA binding deficient mutant of D-AKAP1, fuses mitochondrial networks in the soma (Merrill, et al. 2011) and in dendrites of hippocampal neurons (Dickey, et al. 2011). Enhanced mitochondrial fusion by D-AKAP1 is associated with enhanced resistance of hippocampal neurons to various toxic insults including rotenone, hydrogen peroxide and staurosporine (Merrill, et al. 2011).
As a negative feedback mechanism, D-AKAP1 recruits phosphatases to prevent excessive PKA activity at the mitochondrion that otherwise can be detrimental. For instance, in non-neuronal cells, the catalytic subunit of protein phosphatase 2A (PP2A) interacts with D-AKAP1 in lung tissue to regulate EGF-mediated RSK1 activity in fibroblasts in order to ensure the timely deactivation of PKA-regulated mitochondrial fusion (Chaturvedi, et al. 2009). Although PP2A/Bβ2 translocates to mitochondria to dephosphorylate the same PKA-regulated site in Drp1 (Ser656) to promote mitochondrial fission (Dagda, et al. 2008, Merrill, et al. 2013), it remains to be elucidated whether D-AKAP1 can spatially and simultaneously recruit endogenous PP2A in neurons as a negative feedback mechanism to halt PKA signaling.
Another mechanism that regulates the level of mitochondrial PKA activity at the OMM is through the ubiquitin-proteasome mediated degradation of D-AKAP1. D-AKAP1 levels in the OMM can be rapidly degraded during hypoxic stress in neurons. D-AKAP1 is specifically tagged for ubiquitin proteasome-mediated degradation by the E3 ubiquitin ligase Seven In-Absentia Homolog 2 (Siah2) in neurons during hypoxic stress in vivo. Therefore, Siah2-mediated degradation of D-AKAP1 during hypoxic conditions uncouples PKA signaling at the mitochondrion which leads to a detrimental decrease in mitochondrial respiration in the ischemic brain (Carlucci, et al. 2008). In addition to hypoxia, future endeavors should elucidate whether oxidative stress elicited by mitochondrially-directed toxins (e.g. rotenone and 6-hydroxydopamine) promote mitochondrial dysfunction by enhancing the ubiquitin proteasome-mediated degradation of endogenous D-AKAP1 to uncouple PKA signaling in the mitochondria.
D-AKAP1 can enhance the mitochondrial import of mRNAs that encode mitochondrial antioxidants and metabolic regulators of oxidative phosphorylation. For instance, the KH domains located on the C-terminal region of D-AKAP1 can directly associate with several mRNAs, that encode for manganese superoxide dismutase, the F0-f subunit of ATP synthase, and lipoprotein lipase (LPL) in HeLa cells (Ginsberg, et al. 2003, Unal, et al. 2008). In light of these observations, future studies should address whether enhanced mitochondrial import of mRNA is an additional neuroprotective mechanism of D-AKAP1.
In addition to remodeling mitochondria through the mitochondrial fission/fusion (MFF) machinery, the removal of irreversibly damaged mitochondria through autophagy, also termed mitophagy, is critical for the maintenance of high quality mitochondria in neurons. Emerging evidence suggests that PKA is modulator of autophagy in neurons. In conjunction to PKC (Jiang, et al. 2010), PKA also phosphorylates the autophagy cargo recognition receptor microtubule-associated protein 1A/1B-light chain 3 (LC3) to suppress autophagic flux induced by rapamycin (a pharmacological activator of autophagy) or as induced by various cell culture models of PD (Cherra, et al. 2010, Dagda, et al. 2011). Suggesting a role of autophagy in neurite maintenance, suppression of autophagic flux via PKA-mediated phosphorylation of LC3 is associated with increased neurite outgrowth in primary cortical neurons and blocks autophagy-mediated blunting of neurites induced by overexpression of a PD-associated LRRK2 mutant (G2019S) (Cherra, et al. 2008, Plowey, et al. 2008). Recent evidence suggests that D-AKAP1 is a regulator of mitochondrial turnover by maintaining mitochondrial health (Dagda, et al. 2011). Moreover, enhanced PKA signaling via D-AKAP1 overexpression suppresses autophagy induced by 6-OHDA and by loss of endogenous PTEN-induced kinase 1, a large mitochondria serine/threonine (ser/thr) associated with familial recessive forms of Parkinson’s disease (Dagda, et al. 2011).
D-AKAP1 can associate with the OMM-localized Na/Ca+2 exchanger NcX3 to facilitate the efflux of calcium from mitochondria as suggested by several in vitro models of ischemia (Scorziello, et al. 2013). Hence, it is plausible that D-AKAP1 preserves mitochondrial function by preventing the pathological accumulation of Ca2+ in mitochondria by promoting Ca2+ efflux through NcX3 located in postsynaptic and presynaptic compartments of neurons.
D-AKAP2 is a second dual specificity mitochondrially-targeted AKAP that recruits PKA to the OMM (Huang, et al. 1997, Wang, et al. 2001). However, unlike D-AKAP1, D-AKAP2 is unique in that it contains two RGS domains and exhibits mixed cytosolic and mitochondrial localization. Paradoxically, D-AKAP2 is not a classical RGS protein given the lack of evidence supporting an interaction of D-AKAP2 with Gα subunits. Although much is known about the 3D structure of D-AKAP2 and the molecular mechanism by which it binds with type II R subunits of PKA (Kinderman, et al. 2006), very little is known regarding the physiological function of D-AKAP2 in eukaryotic cells. In non-neuronal cells, D-AKAP2 regulates endosomal trafficking and the recycling of the transferrin receptor (Eggers, et al. 2009) which raises the possibility that D-AKAP2 regulates endosomal trafficking in neurons as well.
In peripheral neurons, immunohistochemical studies in muscle tissue showed that D-AKAP2 localizes as discrete protein clusters at the synaptic terminals of motor axons. Given that PKA is a major regulator of neurotransmitter release (Yoshihara, et al. 2000), it is conceivable that D-AKAP2 regulates the release of acetylcholine in motor neurons that innervate the skeletal muscle. Hence, future endeavors should focus on the possible interplay of D-AKAP1 and D-AKAP2 at presynaptic and postsynaptic sites for regulating different aspects of neurite biology.
Rab32 is a third mitochondrially directed AKAP that associates with type II R subunits of PKA. Rab32, a 25kDa GTPase, was identified as a regulator of mitochondrial dynamics and fusion in non-neuronal cell lines. Like D-AKAP1, Rab32 promotes mitochondrial fusion, regulates mitochondrial movement at the microtubule organizing center, and upregulates survival by phosphorylating Drp1 and BAD in HeLa cells (Alto, et al. 2002). Moreover, this small mitochondrial GTPase regulates calcium handling and translocation of calnexin at the ER (Bui, et al. 2010). However, based on mRNA expression levels alone, it is likely that D-AKAP1 splice variants plays a predominant role in mitochondrial dynamics compared to Rab32 given that endogenous Rab32 is almost undetectable in human brain tissue (Alto, et al. 2002).
3. Role of PKA in regulating dendrite morphology and function
The maintenance of complex dendritic arbors and axons depends on a proper level of neurotrophic support, continuous synaptic input, efficient anterograde movement of mitochondria, and the maintenance of functional mitochondria through the continuous activation of mitochondrial quality control pathways (Reichardt. 2006) (Schwarz. 2013) (Ashrafi, et al. 2014, Wang, et al. 2011). A variety of ser/thr kinases that have been identified to regulate neurite development include extracellular regulated kinase (ERK), PKA, Ca+2/calmodulin-dependent (CAM) kinase, and protein kinase C (PKC) (Dickey, et al., Gomez, et al. 2002, Goshima, et al. 1993, Sarina, et al. 2013, Strack. 2002, Tominaga-Yoshino, et al. 2002). PKA is a bone fide regulator of neurite biogenesis and morphology (Aglah, et al. 2008, Rodger, et al. 2005). Distinct endogenous pools of PKA modulate neurite function including dendrite outgrowth, the activity of L-type Ca+2 channels in postsynaptic densities, dendritic spine morphology, dopamine biosynthesis at synaptic terminals, axonal regeneration, and remodeling (Dagda, et al. 2011, Davare, et al. 1999, Dickey, et al., Merrill, et al. 2011).
In various cell culture models, PKA can indiscriminately remodel both presynaptic and postsynaptic compartments in developing neurons (Aglah, et al. 2008, Kim, et al. 1996, Rydel, et al. 1988). Interestingly, PKA regulates dendritic morphology in a bimodal manner. Paradoxically, while a basal amount of PKA activity is required for promoting neurite outgrowth, high levels of PKA activity simplifies and shortens dendrites in neurons (Copf. 2014) (Xu, et al. 2012). For instance, treating spiral ganglion neurons with a low concentration of cyclic AMP promotes the sprouting of dendrites while high concentrations of cAMP blunts dendrites and promotes apoptosis even in the absence of toxic insults (Xu, et al. 2012). Therefore, a proper balance of local PKA activity is critical for promoting optimal growth of dendritic arbors in developing neurons.
Several distinct AKAPs have been identified to regulate dendrite morphogenesis. For instance, microtubule associated protein 2B (MAP2B), a well characterized AKAP in neurons, forms cross-bridges with microtubules required for neurite outgrowth and for stabilizing dendrite arbors in developing neurons. In vivo, MAP2B knockout mice show a severe loss of microtubule cross-bridges leading to a dramatic shortening of dendrites in the cortex and hippocampus suggesting that type II R-associated PKA pools in dendrites is essential for dendritogenesis (Harada, et al. 2002). Unexpectedly, this severe decrease in endogenous dendritic-localized PKA holoenzymes leads to a robust decrease in global PKA activity in MAP2B knockout mice suggesting that dendritic-localized PKA is the predominant pool of PKA in neurons (Harada, et al. 2002).
A proper level of mitochondrial density at neurites is critical for the structural maintenance and proper development of postsynaptic and presynaptic compartments in neurons. Indeed, a landmark study unequivocally showed that mitochondria are critical for the development of dendritic spines, dendrites, and synapses as expression of dominant negative mutants of the fission promoter Drp1 reduced dendrite complexity and reduced the number of synapses in neurons (Li, et al. 2004). At the postsynaptic site, mitochondria are critical for powering the mobilization and recycling of the long term pool vesicles in Drosophila, an event downstream of PKA signaling (Verstreken, et al. 2005). In vivo, knockdown of endogenous Drp1 depletes mitochondria from neurites and sensitizes midbrain dopaminergic neurons to degeneration (Berthet, et al. 2014). In addition, a deficiency in mitochondrial trafficking to dendrites through RNAi-mediated knock down of the dendrite-specific mitochondrial motor adaptor protein TRAK2 depletes dendrites of mitochondria and shortens dendrites (van Spronsen, et al. 2013). One study raised the possibility that PKA/D-AKAP1 is an essential regulator of dendritic development. Transient expression of D-AKAP1 increased dendritic complexity while reducing synaptic connectivity in hippocampal neurons. D-AKAP1 promotes dendritic outgrowth partly by remodeling mitochondrial morphology as transient expression of a PKA phosphomimetic construct of Drp1(S656D) phenocopied the enhanced dendritic complexity induced by D-AKAP1 overexpression. Mechanistically, D-AKAP1-mediated dendritogenesis involves increased mitochondrial density at dendrites and an enhanced bioenergetics status and optimal calcium buffering activity of dendritic mitochondria (Dickey, et al. 2011). A recent study showed that PINK1, plays a role in neuronal development. Indeed, transient expression of an N-terminal truncated version of PINK1, which lacks the mitochondrial leader sequence and predominantly positions PINK1 at the cytosol, enhances the complexity of dendritic arbors while induces a modest morphological effect in axons suggesting that cytosolic-localized PINK1 plays a neurodevelopmental role. Mitochondrially-localized PKA was identified as the downstream effector of PINK1-mediated dendritic outgrowth as transient co-expression of a mitochondrially-targeted inhibitor of PKA abrogated the ability of PINK1 to enhance neurite outgrowth (Dagda, et al. 2014). Like PKA/D-AKAP1, PINK1 regulates mitochondrial function, morphology, biogenesis, and survival (reviewed in (Cookson. 2005)). Collectively, these observations suggest that mitochondrial PKA and PINK1 participate in a similar prosurvival signaling pathway for regulating dendrite morphogenesis and for the maintenance of dendritic arbors.
PKA regulates cytoskeletal dynamics in vivo. In the adult nervous system, transgenic flies that express a mutant catalytic subunit of PKA leads to a reduction in lengths and complexity of dendritic arbors suggesting that PKA plays a critical role in regulating the basal number of dendritic branch points (Copf. 2014). Furthermore, it is worth noting that PKA in this experimental context is involved in regulating the branching and outgrowth of dendrites without altering the polarity in dendrites as well. Although dynein was demonstrated to be phosphorylated by PKA in vitro, transgenic expression of a PKA phosphomimetic mutant of dynein in vivo was not able to phenocopy the effects of PKA expression for elevating dendritic complexity suggesting that PKA-mediated phosphorylation of dynein is not sufficient for governing dendrite morphogenesis and/or that other unidentified pools of PKA are required for remodeling dendritic arbors (Copf. 2014).
4. PKA dysregulation in Neurodegenerative Disorders
Alzheimer’s disease (AD)
PKA dysregulation in neurodegenerative disorders has been highlighted by several studies over the last 15 years. In particular, there is strong evidence showing that PKA-regulated pathways in the hippocampus contribute to molecular pathogenesis and cognitive decline in AD.
For instance, a study by Tong et al., (Tong, et al. 2001) reported that levels of amyloid beta 1-42, or Aβ(1-42), which do not compromise the survival of cortical neurons, interferes with functions critical for neuronal plasticity. In brief, pretreatment of cells with Aβ(1-42), at sublethal concentrations, suppressed the activation and transcription of brain-derived neurotrophic factor (BDNF) and phosphorylation of cyclic AMP response element binding protein (p-CREB).
Interestingly, in addition to suppressing p-CREB, another study showed that treating neurons with sublethal concentrations of Aβ(1-42) interferes with BDNF-induced activation of other prosurvival ser/thr kinases including the extracellular signal-regulated protein kinase (ERK) and the phosphatidylinositol 3-kinase (PI3-K)/Akt pathways (Tong, et al. 2004). Considering the critical role that CREB and BDNF play in neuronal plasticity and learning and memory, the above observations suggest that Aβ peptides contribute to a gradual cognitive decline by suppressing PKA signaling in AD. Aβ(1-42) can also promote mitochondrial dysfunction and impair mitochondrial trafficking at the neurites of hippocampal neurons (Devi, et al. 2010). For instance, Rui et al. (Rui, et al. 2006) showed that a brief exposure of hippocampal neurons to Aβ(1-40) results in a rapid and severe impairment of mitochondrial trafficking without inducing obvious morphological changes, which was alleviated by stimulation of PKA effectively. In addition to hippocampal neurons, Aβ(1-40) treatment caused a dysregulation of PKA-regulated signaling pathways in cortical neurons. In a recent study, Wang et al. (Wang, et al. 2011) reported that persistent treatment of primary cortical neurons with Aβ(1-40) promotes the desensitization of the β2 adrenergic receptor leading to decreased cAMP levels, reduced PKA phosphorylation of serine 845 on AMPA receptor subunit 1 (GluR1), and decreased AMPA receptor-mediated miniature excitatory postsynaptic currents. Paradoxically, although low amounts of intracellular Aβ drives transcription of CREB-regulated genes, high loads of intracellular Aβ induced by stable overexpression of an APP containing AD-associated mutations results in sustained hyper-phosphorylation of CREB that fails to translocate to the nucleus (Arvanitis, et al. 2007). The failure in CREB nuclear translocation could be a key factor in early synaptic dysfunction and behavioral impairments that are exacerbated by extracellular Aβ accumulation.
Given that PKA signaling appears to be adversely affected in the Aβ(1-42) model of AD, can pharmacological modulation of PKA and CRE-dependent transcription be considered feasible therapeutic targets for early phases of AD? Several studies have highlighted the potential of cyclic-AMP as an experimental therapy. For instance, Vitolo et al. (Vitolo, et al. 2002) have demonstrated in their study that Aβ(1-42) treatment of cultured hippocampal neurons leads to a robust decline of CREB phosphorylation and of long term potentiation (LTP), pathological events that are reversed by rolipram, a specific inhibitor of type IV phosphodiesterases. Conversely, reversal of LTP inhibition by Rolipram and forskolin was blocked by H89, a pharmacological inhibitor of PKA (Vitolo, et al. 2002). Furthermore, treating M17 cells expressing an AD-associated mutant APP with cAMP rescue the deficiencies in mitochondrial biogenesis, mitochondrial dysfunction, and decreased p-CREB suggesting that a decline in mitochondrial PKA signaling is directly associated with AD pathology (Sheng, et al. 2012). Nobiletin, a naturally occurring polymethoxylated flavone from Citrus depressa that enhances PKA/CREB-dependent signaling, has been shown to reverse Aβ(1-42)-induced decreases in CREB phosphorylation in cultured hippocampal neurons (Matsuzaki, et al. 2006). In a more recent study, Wang et al., (Wang, et al. 2009) reported that the inhibition of LTP by β-amyloid is prevented by the activation of β2-adrenoceptors and by pharmacological activation of the cAMP/PKA signaling pathway in rat hippocampal slices. Collectively, these aforementioned observations suggest that Aβ can directly and indirectly negatively impact PKA-regulated signaling pathways while pharmacological agents that enhance the cAMP/PKA/CREB-signaling pathway have the potential for the treatment of AD.
Huntington’s disease (HD)
Several studies have reported mitochondrial abnormalities to be involved in HD progression. However, the evidence for dysregulated PKA signaling is sparse and not very clear in different HD models. For instance, Giralt et al., (Giralt, et al. 2011) observed hippocampal PKA hyperactivation in naive R6/1 mice (mutant huntingtin transgenic mice), compared with wild-type (WT) mice. In conjunction with over-activation of the PKA signaling pathways, the authors observed significant down-regulation of protein expression levels of phosphodiesterase (PDE) 4. Likewise, hippocampal PKA inhibition by infusion of Rp-cAMPS, a reversible PKA inhibitor, restored long-term memory in R6/2 mice, suggesting that overactivation of global PKA signaling in the hippocampus is one of the molecular mechanisms underlying cognitive decline in R6 animals. Furthermore, a recent study by Lin and colleagues (Lin, et al. 2013) demonstrated that lower PKA activity was associated with proteasome impairment in the synaptic fractions from the striatum of two HD mouse models (R6/2 and N171-82Q) and in mutant HTT (mHTT)-expressing striatal cells. Interestingly, in this study, pharmacological activation of PKA enhanced the phosphorylation of Rpt6 (a component of the proteasome), rescued the impaired proteasome activity, and reduced mHTT aggregates. Overall, these published observations suggest that bimodal dysregulation of PKA signaling contributes to cognitive decline and memory impairment in the hippocampus in the case of overactive PKA signaling while decreased PKA signaling contributes to degeneration of medium spiny neurons. Therefore, new therapeutic interventions should be developed and tailored for “normalizing” PKA signaling in hippocampal and striatal neurons to reverse cognitive decline while preventing neurodegeneration.
Parkinson’s disease (PD)
The evidence for a dysregulation of PKA signaling pathways in PD has been observed in in vivo and in vitro models as well as in postmortem PD brain tissue. Indeed, Howells et al. (Howells, et al. 2000) were among the first to report a dysregulation of PKA-regulated genes when the authors of this study showed that mRNA levels of BDNF were significantly reduced in substantia nigra neurons prior to degeneration.
Given that mitochondrial dysfunction and fragmentation contributes to neurodegeneration in various in vitro and in vivo PD models, it is plausible that a decline in mitochondrial-derived PKA signaling contributes to PD etiology. Indeed, decreased PKA-mediated phosphorylation of the mitochondrial fission inducer Drp1 has been observed in cell culture models of PD. In a genetic cell culture model of PD, RNAi-mediated knockdown of endogenous PINK1 led to decreased PKA-mediated phosphorylation of Drp1 and a concomitant increase in calceneurin (PP2B)-mediated Drp1-dependent mitochondrial fission (Sandebring, et al. 2009). Moreover, transient expression of D-AKAP1 or phosphomimetic mutants of Drp1 prevents cell death induced by loss of PINK1 suggesting that loss of PKA-mediated phosphorylation of Drp1 through D-AKAP1 contributes to cellular pathology associated in PINK1 deficient cells (Dagda, et al. 2011). Recently, Parisiadou and colleagues (Parisiadou, et al. 2014) demonstrated that LRRK2, a large ser/thr kinase that is mutated in an autosomal dominant familial form of PD characterized by cortical Lewy body pathology (Poulopoulos, et al. 2012), is a new atypical AKAP-like regulator of PKARIIβ in dendrites of neurons. The PD-associated LRRK2 R1441C missense mutation impaired the interaction of LRRK2 with PKARIIβ and induced excessive PKA activity in the dendritic spines of these neurons which compromised their sensitivity to stimulation via dopamine receptor D1 (Parisiadou, et al. 2014). Furthermore, Chalovich and colleagues showed that 6-hydroxydopamine (6-OHDA) treatment of B65 neuronal cells led to an aberrant mislocalization of pCREB from the nucleus to the cytosol, decreased total PKA signaling which was accompanied by decreased mRNA levels of BDNF and Bcl-2, two PKA-regulated genes. Conversely, cAMP treatment reversed 6-OHDA-induced cell death and restored nuclear localization of p-CREB. There is credence for the concept that mislocalization of p-CREB contributes to PD etiology (Chalovich, et al. 2006). An aberrant accumulation of p-CREB granules has been reported in the cytoplasm of substantia nigra dopaminergic neurons in postmortem PD brain tissue compared to age-matched control cases suggesting that decreased PKA-mediated transcription of prosurvival genes is hampered due to cytosolic sequestration of p-CREB (Chalovich, et al. 2006). In further support of this concept, dysregulation of CREB has been observed in other tissue culture models of PD. Recently, Dagda et al. (Dagda, et al. 2014) reported decreased global PKA activity in stable PINK1 knockdown cells which was correlated with decreased nuclear p-CREB. Recent studies have shown proof-of-concept that pharmacological activators of PKA may pose as a promising therapeutic intervention for ameliorating mitochondrial dysfunction in familial PD. For instance, Dagda et al. (Dagda, et al. 2011) demonstrated that treatment of PINK1 deficient SH-SY5Y cells with pharmacological PKA activators or transient expression of a constitutively active form of mitochondrially-targeted PKA or D-AKAP1 reversed mitochondrial fragmentation. In a recent study, Hwang et al., (Hwang, et al. 2014) reported that the mitochondrial membrane transport protein human uncoupling protein 2 (hUCP2) protected dopamine neurons in adult flies against rotenone -mediated mitochondrial fragmentation and toxicity by increasing intracellular cAMP levels. On the other hand, PKA inhibitors blocked the preserved mitochondrial integrity, movement, and cell survival induced by hUCP2 in hUCP2-expressing dopamine neurons exposed to rotenone. The neuroprotective role of PKA in PD is further highlighted by the study by Yang et al., (Yang, et al. 2008) which showed attenuation of MPTP toxicity by rolipram. Hence, this study shows proof-of-concept that phosphodiesterase inhibitors are promising anti-PD therapeutic agents that can delay degeneration of substantia nigra dopaminergic neurons.
Conclusion
In summary, PKA signaling -as regulated by AKAPs- governs mitochondrial morphology, function, mitochondrial trafficking, and neuronal survival during physiological conditions. However, during conditions of enhanced oxidative stress, dysregulated PKA signaling in various neuronal subcompartments of specific neuronal populations contributes to a converging molecular pathology in a spectrum of brain degenerative diseases including mitochondrial dysfunction, increased ROS, impaired mitochondrial trafficking and unopposed apoptotic signaling (Fig. 1). Hence, understanding how PKA is dysregulated in various neuronal subcompartments will help guide the development of future pharmacological therapies for reversing degeneration in various brain degenerative diseases.
Figure 1. A decline of PKA-protective signaling pathways in brain degenerative diseases leads to a loss of neurites.
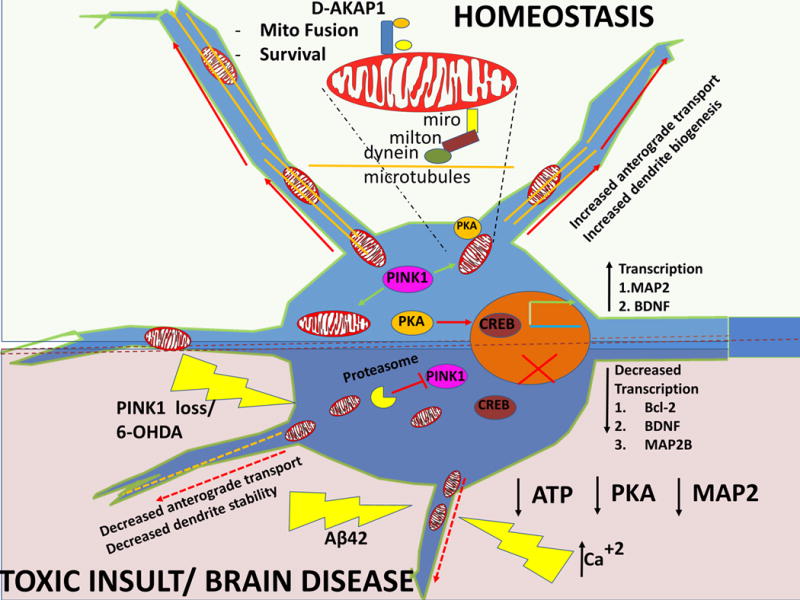
A continuous level of mitochondrial turnover, anterograde trafficking of mitochondria to neurites (shown for dendrites only), neurotrophic support as maintained by protective ser/thr kinases (PINK1 and PKA), and dendrite-remodeling events are required to maintain dendritic arbors and synaptic connectivity in neurons under physiological conditions (top half of the figure). However, loss of protective PKA signaling as induced by oxidative stress and as evidenced in PD and AD models, leads to increased oxidative stress, Drp1-dependent mitochondrial fragmentation, decreased oxidative phosphorylation, impaired mitochondrial trafficking, decreased mitochondrial biogenesis, and altered CREB localization/transcriptional-related activities (bottom half of figure).
Future endeavors should focus on developing therapies with the end-goal of “normalizing” the activities of distinct compartmentalized pools of PKA in neuronal subpopulations affected by brain degenerative diseases. Legend: 1) Yellow thunders: reactive oxygen species/toxic insult. 2) Orange lines in dendrites: microtubules, 3) Green arrow in nucleus: transcriptional activity.
It is worth noting that the use of mitochondrially-directed antioxidants, calcium chelators/metal protein-binding compounds, NMDA receptor inhibitors, and caspase inhibitors have been developed but have demonstrated modest to no benefit in clinical trials for treating various brain degenerative disorders (Snow, et al. 2010) (Johansson, et al. 2011) (Bonelli, et al. 2006) (Regland, et al. 2001) (Sampson, et al. 2012). The development of pharmacologically active compounds that can tout protective signaling in neurons poses a paramount challenge due to the compartmentalized nature of PKA signaling pathways in neurons. Although the use of global pharmacological activators of PKA (cAMP and rolipram) to reverse the loss of neurites as induced by brain degenerative disorders is tractable, serious side effects associated with high doses of global PKA activators include epilepsy and loss of dendrites (Ludvig, et al. 1992) (Xu, et al. 2012). Hence, it is imperative that pharmacological or viral-mediated gene therapy be developed and experimentally tested preclinically for future use in humans to reverse neurodegeneration in PD and AD.
Acknowledgments
This writing of this review was supported by a National Institute of Health grant GM103554, a COBRE grant in “Cell Signaling across Cell Membranes”.
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- Affaitati A, Cardone L, de Cristofaro T, Carlucci A, Ginsberg MD, Varrone S, Gottesman ME, Avvedimento EV, Feliciello A. Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J Biol Chem. 2003;278:4286–4294. doi: 10.1074/jbc.M209941200. [DOI] [PubMed] [Google Scholar]
- Aglah C, Gordon T, Posse de Chaves EI. cAMP promotes neurite outgrowth and extension through protein kinase A but independently of Erk activation in cultured rat motoneurons. Neuropharmacology. 2008;55:8–17. doi: 10.1016/j.neuropharm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Alto NM, Soderling J, Scott JD. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J Cell Biol. 2002;158:659–668. doi: 10.1083/jcb.200204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer CA, Davies P. PKA phosphorylations on tau: developmental studies in the mouse. Dev Neurosci. 2000;22:303–309. doi: 10.1159/000017454. [DOI] [PubMed] [Google Scholar]
- Arvanitis DN, Ducatenzeiler A, Ou JN, Grodstein E, Andrews SD, Tendulkar SR, Ribeiro-da-Silva A, Szyf M, Cuello AC. High intracellular concentrations of amyloid-beta block nuclear translocation of phosphorylated CREB. J Neurochem. 2007;103:216–228. doi: 10.1111/j.1471-4159.2007.04704.x. [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci. 2014;34:14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonelli RM, Wenning GK. Pharmacological management of Huntington’s disease: an evidence-based review. Curr Pharm Des. 2006;12:2701–2720. doi: 10.2174/138161206777698693. [DOI] [PubMed] [Google Scholar]
- Bui M, Gilady SY, Fitzsimmons RE, Benson MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD, Simmen T. Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem. 2010;285:31590–31602. doi: 10.1074/jbc.M110.101584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone L, Carlucci A, Affaitati A, Livigni A, DeCristofaro T, Garbi C, Varrone S, Ullrich A, Gottesman ME, Avvedimento EV, Feliciello A. Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol. 2004;24:4613–4626. doi: 10.1128/MCB.24.11.4613-4626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone L, de Cristofaro T, Affaitati A, Garbi C, Ginsberg MD, Saviano M, Varrone S, Rubin CS, Gottesman ME, Avvedimento EV, Feliciello A. A-kinase anchor protein 84/121 are targeted to mitochondria and mitotic spindles by overlapping amino-terminal motifs. J Mol Biol. 2002;320:663–675. doi: 10.1016/s0022-2836(02)00479-5. [DOI] [PubMed] [Google Scholar]
- Carlucci A, Adornetto A, Scorziello A, Viggiano D, Foca M, Cuomo O, Annunziato L, Gottesman M, Feliciello A. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. Embo J. 2008;27:1073–1084. doi: 10.1038/emboj.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnegie GK, Means CK, Scott JD. A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life. 2009;61:394–406. doi: 10.1002/iub.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai S, Li M, Lan J, Xiong ZG, Saugstad JA, Simon RP. A kinase-anchoring protein 150 and calcineurin are involved in regulation of acid-sensing ion channels ASIC1a and ASIC2a. J Biol Chem. 2007;282:22668–22677. doi: 10.1074/jbc.M703624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem. 2006;281:17870–17881. doi: 10.1074/jbc.M602632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi D, Cohen MS, Taunton J, Patel TB. The PKARIalpha subunit of protein kinase A modulates the activation of p90RSK1 and its function. J Biol Chem. 2009;284:23670–23681. doi: 10.1074/jbc.M109.032813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ, 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190:533–539. doi: 10.1083/jcb.201002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–323. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Copf T. Developmental shaping of dendritic arbors in Drosophila relies on tightly regulated intra-neuronal activity of protein kinase A (PKA) Dev Biol. 2014;393:282–297. doi: 10.1016/j.ydbio.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ, 3rd, Chu CT. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011;18:1914–1923. doi: 10.1038/cdd.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Merrill RA, Cribbs JT, Chen Y, Hell JW, Usachev YM, Strack S. The spinocerebellar ataxia 12 gene product and protein phosphatase 2A regulatory subunit Bbeta 2 antagonizes neuronal survival by promoting mitochondrial fission. J Biol Chem. 2008 doi: 10.1074/jbc.M800989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT. Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem. 2014;128:864–877. doi: 10.1111/jnc.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Dong F, Rubin CS, Hell JW. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem. 1999;274:30280–30287. doi: 10.1074/jbc.274.42.30280. [DOI] [PubMed] [Google Scholar]
- Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochim Biophys Acta. 2010;1802:11–19. doi: 10.1016/j.bbadis.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey AS, Strack S. PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J Neurosci. 2011;31:15716–15726. doi: 10.1523/JNEUROSCI.3159-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers CT, Schafer JC, Goldenring JR, Taylor SS. D-AKAP2 interacts with Rab4 and Rab11 through its RGS domains and regulates transferrin receptor recycling. J Biol Chem. 2009;284:32869–32880. doi: 10.1074/jbc.M109.022582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciello A, Gottesman ME, Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Feliciello A, Jones JK, Avvedimento EV, Gottesman ME. PKA-dependent binding of mRNA to the mitochondrial AKAP121 protein. J Mol Biol. 2003;327:885–897. doi: 10.1016/s0022-2836(03)00173-6. [DOI] [PubMed] [Google Scholar]
- Giralt A, Saavedra A, Carreton O, Xifro X, Alberch J, Perez-Navarro E. Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington’s disease. Hum Mol Genet. 2011;20:4232–4247. doi: 10.1093/hmg/ddr351. [DOI] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell’Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima Y, Ohsako S, Yamauchi T. Overexpression of Ca2+/calmodulin-dependent protein kinase II in Neuro2a and NG108-15 neuroblastoma cell lines promotes neurite outgrowth and growth cone motility. J Neurosci. 1993;13:559–567. doi: 10.1523/JNEUROSCI.13-02-00559.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusdon AM, Chu CT. To eat or not to eat: neuronal metabolism, mitophagy, and Parkinson’s disease. Antioxid Redox Signal. 2010;14:1979–1987. doi: 10.1089/ars.2010.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol. 2002;158:541–549. doi: 10.1083/jcb.200110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci U S A. 1997;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang RD, Wiemerslage L, LaBreck CJ, Khan M, Kannan K, Wang X, Zhu X, Lee D, Fridell YW. The neuroprotective effect of human uncoupling protein 2 (hUCP2) requires cAMP-dependent protein kinase in a toxin model of Parkinson’s disease. Neurobiol Dis. 2014;69:180–191. doi: 10.1016/j.nbd.2014.05.032. [DOI] [PubMed] [Google Scholar]
- Jiang H, Cheng D, Liu W, Peng J, Feng J. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem Biophys Res Commun. 2010;395:471–476. doi: 10.1016/j.bbrc.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson C, Ballard C, Hansson O, Palmqvist S, Minthon L, Aarsland D, Londos E. Efficacy of memantine in PDD and DLB: an extension study including washout and open-label treatment. Int J Geriatr Psychiatry. 2011;26:206–213. doi: 10.1002/gps.2516. [DOI] [PubMed] [Google Scholar]
- Kim YT, Wu CF. Reduced growth cone motility in cultured neurons from Drosophila memory mutants with a defective cAMP cascade. J Neurosci. 1996;16:5593–5602. doi: 10.1523/JNEUROSCI.16-18-05593.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lin JT, Chang WC, Chen HM, Lai HL, Chen CY, Tao MH, Chern Y. Regulation of feedback between protein kinase A and the proteasome system worsens Huntington’s disease. Mol Cell Biol. 2013;33:1073–1084. doi: 10.1128/MCB.01434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livigni A, Scorziello A, Agnese S, Adornetto A, Carlucci A, Garbi C, Castaldo I, Annunziato L, Avvedimento EV, Feliciello A. Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol Biol Cell. 2006;17:263–271. doi: 10.1091/mbc.E05-09-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludvig N, Mishra PK, Jobe PC. Dibutyryl cyclic AMP has epileptogenic potential in the hippocampus of freely behaving rats: a combined EEG-intracerebral microdialysis study. Neurosci Lett. 1992;141:187–191. doi: 10.1016/0304-3940(92)90891-a. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K, Yamakuni T, Hashimoto M, Haque AM, Shido O, Mimaki Y, Sashida Y, Ohizumi Y. Nobiletin restoring beta-amyloid-impaired CREB phosphorylation rescues memory deterioration in Alzheimer’s disease model rats. Neurosci Lett. 2006;400:230–234. doi: 10.1016/j.neulet.2006.02.077. [DOI] [PubMed] [Google Scholar]
- Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, Gold MG, Petrosyan S, Taylor SS, Murphy AN, Ha T, Santana LF, Tasken K, Scott JD. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc Natl Acad Sci U S A. 2011;108:E1227–1235. doi: 10.1073/pnas.1107182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RA, Dagda RK, Dickey AS, Cribbs JT, Green SH, Usachev YM, Strack S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011;9:e1000612. doi: 10.1371/journal.pbio.1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RA, Slupe AM, Strack S. N-terminal phosphorylation of protein phosphatase 2A/Bbeta2 regulates translocation to mitochondria, dynamin-related protein 1 dephosphorylation, and neuronal survival. FEBS J. 2013;280:662–673. doi: 10.1111/j.1742-4658.2012.08631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhall KJ, Criniti AR, Cheah CS, Smith KC, Kafer KE, Burkart AD, McKnight GS. Dynamic anchoring of PKA is essential during oocyte maturation. Curr Biol. 2006;16:321–327. doi: 10.1016/j.cub.2005.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Dixon JE. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Papa S. The NDUFS4 nuclear gene of complex I of mitochondria and the cAMP cascade. Biochim Biophys Acta. 2002;1555:147–153. doi: 10.1016/s0005-2728(02)00270-0. [DOI] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci. 2014;17:367–376. doi: 10.1038/nn.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino C, Feliciello A, Schiattarella GG, Esposito G, Guerriero R, Zaccaro L, Del Gatto A, Saviano M, Garbi C, Carangi R, Di Lorenzo E, Donato G, Indolfi C, Avvedimento VE, Chiariello M. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc Res. 2010;88:101–110. doi: 10.1093/cvr/cvq155. [DOI] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos M, Cortes E, Vonsattel JP, Fahn S, Waters C, Cote LJ, Moskowitz C, Honig LS, Clark LN, Marder KS, Alcalay RN. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J Mol Neurosci. 2012;47:139–143. doi: 10.1007/s12031-011-9696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Wang M, Pham S, Sze CI, Eckman CB. Downregulation of CREB expression in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Mol Neurodegener. 2011;6:60. doi: 10.1186/1750-1326-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG. Treatment of Alzheimer’s disease with clioquinol. Dement Geriatr Cogn Disord. 2001;12:408–414. doi: 10.1159/000051288. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodger J, Goto H, Cui Q, Chen PB, Harvey AR. cAMP regulates axon outgrowth and guidance during optic nerve regeneration in goldfish. Mol Cell Neurosci. 2005;30:452–464. doi: 10.1016/j.mcn.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci U S A. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson EL, Jenagaratnam L, McShane R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst Rev. 2012;5:CD005380. doi: 10.1002/14651858.CD005380.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, Miller DW, Zambrano I, Cowburn RF, Behbahani H, Cedazo-Minguez A, Cookson MR. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One. 2009;4:e5701. doi: 10.1371/journal.pone.0005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarina, Yagi Y, Nakano O, Hashimoto T, Kimura K, Asakawa Y, Zhong M, Narimatsu S, Gohda E. Induction of neurite outgrowth in PC12 cells by artemisinin through activation of ERK and p38 MAPK signaling pathways. Brain Res. 2013;1490:61–71. doi: 10.1016/j.brainres.2012.10.059. [DOI] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorziello A, Savoia C, Sisalli MJ, Adornetto A, Secondo A, Boscia F, Esposito A, Polishchuk EV, Polishchuk RS, Molinaro P, Carlucci A, Lignitto L, Di Renzo G, Feliciello A, Annunziato L. NCX3 regulates mitochondrial Ca(2+) handling through the AKAP121-anchored signaling complex and prevents hypoxia-induced neuronal death. J Cell Sci. 2013;126:5566–5577. doi: 10.1242/jcs.129668. [DOI] [PubMed] [Google Scholar]
- Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120:419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM, Protect Study G. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- Strack S. Overexpression of the protein phosphatase 2A regulatory subunit Bgamma promotes neuronal differentiation by activating the MAP kinase (MAPK) cascade. J Biol Chem. 2002;277:41525–41532. doi: 10.1074/jbc.M203767200. [DOI] [PubMed] [Google Scholar]
- Tominaga-Yoshino K, Kondo S, Tamotsu S, Ogura A. Repetitive activation of protein kinase A induces slow and persistent potentiation associated with synaptogenesis in cultured hippocampus. Neurosci Res. 2002;44:357–367. doi: 10.1016/s0168-0102(02)00155-4. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. Beta -amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival Is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Unal R, Pokrovskaya I, Tripathi P, Monia BP, Kern PA, Ranganathan G. Translational regulation of lipoprotein lipase in adipocytes: depletion of cellular protein kinase Calpha activates binding of the C subunit of protein kinase A to the 3′-untranslated region of the lipoprotein lipase mRNA. Biochem J. 2008;413:315–322. doi: 10.1042/BJ20071559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yuen EY, Zhou Y, Yan Z, Xiang YK. Amyloid beta peptide-(1-42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem. 2011;286:31852–31863. doi: 10.1074/jbc.M111.244335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Sunahara RK, Krumins A, Perkins G, Crochiere ML, Mackey M, Bell S, Ellisman MH, Taylor SS. Cloning and mitochondrial localization of full-length D-AKAP2, a protein kinase A anchoring protein. Proc Natl Acad Sci U S A. 2001;98:3220–3225. doi: 10.1073/pnas.051633398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QW, Rowan MJ, Anwyl R. Inhibition of LTP by beta-amyloid is prevented by activation of beta2 adrenoceptors and stimulation of the cAMP/PKA signalling pathway. Neurobiol Aging. 2009;30:1608–1613. doi: 10.1016/j.neurobiolaging.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Engbers J, Khaja S, Xu L, Clark JJ, Hansen MR. Influence of cAMP and protein kinase A on neurite length from spiral ganglion neurons. Hear Res. 2012;283:33–44. doi: 10.1016/j.heares.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Gotz ME, Ozawa H, Luckhaus C, Saito T, Rosler M, Riederer P. Hippocampal level of neural specific adenylyl cyclase type I is decreased in Alzheimer’s disease. Biochim Biophys Acta. 2000;1535:60–68. doi: 10.1016/s0925-4439(00)00083-1. [DOI] [PubMed] [Google Scholar]
- Yang L, Calingasan NY, Lorenzo BJ, Beal MF. Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp Neurol. 2008;211:311–314. doi: 10.1016/j.expneurol.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Suzuki K, Kidokoro Y. Two independent pathways mediated by cAMP and protein kinase A enhance spontaneous transmitter release at Drosophila neuromuscular junctions. J Neurosci. 2000;20:8315–8322. doi: 10.1523/JNEUROSCI.20-22-08315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]