

Abstract
The cytokines IL-6 and IL-10 are produced by cells of the adaptive and innate arms of the immune system and they appear to play key roles in genetically diverse autoimmune diseases such as relapsing remitting multiple sclerosis (MS), rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE). Whereas previous intense investigations focused on the generation of autoantibodies and their contribution to immune-mediated pathogenesis in these diseases more recent attention has focused on the roles of cytokines such as IL-6 and IL-10. In response to pathogens, antigen presenting cells (APC), including B cells, produce IL-6 and IL-10 in order to up- or down-regulate immune cell activation and effector responses. Evidence of elevated levels of the proinflammatory cytokine IL-6 has been routinely observed during inflammatory responses and in a number of autoimmune diseases. Our recent studies suggest that MS peripheral blood B cells secrete higher quantities of IL-6 and less IL-10 than B cells from healthy controls. Persistent production of IL-6, in turn, contributes to T cell expansion and the functional hyperactivity of APC such as MS B cells. Altered B cell activity can have a profound impact on resultant T cell effector functions. Enhanced signaling through the IL-6 receptor can effectively inhibit cytolytic activity, induce T cell resistance to IL-10-mediated immunosuppression and increase skewing of autoreactive T cells to a pathogenic Th17 phenotype. Our recent findings and studies by others support a role for the indirect attenuation of B cell responses by Glatiramer acetate (GA) therapy. Our studies suggest that GA therapy temporarily permits homeostatic regulatory mechanisms to be reinstated. Future studies of mechanisms underlying dysregulated B cell cytokine production could lead to the identification of novel targets for improved immunoregulatory therapies for autoimmune diseases.
Keywords: Multiple sclerosis, interleukin-6, interleukin-10, Glatiramer acetate, B lymphocytes
1. Introduction
Autoimmune diseases, including relapsing-remitting multiple sclerosis (MS), rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) demonstrate abnormalities in both innate and adaptive immune responses. In spite of the fact that MS has a distinct genetic susceptibility profile in comparison to RA and SLE, these diseases display altered regulation in similar mediators, such as the cytokines interleukin-6 (IL-6) and interleukin-10 (IL-10), involved in controlling inflammation and immunity [1, 2]. Therapies modulating IL-6 and IL-10 activity have been examined in RA and models of SLE for two decades [3–8]. Anti-IL-6 targeted therapies have been FDA approved for the treatment of rheumatic diseases including RA, SLE, Castleman’s disease and there are ongoing clinical trials for other autoimmune diseases [3, 5, 9]. Anti-IL-10 therapy is in clinical trials for RA [6]. Here, we review recent studies and our own work exploring the roles of IL-6 and IL-10 in MS [10]. These studies suggest that despite the differences in clinical and genetic profiles, understanding the mechanisms influencing IL-6 and IL-10 cytokine programs could lead to improved targeted therapies for MS and a number of other autoimmune diseases.
2. Interleukin-6
Both IL-6 and IL-10 can be produced at inflammatory sites and several recent reviews have described the pleiotropic effects of IL-6 and IL-10 in detail [3–5, 9]. IL-6 and the IL-6 receptor alpha chain (IL- 6R) are expressed by monocytes, lymphocytes, fibroblasts, endothelial cells and a number of other cell types [11–14]. After IL-6 ligation, the IL-6R signals through homodimerization of gp130 (CD130) that is a ubiquitous common signal transducer for several cytokines. IL-6 signaling through gp130 activates the protein-tyrosine kinase, Janus kinase 1 (JAK1), and transiently activates signal transducer and activator of transcription 3 (STAT3) dimerization and phosphorylation, followed by STAT3 nuclear translocation and target gene transcription. IL-6 activates the mitogen activated protein kinase (MAPK) pathway, resulting in transcription of other genes. For example, in human monocyte-derived macrophages 39 genes were induced by exposure to IL-6 while 31 transcripts were down-modulated [15]. IL-6 activates the tyrosine phosphatase, SHP-2, while STAT3 induces protein inhibitors of activated STATs (PIAS), suppressor of cytokine signaling 1 (SOCS1) and SOCS3 as part of a complex auto- regulatory negative feedback loop. A soluble form of the IL-6R (sIL-6R) can be produced by enzymatic cleavage of the membrane form or by alternative splicing of the IL-6 receptor alpha transcript. IL-6 bound to the sIL-6R can form a complex with gp130 on cells and transduce a signal to the cell. Such IL-6 “trans-signaling” has been implicated in lupus renal pathology [13, 14, 16]. IL-6 has important roles in homeostatic mechanisms, such as inducing hepcidin which reduces serum iron levels and enhances the hepatocyte inflammatory response, inducing acute phase proteins, cell growth, cell survival and promotes differentiation of lymphocytes and many other cells [7, 9].
3. Interleukin-10
IL-10 is predominantly produced by stimulated myeloid cells and lymphocytes and can be produced at lower levels by other cells during inflammation [17]. IL-10 dimerizes and binds to a tetramer consisting of two IL-10 receptor (IL-10R)1 subunits and two IL-10R2 subunits and with JAK1 and Tyk2. This induces the phosphorylation and activation of STAT1, STAT3, and, in some cells, STAT5 leading to an inhibition of NFκB-mediated signal transduction [17]. IL-10 induces a sustained activation of STAT3 and SOCS3 transcription which contribute to transcriptional regulation of pro-apoptotic genes, inhibition of cell activation, cytokine production and proliferation [18–21]. IL-10 inhibits production of a number of proinflammmatory cytokines, including IL-1β, IL-6, IL-12, IL-18, GM-CSF and TNFα and IL-10 promotes the production of other anti-inflammatory mediators, such as the IL-1β receptor antagonist and soluble TNFα receptors p55 and p75 [17]. The anti-inflammatory effects of IL-10 have been demonstrated in models of MS, RA, SLE, diabetes, inflammatory bowel disease and other autoimmune disorders [17]. In macrophages, IL-10 is expressed as a means to inhibit immune responses [22–24]. Although IL-10 has been reported to suppress CD40, CD80, CD86, IL-12 and T effector functions, IL-10 in the presence of other cytokines, enhances human B cell activation, proliferation and differentiation into immunoglobulin (Ig) secreting cells [22, 25–28] Importantly, IL-6 and IL-10 expression and function can be regulated by a number of factors including other cytokines, genetic polymorphisms and noncoding RNAs as described below.
4. Cytokine regulation by genetic elements
4.1 Noncoding RNAs
MicroRNAs (miRNAs) and long noncoding RNAs (lncRNAs) can influence a single cytokine pathway or regulate a cassette of cytokines en bloc [29–32]. Multiple studies have identified dysregulated miRNAs in MS patients and their potential roles in controlling gene expression in lymphocytes and CNS tissues [31–34]. In particular, miRNA-155 has been the most thoroughly studied and was found to confer susceptibility to experimental autoimmune encephalomyelitis (EAE) through regulation of dendritic cells (DC) cytokines, including IL-6, and T-cell intrinsic mechanisms that support Th17 development [34]. In humans, several observations support a role for miRNAs in controlling cytokine production. For instance, overexpression of miRNA-132, that targets surtuin-1, in B cells from MS patients is responsible for elevated TNFα and LTβ but had no impact on IL-10 secretion [35]. miRNA- 142-3p inhibits IL-10 expression and is elevated in PBMC from MS patients [36]. In the same study, they found that the immunomodulatory therapeutic glatiramer acetate (GA) reduced levels of miR-146a and miR-142-3p observed in peripheral blood mononuclear cells (PBMC) from MS patients [36, 37]. However, miR-146a negatively regulates IL-6 [38]. Other studies have demonstrated modulation of miRNAs by MS therapies such as IFN-β and Natilizumab [39, 40].
4.2 Genetics
Beyond the known HLA risk loci, such as the HLA-DRB1 locus, approximately 52 risk loci for MS have been identified in GWAS studies, many known as key regulators of the immune response [41]. For example, notable Single Nucleotide Polymorphisms (SNPs) in costimulatory molecules, cytokines, cytokine receptors and signaling pathways that impact APC function and T cell migration, expansion and differentiation such as CD40, CD58, CD69, CD86, MERTK, CCR4, CXCR5, IL12A, IL12B, IL2RA, IL22RA, TYK2, STAT3, STAT4, NFKBI, EOMES, IRF8, BCL10 and BATF were identified and could be important in grouping MS patients into subsets for treatment [41–43]. Recent pathway analysis of GWAS datasets have identified cell adhesion molecules such as ITGAL, ICAM1, ICAM3 and validated VCAM1 as important potential targets for novel therapies [44]. IL-6, IL-10 and their downstream effectors can impact expression of many of the cell adhesion molecules, chemokine receptors and signaling pathways that have been associated with MS.
Although SNPs for IL-10, IL-6 and their receptors have not been identified in GWAS studies to date as risk alleles for MS, it is clear that some MS patients harbor systemic dysregulation of these cytokines. Of note, IL-6 cytokine responsiveness and inflammation are impaired in RA patients with a common IL-6R polymorphism [45]. Thus, it is likely that future studies of the IL-6R common variants along with other select genes will distinguish aggressive from indolent disease subgroups of MS patients. Importantly, SNPs in the STAT3 pathway have been described in MS and additional studies are needed to determine the impact of these variants on disease [41, 42, 46].
A recent large study of candidate causal genetic variants for 21 autoimmune diseases, including MS, indicated that approximately 90% of the causal variants were non-coding SNPs and that 60% could be mapped to immune-related enhancer-like elements, while only 10–20% appeared to alter known transcription factor binding motifs. These authors concluded that the non-coding variants might cause subtle key differences in transcription or epigenetics that influence immune responses, allowing variants to escape pressure from negative selection [47]. Of interest, some originally predicted “non-coding RNAs” have been found to code for small peptides with powerful influences on cell regulation [48].
5. A Cytokine Imbalance in MS
Relapsing-remitting MS is an immune-mediated inflammatory demyelinating disorder of the central nervous system [49–51]. In MS, cells producing IL-10 are protective whereas those that produce IL-6 appear to contribute to inflammation related to disease pathogenesis. Upon ex vivo stimulation, PBMC isolated from MS patients produced decreased levels of IL- 10 and elevated levels of IL-6 compared to healthy controls [52–54]. The exact mechanism of the enhanced IL-6 secretion and deficiency in IL-10 production in MS PBMC is not clear. Recent studies have begun to uncover perturbations in the response to both IL-6 and IL-10 that might further contribute to the dysregulated axis of IL-6 and IL-10 in MS. Here we will discuss IL-10 and IL-6 production, differential responses of MS immune cells to stimulation by these cytokines, and, how therapeutic intervention with glatiramer acetate (GA) alters dysregulated IL-6 and IL-10 in MS.
6. Lessons from EAE Murine Models
Both T helper-1 (Th1) cells that produce interferon-γ and Th17 cells that secrete IL-17 contribute to disease pathogenesis in MS and EAE. The discovery that IL-6 promoted Th17 development prompted further investigations into the role of IL-6 in EAE and MS. Indeed, IL-6-deficient mice were resistant to EAE [55, 56] because they failed to induce central nervous system (CNS) specific Th1 and Th17 cells [57]. Treatment of EAE mice with a neutralizing antibody to IL-6 diminished EAE severity, but increased IL-6 levels in the CNS [58]. Furthermore, IL-6 deficient T cells could not induce EAE upon adoptive transfer to wild type mice [59]. However, wild type DC loaded with CNS antigens rescued susceptibility to EAE in IL-6-deficient recipient mice [60]. These studies highlight the importance of IL-6 in the establishment of EAE. The role of IL-6 in ongoing EAE and MS is less clear. Surprisingly, B cell-specific MHC class II and IL-6 knockout mice demonstrated decreased disease severity in a B cell-dependent EAE model, suggesting a role for antigen presenting cell (APC) function and IL-6 production by B cells in the establishment and progression to symptomatic disease [61].
Most studies supporting the roles of IL-10 as protective and IL-6 in promoting disease were carried out in the mouse model of MS, EAE. In this model, IL-10- deficient mice developed more severe EAE compared to wild type mice, whereas mice overexpressing IL-10 were resistant to EAE [62, 63]. The ability of recombinant IL-10 or IL-10-transduced cells to control disease once disease was established yielded inconsistent results [64–67]. Yet the adoptive transfer of IL- 10-expressing B regulatory cells (Breg) at the onset of disease, or adoptive transfer of T regulatory cells (Treg) reduced EAE severity through an IL-10-dependent mechanism [68–71]. These results suggest that it was not simply the presence of IL-10 that limited disease, but rather regulatory cells that expressed IL-10 were key to controlling aberrant immune responses.
Of note, innate and adaptive regulatory cells control inflammatory processes by secretion of cytokines, including IL-10, transforming growth factor-β (TGFβ) and IL-35 and through cell contact dependent mechanisms [72–74]. Both cytokines and cell contact dependent mechanisms play a role in the promotion or suppression of EAE and these observations have been recently reviewed [75, 76]. For example, TGFβ alone or in combination with IL-10 acts to restrain the immune response. Alternatively, TGFβ acts in concert with IL-6 and other factors to drive the development of Th17 cells and potentiate EAE [75]. Although IL-12 promotes Th1 cell differentiation and Th1 secreted IFNγ acts as a positive feedback to upregulate the IL12Rβ2, the activation of the transcription factor STAT1 by IFNγ as well as IFNγ itself are dispensable for EAE induction [76]. By contrast, mice deficient in IL-17, IL-23, the IL-12 induced transcription factor STAT4, the Th1-associated transcription factor T-bet or the Th17-associated transcription factors RORγt and RORα appear to have varying levels of resistance to EAE [76, 77]. Mice deficient in IRF8, a GWAS identified MS susceptibility gene, were resistant to EAE and unable to generate Th1, Th17 or Treg cells [77]. IRF8 promoted EAE development through several mechanisms. As an example, IRF8 was required for expression of APC αvβ8 integrin which facilitated delivery of TGFβ to naïve cells that was required for Th17 differentiation. Interestingly, IRF8 deficiency also led to elevated IL-27 levels. IL-27 acts to suppress EAE. With regard to B cell regulation of EAE, mice with plasma cells that could not produce IL-35 in addition to IL-10 were unable to recover from EAE suggesting that these suppressive effectors act in a coordinated manner to restrain hyperinflammatory responses [74].
7. Immunomodulation by MS therapies
7.1 Glatiramer Acetate
Glatiramer acetate (GA) is an analog of myelin basic protein (MBP), a candidate MS antigen and GA is an FDA approved therapy for MS. GA is a synthetic copolymer consisting of four random amino acids (L- alanine, L-lysine, L-glutamic acid and L-tyrosine) in a similar ratio to MBP. GA impacts the immune system in a variety of ways [78], including promiscuous binding to HLA molecules [79, 80], interfering with the activation of antigen-specific T cells [81, 82] and induction of Th2 cells responses [83, 84].
GA is associated with increased IL-10 production [85]; however, isolated GA-specific T cell clones secreted IL-6 [86]. The role of IL-6 in GA therapy has not been resolved because IL-6 can also support Th2 differentiation in T cells by inducing expression of IL-4 and in murine models, blocking IFN-γ signaling [87, 88]. This makes GA a particularly interesting therapy to study the dual role of IL-6 in supporting both Th2 and Th17 responses that are beneficial and detrimental in MS respectively, while suppressing the also detrimental Th1 cells.
In mice, GA increases DC that produce IL-10 and IL-10-secreting type 2 monocytes that suppress EAE by inducing Treg cells [89, 90]. GA treatment also reduces the expression of IL-6 in the CNS of EAE mice [91]. Perhaps most surprisingly, B cells from GA-treated EAE-naïve mice secrete more IL-10 and suppress EAE upon adoptive transfer [92]. B cells from mice with EAE that are treated with GA produce less IL-6 and more IL-10 and exhibit enhanced Breg function in suppressing EAE compared to untreated B cells [93, 94].
7.2. Other MS therapies modulate IL-6 and IL-10
Other therapies for MS have been reported to modulate cytokine production. For example, therapeutic benefits that might be partially derived from increased IL-10 production and restoration of Treg function have been suggested for IFN-βs. IFN-βs, including IFN-β1a and IFN-β1b, currently comprise 5 of the 12 FDA approved therapies for MS. IFN-β therapy transiently increased IL-10 production by monocytes in vivo and increased the total frequency of PBMC that spontaneously secreted IL-10 [95, 96]. Moreover, B cells from patients treated with IFN-β secreted increased amounts of IL-10 [97]. IFN-β also induced increased regulatory T cells including Tr1 [98]. Finally, IFN-β has been reported to enhance IL-10R1 expression and IL-10 signaling via STAT3 [99].
Therapies that might not directly modulate APC cytokine production, altered secretion of IL-6 and IL-10. Fingolimod, a sphingosine 1-phosphate receptor targeting agent, increased the percentage of circulating B cells producing IL-10 and IL-10 production by DC in vitro but did not modulate IL-6 secretion [100, 101]. Teriflunomide, which inhibits dihydroorotate dehydrogenase resulting in inhibition of pyrimidine synthesis, blocked rapidly dividing B and T cells and inhibited IL-6 production by a mechanism reported to be independent from pyrimidine synthesis [102]. Another therapeutic agent that blocks cell division, mitoxantrone, impacts cytokine production in mice [103, 104]. However, no similar effects were observed in mitoxantrone-treated MS patients [105, 106]. Dimethyl fumarate (DMF) transiently induced Th2 cells and reduced psoriasis patients’ IFN-γ+ CD4+ cells in vivo, and induced type II DC secreting IL-10 in vitro in cultures of both human and murine cells [107]. Type II DMF-induced DC were protective in an EAE model upon adoptive transfer in mice [107]. However, it is unknown whether DMF acts primarily by modulating DC in MS patients. Moreover it is not entirely clear how MS therapies remodel the immune system components that contribute to differential cytokine secretion.
8. Cytokine Production and Responses in the Periphery
8.1 Innate Immune Cells and APC
Myeloid cells act as APC and secrete an array of cytokines that influence T cell polarization, B cell activation, proliferation and differentiation into plasma cells and myeloid cells orchestrate tissue inflammation and repair. Of interest, with regard to innate immune cells, differences in cytokine profiles obtained after stimulation through the highly conserved pattern recognition receptors (PRR) including the toll-like receptors (TLR) have been observed between MS and healthy individuals. The degree to which the autoimmune environment influences innate immune cells has not been elucidated, but multiple studies support the existence of subtypes of monocytes and DC that are programmed by their microenvironment and related stimuli to become polarized to secrete specific cytokines that have been generally observed in in vitro studies [108–110]. For example, macrophages have been categorized into three major phenotypes: a) classical or inflammatory, b) alternatively activated or regulatory and c) cells that contribute to tissue repair and wound healing [111].
Monocytes from MS patients express less TLR7 and subsequently secrete less IL-6 upon TLR7 stimulation [112]. CD14+CD16+ inflammatory monocytes are preferentially expanded in MS patients and express higher levels of activation markers compared to healthy control monocytes [113]. In the same study, both CD14+CD16+ inflammatory and CD14+ classical monocytes from MS patients secreted more IL-6 compared to healthy control monocytes when stimulated with the TLR4 ligand, LPS. However, CD14+CD16+ inflammatory monocytes also secreted significantly more IL-10 compared to healthy control monocytes. This was surprising because CD14+CD16+ monocytes have been associated with inflammatory cytokine production and often produce less IL-10 than the classical CD14+CD16− monocytes [114]. These studies also highlight alterations in activated myeloid cell TLR expression that are likely tied to differential expression of cytokines in MS patients.
Unlike for B cells, GA therapy directly impacts monocyte activation to a variety of cytokine and TLR stimuli [115]. When monocytes were cultured with GA, IL-10 levels were increased and IL-6 secretion was diminished [116, 117]. This is in line with previous studies showing that recombinant IL-10 suppressed IL-6 production by monocytes in both healthy donors and MS patients [118]. However, whether the suppression of the IL-6 response is the result of increased IL-10 production is not known. In cultures stimulated with MBP and polyclonal T cells, monocytes from untreated MS patients produced more IL-6 [119]. However, T cells from GA-treated MS patients induced monocytes to secrete IL-10 and subsequently promoted Th2 differentiation [120].
DC are highly potent APC that exert profound effects on T cell polarization. In MS patients the frequency of myeloid DC producing IL-6 was significantly elevated, while those producing IL-10 were similar to healthy controls [121]. However, spontaneous secretion of IL-6 and IL-10 was lower in MS patients’ plasmacytoid DC and after stimulation with the double stranded DNA virus, HSV1 compared to healthy donors [122]. This may be attributed to the observation that, despite the lower plasmacytoid DC cytokine production, the frequency of myeloid DC that secreted IL-6 upon TLR7 stimulation was increased in MS patients [123].
Myeloid DC incubated with GA produced less IL-12, more IL-10 and induced more Th2 cells compared to myeloid DC from healthy controls [124, 125]. These observations appear consistent with IL-12 deficient asthma models where Th2 differentiation was observed with lower doses of antigen, OX40L, IL-4 and IL-33, whereas Th17 differentiation was found with higher doses of antigen, and correlated with APC expression of CD40, CD86, IL-6 and TGF-β production [126]. APC cytokine production was correlated with the strength of the stimulation which directly influenced the strength of NFκB signaling [126]. The importance of a similar threshold effect has recently been described for the activation of B cell receptor induced NFκB activation which can be prolonged by a positive feedback loop [127]. Thus one possibility is that GA therapy attenuates antigen-induced myeloid APC and B cell activation and function allowing induction of negative feedback mechanisms. Such a tempered B cell response allows for skewing of T cells undergoing differentiation to switch from a Th17 to a Th2 differentiation pathway.
8.2 B Lymphocytes
B lymphocytes secrete IL-6 and IL-10 in response to stimulation by TLR agonists, CD40 stimulation, and BCR ligation [128, 129]. B cells producing IL-10 (B10 cells) are identifiable by intracellular staining for IL- 10 after a brief ex vivo stimulation whereas B10 progenitor (B10PRO) cells require additional activation signals over 48–72 h in culture [130]. There are divergent reports describing the phenotype of B10 cells that include CD24hi CD27+ [130], CD19+CD25+Cd71+CD73lo [131], and CD19+ CD24hi CD38hi [132, 133], yet B cells that make IL-10 are not exclusively confined within these phenotypes. In addition to phenotypic studies, one study comparing IL-10+ and IL-10− B cells demonstrated over 99 percent similarity in gene expression [134] whereas another study identified changes in B10 and non-B10 gene expression [135]. Despite their incomplete phenotypic profiles, human B10 cells or a subset of IL-10 producing B cells appears to be similar to their murine counterparts in their ability to dampen T cell responses [133, 135].
The frequency of B10 cells in the blood was similar between healthy donors (HD) and MS patients but the frequency of MS B10PRO cells was dependent on the stimuli in cultures. When cultured in vitro, B cells from MS patients exhibited a striking defect in their ability to secrete IL-10 or to do so in a normal ratio to proinflammmatory cytokines [52, 136]. This may be partially explained by the reduced level of TLR9 expression in memory B cells from MS patients as B cells were exposed to CpG, which is commonly used to induce maximal IL-10 production by B cells, however, this deficit in IL-10 production was also observed independently of TLR9 stimulation. It is not yet clear how to correlate human B10 and B10PRO cells with IL-10 production after in vitro stimulation cultures and whether these assays translate to B cell activity in vivo.
More recently the fate of murine B10 cells was investigated utilizing a GFP reporter driven by an IL-10 promoter. Using this system, B cells secreting IL-10 were found to differentiate into antibody secreting plasmablasts/plasma cells [137]. Human IL-10+ B cells are more difficult to study because of their phenotypic heterogeneity and requirement for stimulation in detection assays. Using IL-10 secretion capture assays two studies have approached the question of B10 cell fate in humans. One study found that IL-10+ B cells secreted less IgG in culture compared to IL-10− B cells [134]. However, another study found that IL-10+ naïve B cells are primary producers of IgG4 [131]. Further studies are warranted to dissect the capabilities of IL-10 producing B cells in humans and whether their functions parallels B10 or Breg cells in mice.
In addition to the underproduction of IL-10, B cells isolated from MS patients also secrete excessive levels of IL-6 as demonstrated by our laboratory and others [10, 138]. The combined effect results in a significant imbalance in the ratio of IL-6 to IL-10 from polyclonal activated memory and naïve B cells from MS patients [10]. It remains to be seen how B cell derived IL-6 and IL-10 play a role in MS; however, from studies carried out in mice, B cell-derived IL-6 directs the transition to Th17 cells which results in disease pathogenesis [61]. Furthermore IL-6 production by B cells was reduced in mice treated with GA and IL- 10 was enhanced [94]. The results in murine models led us to study whether a similar alteration of the IL-6:IL-10 ratio could be observed in B cells isolated from GA-treated MS patients.
We hypothesized that GA could modulate the ability of B cells to secrete IL-6 and IL-10. While we found no direct impact of GA on B cells [10], B cells from GA- treated MS patients secreted IL-10 at levels similar or in some cases greater than that produced by B cells isolated from healthy donors. There has been considerable interest in defining the regulatory role of B cells and possibly that of Ig-secreting cells in the context of autoimmune disease based on observations in mice [142]. We tested whether B cells from GA-treated MS patients secreted more antibodies than untreated MS patients and found that both IgG and IgM secretion was significantly higher in GA-treated MS patients [10]. We did not determine whether these antibodies were GA specific or of the IgG4 isotype, as reported by van de Veen et al. from IL-10 secreting naïve B cells or IgG4 GA-specific antibodies that have been observed to develop during GA therapy [133, 143]. It is important to delineate the fate of the IL-10 producing cells induced by GA or other MS therapies given their potential impact on disease.
In contrast to the GA-mediated enhanced production of IL-10 by MS B cells, we found that the secretion of IL-6 by B cells from GA-treated MS patients was transiently diminished during the first 32 months of therapy and increased thereafter to levels equal to, or in excess of, treatment-naïve patients [10]. This finding was similar to our B cell cytokine production data from two GA-treated MS patients with disease exacerbations (unpublished data). Although it is premature to speculate whether decreased levels of ex vivo B cell IL-6 production is predictive of a positive clinical response to GA therapy, these results do raise important questions as to the dual role of IL-6 in promoting pathogenic Th17 and protective Th2 effector cells during GA therapy.
8.3 T Lymphocytes
The evaluation of regulatory T cells in MS has been a long-standing interest in the field [139–141]. More recent studies using techniques such as intracellular staining have allowed for the identification of Treg cells that mediate cytokine-dependent suppression (Tr1). However, Tr1 cells that produce Stat-3 induced IL-10 early after activation, do not fall into a single phenotypic category [140–142]. The suppressive capabilities of Tr1 cells are dependent on IL-10 and other soluble mediators, such as TGF-β. These cells are distinct from CD4+CD25+/hi FoxP3+ Treg cells that are also capable of secreting IL-10 but exert suppression through a cell contact-dependent mechanism [143].
Naturally occurring Treg cells have been more extensively studied in MS and appear to have a similar frequency compared to healthy individuals in most cohorts but diminished suppressive capacity [144–146]. Like IL-10 producing human B cells there are limited studies focused on Tr1 cells because IL-10 secretion is the defining characteristic. To determine whether the induction of Tr1 cells was altered in MS patients, T cells were stimulated with anti-CD3 and anti-CD46 monoclonal antibodies. The Tr1 cells derived from MS patients secreted less IL-10 compared to Tr1 from healthy donors [147]. Similarly, stimulation with anti-CD28, anti-CD3 and anti-CD46 antibodies combined resulted in a lower frequency of IL-10+ T cells and lower secretion of IL-10 by MS T cells [148]. Together these studies showed that in vitro differentiation of MS Tr1 was defective and MS Tr1 cells failed to secrete high levels of IL-10. It remains to be seen whether these in vitro defects are reflected by impaired Tr1 function of MS T cells in vivo.
CD8+ T cells are also capable of producing IL-10 [149] and have been described in the peripheral blood and CSF of MS patients as compared to healthy controls. CD8+ IL-10-producing cells were similar in phenotype to CD4+ Treg cells, characterized by the expression of CD25+ FoxP3+ and low levels of CD127 [150]. The frequency of IL-10+ CD8+ T cells was increased in the periphery of MS patients and was not impacted by GA therapy [151, 152]. Although CD8+ Treg cells secreted IL- 10, their suppressive capacity in MS was thought to be carried out predominately through cell contact dependent mechanisms [150].
Although T cells have the ability to secrete low levels of IL-6, there are few reports on T cell derived IL-6 as the focus of most research has been on the ability of APC to produce sufficient IL-6 to drive Th17 differentiation. In one study researchers found that ex vivo stimulation of MS patients’ T cells with concanavalin A (ConA) or anti-CD3 produced less IL-6 than controls [153]. In contrast, more recent studies have found that CD4+ and CD8+ T cells from MS patients produced more IL-6 when stimulated in the presence of MBP and polyclonal stimulation [119]. Not only were the levels of T cell IL-6 elevated, but a separate study also found that the temporal control over T cell IL-6 production was altered in MS. MS CD4+ and CD8 T cells produced IL-6 at an accelerated rate compared to control T cells, after only a few hours of anti-CD3 stimulation, while T cells from healthy donors did not produce detectable IL-6 until after 24 h of stimulation [154].
9. Cytokines in the central nervous system
A multitude of studies have focused on serum, plasma or cerebrospinal fluid (CSF) cytokine levels to predict conversion to MS, relapses or as biomarkers for disease. Some studies have identified elevated IL-6 in the CSF and IL-10 in the serum of MS patients compared to controls; however these results have not been consistent [155–161]. Interestingly, both sIL-6R and sgp130 are significantly elevated in the serum of MS patients but sgp130 was diminished in the CSF of MS patients[162, 163]. Another study corroborated the results for elevated sIL-6R in only RRMS patients [164].
The CSF gives researchers a window into the CNS (central nervous system) compartment of MS patients but may not be representative of the site of demyelination lesions. While most studies focus on the potential impact of IL-6 and IL-10 on immune cells, resident CNS cells are also capable of secreting and responding to cytokines. In order to determine whether IL-6 and IL-10 are features of MS lesion pathology post mortem brain tissues can be stained by immunohistochemistry or interrogated to determine gene expression. Using qPCR IL-6 transcripts were identified as significantly elevated in MS lesions compared to non-MS brain tissue whereas IL-10 was not detected in either MS or control tissues [165]. However another immunofluorescence study found that IL-10 and the IL-10R were localized both to astrocytes and macrophages in MS lesions [165]. In a separate study IL-6 co-localized with GFAP+ astrocytes and in a few cases with CD68+ macrophages [166]. Interestingly, in another study the percentage of IL-6+ cells increased with the severity of demyelination, was primarily in astrocytes and macrophages but was more prevalent in inactive lesions and those lesions with less oligodendrocyte loss [167]. These observations collectively suggest that IL-6 and IL-10 are detectable, albeit by varying methods, are features of MS lesions and both resident CNS cells and immune cells are capable of producing IL-10 and IL-6 and sensing these cytokines.
10. Resistance to Cytokine Regulation
What is perhaps the most surprising with regard to IL-6 is that exposure to this cytokine appears to render MS T cells resistant to Treg/Tr1-mediated suppression. This effect might be compounded by the diminished ability for T cell, B cell, and innate immune cells to secrete sufficient IL-10 and further preclude naturally occurring regulatory cells from controlling encephalitogenic T cells. IL-6R in complex with gp130 and the IL-10R signal by activation of STAT3 through phosphorylation (p- STAT3) and homodimerization of the STAT3 transcription factor. The activation of STAT3 also induces a potent negative regulatory feedback mechanism resulting in the expression of suppressor of cytokine signaling 3 (SOCS3), which acts as a strong negative regulator of the IL-6 signaling pathway.
Evidence for IL-6 resistance to Treg cell-mediated suppression was first supported by studies demonstrating the elevated expression of the IL-6R on CD4+ T cells [163, 168], CD8+ T cells, B cells, and monocytes from MS patients compared to healthy donors [154]. It is also important to note that IL-10 receptor (IL-10R) levels on total PBMC were similar between healthy donors and MS patients [148]. The baseline level of pSTAT3 is elevated in monocytes, CD4+ and CD8+ T cells from MS patients or patients at high risk to convert to MS and was correlated with disease activity [169, 170]. However, studies have also detected elevated SOCS3 transcripts in PBMC from MS patients [168, 171].
To understand how IL-6 impacts T cell responses in MS patients several studies have isolated T effector cells and incubated them with Treg or Tr1 cells. In one such report, CD4+ T cells and PBMC from MS patients had elevated p-STAT3 after exposure to IL-6, but not IL-10, as compared to CD4+ T cells from healthy donors [168]. The level of pSTAT3 and IL-6R, but not gp130, correlated with the resistance to Treg mediated inhibition in cultures stimulated with anti-CD3 and anti- CD28 antibodies. This resistance to Treg-mediated suppression was reversed by an inhibitor of STAT3 and unaffected by trans-signaling mediated by soluble IL-6R in complex with IL-6 and gp130 [168]. Furthermore, MS patients who were resistant to dexamethasone, a corticosteroid, continued to produce IL-6 after stimulation with LPS in the presence of dexamethasone in vitro [172]. CD4+ and CD8+ T effector cells from MS patients continued to produce higher levels of IL-6 and proliferate when stimulated by anti-CD3 antibody in the presence of CD4+CD25+FoxP3+ Treg cells [154]. Resistance to Treg suppression was reversible upon the addition of a blocking antibody for the IL-6R or a PKB/c-Akt inhibitor that partially blocked the IL-6R signaling pathway [154]. Interestingly, the presence of IL- 6, which was provided by MS T effector cells in a transwell assay or exogenous IL-6, induced expression of the IL-6R in healthy donor T effector cells and subsequently conferred a transient resistance to inhibition by Treg cells. These studies support a role for IL-6 in STAT3-mediated resistance to Treg suppression.
There is also a defect in the ability of MS T cells to differentiate into Tr1 in vitro, and also to respond to IL-10 [148]. T effector cells from MS patients that were incubated with supernatants from Tr1 cultures or recombinant IL-10 were insensitive to IL-10-mediated inhibition as measured by proliferation. Further, CD4+ and CD8+ T cells failed to activate pSTAT3 to the level of healthy donor T cells after incubation with IL-10 [148]. Despite this reduced response to IL-10, IL-10R and STAT3 transcripts were significantly induced in PBMC after 24 hours of stimulation with IL-10. In vitro treatment of T cells with GA inhibited p-STAT3 and Th17 differentiation under Th17-polarizing conditions [117]. Interestingly, these studies were similar to IL-6-mediated resistance to IL-10 in RA patients [173]. Taken together these studies highlight the potential deficiencies in IL-6 function and lymphocyte responsiveness to the inhibitory effects of IL-10 in the presence of increased levels of IL-6.
11. Summary
Our recent studies suggest that the hyperactivity observed in MS peripheral lymphocyte responses could be partly the result of an imbalance in IL-6 and IL-10 regulation [10, 73]. Studies in mice have elucidated a role for IL-10 in the prevention of EAE and for IL-10-expressing regulatory cells in controlling established disease. By contrast, IL-6 is indispensible for the induction of EAE. The roles of these cytokines in ongoing disease are less well defined, but IL-6 appears to promote inflammation in EAE models. In MS, the focus has been on the prevention of clinical symptoms. Immune cells from MS patients exhibited defective IL-10 production and defective responses to Tr1 regulatory cells. IL-10 responsiveness could be inhibited by the over-production of IL-6. Enhanced IL-6R signaling appears to establish an effective blockade in suppression of leukocyte responses by IL-10 producing regulatory cells. Although GA and other immunomodulatory therapies, such as IFN-β, have been shown to promote IL-10 production and dampen IL-6 secretion, it is not known if modulation of cytokine production underlies the efficacy of these therapies in disease as shown in Figure 1. These observations underscore the pleiotropic nature and cross-regulation of cytokines and the likelihood that multiple mechanisms contribute to their involvement in MS disease pathogenesis. Future studies focused on the mechanisms, such as signaling pathways and transcriptional programs that regulate key cytokine pathways could substantially improve therapeutic options for patients with autoimmune diseases.
Figure 1. Model of IL-6 over-production in MS.
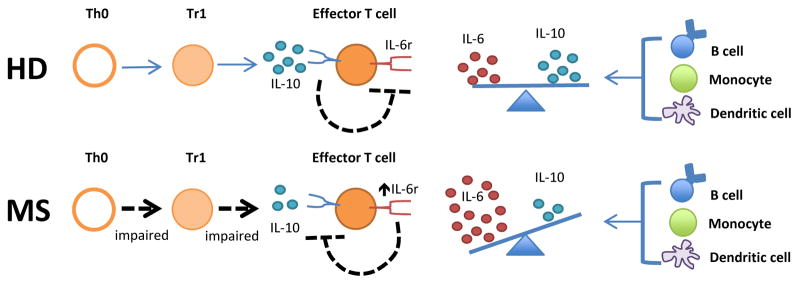
Increased IL-6 promotes inflammatory responses through a number of mechanisms. For example, increased IL-6 production in MS can result in T effector cell resistance to suppression by normal negative feedback signaling induced by IL-10. Increased IL-6 promotes inflammatory responses through a number of other mechanisms.
Highlights.
MS B cells secrete more IL-6 and less IL-10 than B cells from healthy donors
B cell depletion therapy, which ameliorates disease, diminishes IL-6 levels
GA therapy temporarily restores immune regulation, as detected by IL-6 levels
IL-10 secreted by T regulatory cells and monocytes contribute to immune regulation
Agents that enhance IL-10 and immunoregulation are of therapeutic benefit in MS
Acknowledgments
Dr. Ireland is supported by the National Institutes of Health (NIH), National Research Service Award 5 T32 AI005284-34 from the National Institute of Allergy and Infectious Disease (PI, Monson), Dr. Monson is supported by a grant from TEVA Neuroscience, MedImmune, the National MS Society (RG 4944) and Dr. Davis was supported by NIH grants R01DK081872 (PI, Mohan) and P50-AR055503 (PI, Mohan), a grant from the Alliance for Lupus Research and MedImmune/Amplimmune. TEVA Neuroscience, MedImmune, and other funding sources did not contribute to the preparation or approval of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobson R, Giovannoni G. Autoimmune disease in people with multiple sclerosis and their relatives: a systematic review and meta-analysis. J Neurol. 2013;260(5):1272–85. doi: 10.1007/s00415-012-6790-1. [DOI] [PubMed] [Google Scholar]
- 2.Somers EC, et al. Are individuals with an autoimmune disease at higher risk of a second autoimmune disorder? Am J Epidemiol. 2009;169(6):749–55. doi: 10.1093/aje/kwn408. [DOI] [PubMed] [Google Scholar]
- 3.Yao X, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125–39. doi: 10.1016/j.pharmthera.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Davis LS, Hutcheson J, Mohan C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31(10):781–9. doi: 10.1089/jir.2011.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol. 2014 doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian G, et al. Targeting IL-10 in auto-immune diseases. Cell Biochem Biophys. 2014;70(1):37–49. doi: 10.1007/s12013-014-9903-x. [DOI] [PubMed] [Google Scholar]
- 7.Nishimoto N, Kishimoto T, Yoshizaki K. Anti-interleukin 6 receptor antibody treatment in rheumatic disease. Ann Rheum Dis. 2000;59(Suppl 1):i21–7. doi: 10.1136/ard.59.suppl_1.i21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Shakarchi I, Gullick NJ, Scott DL. Current perspectives on tocilizumab for the treatment of rheumatoid arthritis: a review. Patient Prefer Adherence. 2013;7:653–66. doi: 10.2147/PPA.S41433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2(11):619–26. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- 10.Ireland SJ, et al. The Effect of Glatiramer Acetate Therapy on Functional Properties of B Cells From Patients With Relapsing-Remitting Multiple Sclerosis. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naka T, Nishimoto N, Kishimoto T. The paradigm of IL-6: from basic science to medicine. Arthritis Res. 2002;4(Suppl 3):S233–42. doi: 10.1186/ar565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16(3–4):249–84. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 13.Dayer JM, Choy E. Therapeutic targets in rheumatoid arthritis: the interleukin-6 receptor. Rheumatology (Oxford) 2010;49(1):15–24. doi: 10.1093/rheumatology/kep329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose-John S, et al. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80(2):227–36. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 15.Jura J, et al. Identification of interleukin-1 and interleukin-6-responsive genes in human monocyte-derived macrophages using microarrays. Biochim Biophys Acta. 2008;1779(6–7):383–9. doi: 10.1016/j.bbagrm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Tsantikos E, et al. Interleukin-6 trans-signaling exacerbates inflammation and renal pathology in lupus-prone mice. Arthritis Rheum. 2013;65(10):2691–702. doi: 10.1002/art.38061. [DOI] [PubMed] [Google Scholar]
- 17.Moore KW, et al. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 18.Hu X, et al. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol. 2007;82(2):237–43. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- 19.Lai CF, et al. Receptors for interleukin (IL)-10 and IL-6-type cytokines use similar signaling mechanisms for inducing transcription through IL-6 response elements. J Biol Chem. 1996;271(24):13968–75. doi: 10.1074/jbc.271.24.13968. [DOI] [PubMed] [Google Scholar]
- 20.Takagi H, et al. Regulation of cytokine and toll-like receptor signaling by SOCS family genes. Nihon Rinsho. 2004;62(12):2189–96. [PubMed] [Google Scholar]
- 21.Williams LM, et al. Interleukin-10 suppression of myeloid cell activation--a continuing puzzle. Immunology. 2004;113(3):281–92. doi: 10.1111/j.1365-2567.2004.01988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilgenberg E, et al. Interleukin-10-producing B cells and the regulation of immunity. Curr Top Microbiol Immunol. 2014;380:69–92. doi: 10.1007/978-3-662-43492-5_4. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann SR, et al. Biological properties and regulation of IL-10 related cytokines and their contribution to autoimmune disease and tissue injury. Clin Immunol. 2012;143(2):116–27. doi: 10.1016/j.clim.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Koch N, et al. IL-10 protects monocytes and macrophages from complement-mediated lysis. J Leukoc Biol. 2009;86(1):155–66. doi: 10.1189/jlb.0708443. [DOI] [PubMed] [Google Scholar]
- 25.Sabat R. IL-10 family of cytokines. Cytokine Growth Factor Rev. 2010;21(5):315–24. doi: 10.1016/j.cytogfr.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Sabat R, et al. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21(5):331–44. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Clatworthy MR. B-cell regulation and its application to transplantation. Transpl Int. 2014;27(2):117–28. doi: 10.1111/tri.12160. [DOI] [PubMed] [Google Scholar]
- 28.Itoh K, Hirohata S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J Immunol. 1995;154(9):4341–50. [PubMed] [Google Scholar]
- 29.Carpenter S, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341(6147):789–92. doi: 10.1126/science.1240925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carpenter S, Fitzgerald KA. Transcription of Inflammatory Genes: Long Noncoding RNA and Beyond. J Interferon Cytokine Res. 2014 doi: 10.1089/jir.2014.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerau-de-Arellano M, et al. miRNA profiling for biomarker discovery in multiple sclerosis: from microarray to deep sequencing. J Neuroimmunol. 2012;248(1–2):32–9. doi: 10.1016/j.jneuroim.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thamilarasan M, et al. MicroRNAs in multiple sclerosis and experimental autoimmune encephalomyelitis. Autoimmun Rev. 2012;11(3):174–9. doi: 10.1016/j.autrev.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Eisele S, et al. Prospects of transcript profiling for mRNAs and MicroRNAs using formalin-fixed and paraffin-embedded dissected autoptic multiple sclerosis lesions. Brain Pathol. 2012;22(5):607–18. doi: 10.1111/j.1750-3639.2012.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Connell RM, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33(4):607–19. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazaki Y, et al. A novel microRNA-132-surtuin-1 axis underlies aberrant B-cell cytokine regulation in patients with relapsing-remitting multiple sclerosis. PLoS One. 2014;9(8):e105421. doi: 10.1371/journal.pone.0105421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding S, et al. Decreased microRNA-142-3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. 2012;64(9):2953–63. doi: 10.1002/art.34505. [DOI] [PubMed] [Google Scholar]
- 37.Waschbisch A, et al. Glatiramer acetate treatment normalizes deregulated microRNA expression in relapsing remitting multiple sclerosis. PLoS One. 2011;6(9):e24604. doi: 10.1371/journal.pone.0024604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He Y, et al. MiR-146a regulates IL-6 production in lipopolysaccharide-induced RAW264.7 macrophage cells by inhibiting Notch1. Inflammation. 2014;37(1):71–82. doi: 10.1007/s10753-013-9713-0. [DOI] [PubMed] [Google Scholar]
- 39.Hecker M, et al. MicroRNA expression changes during interferon-beta treatment in the peripheral blood of multiple sclerosis patients. Int J Mol Sci. 2013;14(8):16087–110. doi: 10.3390/ijms140816087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meira M, et al. MiR-126: a novel route for natalizumab action? Mult Scler. 2014;20(10):1363–70. doi: 10.1177/1352458514524998. [DOI] [PubMed] [Google Scholar]
- 41.Field J, et al. The genetics of multiple sclerosis: an up-to-date review. Nat Genet. 2012;248(1):87–103. doi: 10.1111/j.1600-065X.2012.01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beecham AH, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. 2013;45(11):1353–60. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patsopoulos NA, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70(6):897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damotte V, et al. A gene pathway analysis highlights the role of cellular adhesion molecules in multiple sclerosis susceptibility. Genes Immun. 2014;15(2):126–32. doi: 10.1038/gene.2013.70. [DOI] [PubMed] [Google Scholar]
- 45.Ferreira RC, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013;9(4):e1003444. doi: 10.1371/journal.pgen.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jakkula E, et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet. 2010;86(2):285–91. doi: 10.1016/j.ajhg.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farh KK, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014 doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Millay DP, et al. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature. 2013;499(7458):301–5. doi: 10.1038/nature12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lassmann H. Multiple sclerosis: Lessons from molecular neuropathology. Exp Neurol. 2013 doi: 10.1016/j.expneurol.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Noyes K, Weinstock-Guttman B. Impact of diagnosis and early treatment on the course of multiple sclerosis. Am J Manag Care. 2013;19(17 Suppl):s321–31. [PubMed] [Google Scholar]
- 51.Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89(2):225–40. doi: 10.1016/j.mayocp.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 52.Ireland SJ, et al. Antibody-independent B cell effector functions in relapsing remitting multiple sclerosis: clues to increased inflammatory and reduced regulatory B cell capacity. Autoimmunity. 2012;45(5):400–14. doi: 10.3109/08916934.2012.665529. [DOI] [PubMed] [Google Scholar]
- 53.Romme Christensen J, et al. Cellular sources of dysregulated cytokines in relapsing-remitting multiple sclerosis. J Neuroinflammation. 2012;9:215. doi: 10.1186/1742-2094-9-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patanella AK, et al. Correlations between peripheral blood mononuclear cell production of BDNF, TNF-alpha, IL-6, IL-10 and cognitive performances in multiple sclerosis patients. J Neurosci Res. 2010;88(5):1106–12. doi: 10.1002/jnr.22276. [DOI] [PubMed] [Google Scholar]
- 55.Eugster HP, et al. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol. 1998;28(7):2178–87. doi: 10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 56.Okuda Y, et al. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int Immunol. 1998;10(5):703–8. doi: 10.1093/intimm/10.5.703. [DOI] [PubMed] [Google Scholar]
- 57.Serada S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105(26):9041–6. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gijbels K, et al. Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated levels of IL-6 bioactivity in central nervous system and circulation. Mol Med. 1995;1(7):795–805. [PMC free article] [PubMed] [Google Scholar]
- 59.Mendel I, et al. Interleukin-6 functions in autoimmune encephalomyelitis: a study in gene-targeted mice. Eur J Immunol. 1998;28(5):1727–37. doi: 10.1002/(SICI)1521-4141(199805)28:05<1727::AID-IMMU1727>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 60.Leech MD, et al. Cutting edge: IL-6-dependent autoimmune disease: dendritic cells as a sufficient, but transient, source. J Immunol. 2013;190(3):881–5. doi: 10.4049/jimmunol.1202925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Molnarfi N, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210(13):2921–37. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bettelli E, et al. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol. 1998;161(7):3299–306. [PubMed] [Google Scholar]
- 63.Samoilova EB, Horton JL, Chen Y. Acceleration of experimental autoimmune encephalomyelitis in interleukin-10-deficient mice: roles of interleukin-10 in disease progression and recovery. Cell Immunol. 1998;188(2):118–24. doi: 10.1006/cimm.1998.1365. [DOI] [PubMed] [Google Scholar]
- 64.Croxford JL, et al. Different therapeutic outcomes in experimental allergic encephalomyelitis dependent upon the mode of delivery of IL-10: a comparison of the effects of protein, adenoviral or retroviral IL-10 delivery into the central nervous system. J Immunol. 2001;166(6):4124–30. doi: 10.4049/jimmunol.166.6.4124. [DOI] [PubMed] [Google Scholar]
- 65.Cua DJ, et al. Central nervous system expression of IL-10 inhibits autoimmune encephalomyelitis. J Immunol. 2001;166(1):602–8. doi: 10.4049/jimmunol.166.1.602. [DOI] [PubMed] [Google Scholar]
- 66.Cannella B, et al. IL-10 fails to abrogate experimental autoimmune encephalomyelitis. J Neurosci Res. 1996;45(6):735–46. doi: 10.1002/(SICI)1097-4547(19960915)45:6<735::AID-JNR10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 67.Nagelkerken L, Blauw B, Tielemans M. IL-4 abrogates the inhibitory effect of IL-10 on the development of experimental allergic encephalomyelitis in SJL mice. Int Immunol. 1997;9(9):1243–51. doi: 10.1093/intimm/9.9.1243. [DOI] [PubMed] [Google Scholar]
- 68.Fillatreau S, et al. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–50. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 69.Matsushita T, et al. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185(4):2240–52. doi: 10.4049/jimmunol.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsushita T, et al. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118(10):3420–30. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang X, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16(2):249–56. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 72.Ray A, et al. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188(7):3188–98. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yoshizaki A, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature. 2012;491(7423):264–8. doi: 10.1038/nature11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen P, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–70. doi: 10.1038/nature12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raphael I, et al. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2014 doi: 10.1016/j.cyto.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta. 2011;1812(2):246–51. doi: 10.1016/j.bbadis.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshida Y, et al. The transcription factor IRF8 activates integrin-mediated TGF-beta signaling and promotes neuroinflammation. Immunity. 2014;40(2):187–98. doi: 10.1016/j.immuni.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aharoni R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun Rev. 2013;12(5):543–53. doi: 10.1016/j.autrev.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 79.Fridkis-Hareli M, et al. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells--specificity and promiscuity. Proc Natl Acad Sci U S A. 1994;91(11):4872–6. doi: 10.1073/pnas.91.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fridkis-Hareli M, et al. Synthetic copolymer 1 and myelin basic protein do not require processing prior to binding to class II major histocompatibility complex molecules on living antigen-presenting cells. Cell Immunol. 1995;163(2):229–36. doi: 10.1006/cimm.1995.1121. [DOI] [PubMed] [Google Scholar]
- 81.Aharoni R, et al. Copolymer 1 acts against the immunodominant epitope 82–100 of myelin basic protein by T cell receptor antagonism in addition to major histocompatibility complex blocking. Proc Natl Acad Sci U S A. 1999;96(2):634–9. doi: 10.1073/pnas.96.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Racke MK, et al. Copolymer-1-induced inhibition of antigen-specific T cell activation: interference with antigen presentation. J Neuroimmunol. 1992;37(1–2):75–84. doi: 10.1016/0165-5728(92)90157-g. [DOI] [PubMed] [Google Scholar]
- 83.Aharoni R, et al. Copolymer 1 induces T cells of the T helper type 2 that crossreact with myelin basic protein and suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 1997;94(20):10821–6. doi: 10.1073/pnas.94.20.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Duda PW, et al. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105(7):967–76. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Putheti P, et al. Effect of glatiramer acetate (Copaxone) on CD4+CD25high T regulatory cells and their IL-10 production in multiple sclerosis. J Neuroimmunol. 2003;144(1–2):125–31. doi: 10.1016/j.jneuroim.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Dabbert D, et al. Glatiramer acetate (copolymer-1)-specific, human T cell lines: cytokine profile and suppression of T cell lines reactive against myelin basic protein. Neurosci Lett. 2000;289(3):205–8. doi: 10.1016/s0304-3940(00)01289-1. [DOI] [PubMed] [Google Scholar]
- 87.Diehl S, et al. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 2000;13(6):805–15. doi: 10.1016/s1074-7613(00)00078-9. [DOI] [PubMed] [Google Scholar]
- 88.Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002;39(9):531–6. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 89.Begum-Haque S, et al. Glatiramer acetate biases dendritic cells towards an anti-inflammatory phenotype by modulating OPN, IL-17, and RORgammat responses and by increasing IL-10 production in experimental allergic encephalomyelitis. J Neuroimmunol. 2013;254(1–2):117–24. doi: 10.1016/j.jneuroim.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 90.Weber MS, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. 2007;13(8):935–43. doi: 10.1038/nm1620. [DOI] [PubMed] [Google Scholar]
- 91.Begum-Haque S, et al. Downregulation of IL-17 and IL-6 in the central nervous system by glatiramer acetate in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;204(1–2):58–65. doi: 10.1016/j.jneuroim.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 92.Kala M, et al. B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp Neurol. 2010;221(1):136–45. doi: 10.1016/j.expneurol.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 93.Begum-Haque S, et al. Augmentation of regulatory B cell activity in experimental allergic encephalomyelitis by glatiramer acetate. J Neuroimmunol. 2011;232(1–2):136–44. doi: 10.1016/j.jneuroim.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Begum-Haque S, et al. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;219(1–2):47–53. doi: 10.1016/j.jneuroim.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 95.Mirowska D, et al. Changes of percentages in immune cells phenotypes and cytokines production during two-year IFN-beta-1a treatment in multiple sclerosis patients. J Neurol. 2003;250(10):1229–36. doi: 10.1007/s00415-003-0170-9. [DOI] [PubMed] [Google Scholar]
- 96.Kvarnstrom M, et al. Longitudinal interferon-beta effects in multiple sclerosis: differential regulation of IL-10 and IL-17A, while no sustained effects on IFN-gamma, IL-4 or IL-13. J Neurol Sci. 2013;325(1–2):79–85. doi: 10.1016/j.jns.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 97.Ramgolam VS, et al. B cells as a therapeutic target for IFN-beta in relapsing-remitting multiple sclerosis. J Immunol. 2011;186(7):4518–26. doi: 10.4049/jimmunol.1000271. [DOI] [PubMed] [Google Scholar]
- 98.Chiarini M, et al. Modulation of the central memory and Tr1-like regulatory T cells in multiple sclerosis patients responsive to interferon-beta therapy. Mult Scler. 2012;18(6):788–98. doi: 10.1177/1352458511427720. [DOI] [PubMed] [Google Scholar]
- 99.Liu BS, Janssen HL, Boonstra A. Type I and III interferons enhance IL-10R expression on human monocytes and macrophages, resulting in IL-10-mediated suppression of TLR-induced IL-12. Eur J Immunol. 2012;42(9):2431–40. doi: 10.1002/eji.201142360. [DOI] [PubMed] [Google Scholar]
- 100.Miyazaki Y, et al. Suppressed pro-inflammatory properties of circulating B cells in patients with multiple sclerosis treated with fingolimod, based on altered proportions of B-cell subpopulations. Clin Immunol. 2014;151(2):127–35. doi: 10.1016/j.clim.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 101.Muller H, et al. The immunomodulator FTY720 interferes with effector functions of human monocyte-derived dendritic cells. Eur J Immunol. 2005;35(2):533–45. doi: 10.1002/eji.200425556. [DOI] [PubMed] [Google Scholar]
- 102.Li L, et al. The effects of teriflunomide on lymphocyte subpopulations in human peripheral blood mononuclear cells in vitro. J Neuroimmunol. 2013;265(1–2):82–90. doi: 10.1016/j.jneuroim.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 103.Fidler JM, DeJoy SQ, Gibbons JJ., Jr Selective immunomodulation by the antineoplastic agent mitoxantrone. I. Suppression of B lymphocyte function. J Immunol. 1986;137(2):727–32. [PubMed] [Google Scholar]
- 104.Fidler JM, et al. Selective immunomodulation by the antineoplastic agent mitoxantrone. II. Nonspecific adherent suppressor cells derived from mitoxantrone-treated mice. J Immunol. 1986;136(8):2747–54. [PubMed] [Google Scholar]
- 105.Gbadamosi J, et al. Effects of mitoxantrone on multiple sclerosis patients’ lymphocyte subpopulations and production of immunoglobulin, TNF-alpha and IL-10. Eur Neurol. 2003;49(3):137–41. doi: 10.1159/000069082. [DOI] [PubMed] [Google Scholar]
- 106.Vogelgesang A, et al. Mitoxantrone treatment in multiple sclerosis induces TH2-type cytokines. Acta Neurol Scand. 2010;122(4):237–43. doi: 10.1111/j.1600-0404.2009.01295.x. [DOI] [PubMed] [Google Scholar]
- 107.Ghoreschi K, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208(11):2291–303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Merad M, et al. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hashimoto D, Miller J, Merad M. Dendritic cell and macrophage heterogeneity in vivo. Immunity. 2011;35(3):323–35. doi: 10.1016/j.immuni.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kawakami T, et al. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J Immunol. 2013;191(6):3358–72. doi: 10.4049/jimmunol.1300342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cohen HB, Mosser DM. Extrinsic and intrinsic control of macrophage inflammatory responses. J Leukoc Biol. 2013;94(5):913–9. doi: 10.1189/jlb.0413236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giacomini E, et al. IFN-beta therapy modulates B-cell and monocyte crosstalk via TLR7 in multiple sclerosis patients. Eur J Immunol. 2013;43(7):1963–72. doi: 10.1002/eji.201243212. [DOI] [PubMed] [Google Scholar]
- 113.Chuluundorj D, et al. Expansion and preferential activation of the CD14CD16 monocyte subset during multiple sclerosis. Immunol Cell Biol. 2014 doi: 10.1038/icb.2014.15. [DOI] [PubMed] [Google Scholar]
- 114.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81(3):584–92. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 115.Weber MS, et al. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain. 2004;127(Pt 6):1370–8. doi: 10.1093/brain/awh163. [DOI] [PubMed] [Google Scholar]
- 116.Ayers CL, et al. Modulation of immune function occurs within hours of therapy initiation for multiple sclerosis. Clin Immunol. 2013;147(2):105–19. doi: 10.1016/j.clim.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen C, et al. Regulatory properties of copolymer I in Th17 differentiation by altering STAT3 phosphorylation. J Immunol. 2009;183(1):246–53. doi: 10.4049/jimmunol.0900193. [DOI] [PubMed] [Google Scholar]
- 118.Filion LG, et al. Monocyte-derived cytokines in multiple sclerosis. Clin Exp Immunol. 2003;131(2):324–34. doi: 10.1046/j.1365-2249.2003.02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clerici M, et al. Single-cell analysis of cytokine production shows different immune profiles in multiple sclerosis patients with active or quiescent disease. J Neuroimmunol. 2001;121(1–2):88–101. doi: 10.1016/s0165-5728(01)00431-3. [DOI] [PubMed] [Google Scholar]
- 120.Kim HJ, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol. 2004;172(11):7144–53. doi: 10.4049/jimmunol.172.11.7144. [DOI] [PubMed] [Google Scholar]
- 121.Huang YM, et al. Multiple sclerosis is associated with high levels of circulating dendritic cells secreting pro-inflammatory cytokines. J Neuroimmunol. 1999;99(1):82–90. doi: 10.1016/s0165-5728(99)00106-x. [DOI] [PubMed] [Google Scholar]
- 122.Sanna A, et al. Multiple sclerosis: reduced proportion of circulating plasmacytoid dendritic cells expressing BDCA-2 and BDCA-4 and reduced production of IL-6 and IL-10 in response to herpes simplex virus type 1. Mult Scler. 2008;14(9):1199–207. doi: 10.1177/1352458508094401. [DOI] [PubMed] [Google Scholar]
- 123.Chiurchiu V, et al. Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann Neurol. 2013;73(5):626–36. doi: 10.1002/ana.23875. [DOI] [PubMed] [Google Scholar]
- 124.Vieira PL, et al. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol. 2003;170(9):4483–8. doi: 10.4049/jimmunol.170.9.4483. [DOI] [PubMed] [Google Scholar]
- 125.Sanna A, et al. Glatiramer acetate reduces lymphocyte proliferation and enhances IL-5 and IL-13 production through modulation of monocyte-derived dendritic cells in multiple sclerosis. Clin Exp Immunol. 2006;143(2):357–62. doi: 10.1111/j.1365-2249.2006.02997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vroman H, van den Blink B, Kool M. Mode of dendritic cell activation: The decisive hand in Th2/Th17 cell differentiation. Implications in asthma severity? Immunobiology. 2014 doi: 10.1016/j.imbio.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 127.Shinohara H, et al. Positive feedback within a kinase signaling complex functions as a switch mechanism for NF-kappaB activation. Science. 2014;344(6185):760–4. doi: 10.1126/science.1250020. [DOI] [PubMed] [Google Scholar]
- 128.Agrawal S, Gupta S. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J Clin Immunol. 2011;31(1):89–98. doi: 10.1007/s10875-010-9456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Baccam M, et al. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-kappa B, AP-1, and C/EBP. J Immunol. 2003;170(6):3099–108. doi: 10.4049/jimmunol.170.6.3099. [DOI] [PubMed] [Google Scholar]
- 130.Iwata Y, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011;117(2):530–41. doi: 10.1182/blood-2010-07-294249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.van de Veen W, et al. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J Allergy Clin Immunol. 2013;131(4):1204–12. doi: 10.1016/j.jaci.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 132.Quan C, et al. Impaired regulatory function and enhanced intrathecal activation of B cells in neuromyelitis optica: distinct from multiple sclerosis. Mult Scler. 2013;19(3):289–98. doi: 10.1177/1352458512454771. [DOI] [PubMed] [Google Scholar]
- 133.Flores-Borja F, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. 2013;5(173):173ra23. doi: 10.1126/scitranslmed.3005407. [DOI] [PubMed] [Google Scholar]
- 134.Lin W, et al. Human Regulatory B Cells Combine Phenotypic and Genetic Hallmarks with a Distinct Differentiation Fate. J Immunol. 2014 doi: 10.4049/jimmunol.1303214. [DOI] [PubMed] [Google Scholar]
- 135.Bouaziz JD, et al. IL-10 produced by activated human B cells regulates CD4(+) T-cell activation in vitro. Eur J Immunol. 2010;40(10):2686–91. doi: 10.1002/eji.201040673. [DOI] [PubMed] [Google Scholar]
- 136.Bar-Or A, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67(4):452–61. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 137.Maseda D, et al. Regulatory B10 cells differentiate into antibody-secreting cells after transient IL-10 production in vivo. J Immunol. 2012;188(3):1036–48. doi: 10.4049/jimmunol.1102500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barr TA, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209(5):1001–10. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reinherz EL, et al. Loss of suppressor T cells in active multiple sclerosis. Analysis with monoclonal antibodies. N Engl J Med. 1980;303(3):125–9. doi: 10.1056/NEJM198007173030303. [DOI] [PubMed] [Google Scholar]
- 140.Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev. 2014;259(1):231–44. doi: 10.1111/imr.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gregori S, Goudy KS, Roncarolo MG. The cellular and molecular mechanisms of immuno-suppression by human type 1 regulatory T cells. Front Immunol. 2012;3:30. doi: 10.3389/fimmu.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Roncarolo MG, et al. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 143.Roncarolo MG, et al. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. doi: 10.1034/j.1600-065x.2001.1820105.x. [DOI] [PubMed] [Google Scholar]
- 144.Feger U, et al. Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol. 2007;147(3):412–8. doi: 10.1111/j.1365-2249.2006.03271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Haas J, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35(11):3343–52. doi: 10.1002/eji.200526065. [DOI] [PubMed] [Google Scholar]
- 146.Viglietta V, et al. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Astier AL, et al. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest. 2006;116(12):3252–7. doi: 10.1172/JCI29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Martinez-Forero I, et al. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur J Immunol. 2008;38(2):576–86. doi: 10.1002/eji.200737271. [DOI] [PubMed] [Google Scholar]
- 149.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med. 2002;195(6):695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. 2010;67(5):625–38. doi: 10.1002/ana.21944. [DOI] [PubMed] [Google Scholar]
- 151.Peelen E, et al. Fraction of IL-10+ and IL-17+ CD8 T cells is increased in MS patients in remission and during a relapse, but is not influenced by immune modulators. J Neuroimmunol. 2013;258(1–2):77–84. doi: 10.1016/j.jneuroim.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 152.Tennakoon DK, et al. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol. 2006;176(11):7119–29. doi: 10.4049/jimmunol.176.11.7119. [DOI] [PubMed] [Google Scholar]
- 153.Brod SA, et al. Increased in vitro induced CD4+ and CD8+ T cell IFN-gamma and CD4+ T cell IL-10 production in stable relapsing multiple sclerosis. Int J Neurosci. 1997;90(3–4):187–202. doi: 10.3109/00207459709000638. [DOI] [PubMed] [Google Scholar]
- 154.Trinschek B, et al. Kinetics of IL-6 production defines T effector cell responsiveness to regulatory T cells in multiple sclerosis. PLoS One. 2013;8(10):e77634. doi: 10.1371/journal.pone.0077634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vladic A, et al. Cerebrospinal fluid and serum protein levels of tumour necrosis factor-alpha (TNF-alpha) interleukin-6 (IL-6) and soluble interleukin-6 receptor (sIL-6R gp80) in multiple sclerosis patients. Cytokine. 2002;20(2):86–9. doi: 10.1006/cyto.2002.1984. [DOI] [PubMed] [Google Scholar]
- 156.Malmestrom C, et al. IL-6 and CCL2 levels in CSF are associated with the clinical course of MS: implications for their possible immunopathogenic roles. J Neuroimmunol. 2006;175(1–2):176–82. doi: 10.1016/j.jneuroim.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 157.Martins TB, et al. Analysis of proinflammatory and anti-inflammatory cytokine serum concentrations in patients with multiple sclerosis by using a multiplexed immunoassay. Am J Clin Pathol. 2011;136(5):696–704. doi: 10.1309/AJCP7UBK8IBVMVNR. [DOI] [PubMed] [Google Scholar]
- 158.Miljkovic D, et al. Nitric oxide metabolites and interleukin-6 in cerebrospinal fluid from multiple sclerosis patients. Eur J Neurol. 2002;9(4):413–8. doi: 10.1046/j.1468-1331.2002.00437.x. [DOI] [PubMed] [Google Scholar]
- 159.Uzawa A, et al. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol. 2009;256(12):2082–4. doi: 10.1007/s00415-009-5274-4. [DOI] [PubMed] [Google Scholar]
- 160.Matejcikova Z, et al. Cerebrospinal fluid inflammatory markers in patients with multiple sclerosis: a pilot study. J Neural Transm. 2014 doi: 10.1007/s00702-014-1244-9. [DOI] [PubMed] [Google Scholar]
- 161.Kleine TO, et al. Approach to discriminate subgroups in multiple sclerosis with cerebrospinal fluid (CSF) basic inflammation indices and TNF-alpha, IL-1beta, IL-6, IL-8. Brain Res Bull. 2003;61(3):327–46. doi: 10.1016/s0361-9230(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 162.Padberg F, et al. No association between anti-myelin oligodendrocyte glycoprotein antibodies and serum/cerebrospinal fluid levels of the soluble interleukin-6 receptor complex in multiple sclerosis. Neurosci Lett. 2001;305(1):13–6. doi: 10.1016/s0304-3940(01)01792-x. [DOI] [PubMed] [Google Scholar]
- 163.Padberg F, et al. CSF and serum levels of soluble interleukin-6 receptors (sIL-6R and sgp130), but not of interleukin-6 are altered in multiple sclerosis. J Neuroimmunol. 1999;99(2):218–23. doi: 10.1016/s0165-5728(99)00120-4. [DOI] [PubMed] [Google Scholar]
- 164.Michalopoulou M, et al. Soluble interleukin-6 receptor (sIL-6R) in cerebrospinal fluid of patients with inflammatory and non inflammatory neurological diseases. Immunol Lett. 2004;94(3):183–9. doi: 10.1016/j.imlet.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 165.Baranzini SE, et al. Transcriptional analysis of multiple sclerosis brain lesions reveals a complex pattern of cytokine expression. J Immunol. 2000;165(11):6576–82. doi: 10.4049/jimmunol.165.11.6576. [DOI] [PubMed] [Google Scholar]
- 166.Maimone D, Guazzi GC, Annunziata P. IL-6 detection in multiple sclerosis brain. J Neurol Sci. 1997;146(1):59–65. doi: 10.1016/s0022-510x(96)00283-3. [DOI] [PubMed] [Google Scholar]
- 167.Schonrock LM, Gawlowski G, Bruck W. Interleukin-6 expression in human multiple sclerosis lesions. Neurosci Lett. 2000;294(1):45–8. doi: 10.1016/s0304-3940(00)01543-3. [DOI] [PubMed] [Google Scholar]
- 168.Schneider A, et al. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med. 2013;5(170):170ra15. doi: 10.1126/scitranslmed.3004970. [DOI] [PubMed] [Google Scholar]
- 169.Frisullo G, et al. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J Neurosci Res. 2006;84(5):1027–36. doi: 10.1002/jnr.20995. [DOI] [PubMed] [Google Scholar]
- 170.Frisullo G, et al. The persistency of high levels of pSTAT3 expression in circulating CD4+ T cells from CIS patients favors the early conversion to clinically defined multiple sclerosis. J Neuroimmunol. 2008;205(1–2):126–34. doi: 10.1016/j.jneuroim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 171.Sedeno-Monge V, et al. Quantitative analysis of the suppressors of cytokine signaling 1 and 3 in peripheral blood leukocytes of patients with multiple sclerosis. J Neuroimmunol. 2014 doi: 10.1016/j.jneuroim.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 172.DeRijk RH, Eskandari F, Sternberg EM. Corticosteroid resistance in a subpopulation of multiple sclerosis patients as measured by ex vivo dexamethasone inhibition of LPS induced IL-6 production. J Neuroimmunol. 2004;151(1–2):180–8. doi: 10.1016/j.jneuroim.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 173.Yamana J, et al. Resistance to IL-10 inhibition of interferon gamma production and expression of suppressor of cytokine signaling 1 in CD4+ T cells from patients with rheumatoid arthritis. Arthritis Res Ther. 2004;6(6):R567–77. doi: 10.1186/ar1445. [DOI] [PMC free article] [PubMed] [Google Scholar]