

Abstract
Synthesizing, localizing, and stabilizing new protein copies at synapses are crucial factors in maintaining the synaptic changes required for storing long-term memories. PKMζ recently emerged as a molecule putatively responsible for maintaining encoded memories over time because its presence correlates with late LTP and because its inhibition disrupts LTP in vitro and long-term memory storage in vivo. Here we investigated PKMζ stability in rat neurons to better understand its role during information encoding and storage. We used TimeSTAMP reporters to track the synthesis and degradation of PKMζ as well as a related atypical PKC, PKCλ. These reporters revealed that both PKMζ and PKCλ were upregulated after chemical LTP induction; however, these new PKMζ copies exhibited more rapid turnover than basally produced PKMζ, particularly in dendritic spines. In contrast to PKMζ, new PKCλ copies exhibited elevated stability. Stable information storage over long periods of time is more challenging the shorter the metabolic lifetime of the candidate molecules.
Keywords: long-term potentiation, memory, protein kinases, protein synthesis, protein turnover, synaptic plasticity
Introduction
Human memory can persist for a lifetime, yet the majority of synaptic proteins in the brain turn over on the scale of hours to days (Cohen et al., 2013). The mechanisms by which long-lasting memories are retained in the face of the dynamic synaptic environment still remain unknown. New protein synthesis is crucial for both long-term potentiation (LTP) and long-term memory (LTM) (Martin et al., 1997; Abraham and Williams, 2008). PKMζ, recently proposed as the primary molecule sustaining LTP and memory, is synthesized from a unique mRNA derived from an internal promoter of the Prkcz gene. PKMζ mRNA is continually transcribed and transported to dendritic spines; however, PKMζ protein is upregulated only during late LTP (Muslimov et al., 2004; Sacktor, 2011). PKMζ lacks the regulatory domain present in full-length PKCζ and is constitutively active once phosphorylated (Sacktor, 2011). Inhibiting PKMζ with the ζ-inhibitory peptide (ZIP), a myristoylated pseudosubstrate peptide derived from the autoinhibitory region of PKCζ, disrupts both LTP in vitro and LTM in vivo (Ling et al., 2002; Pastalkova et al., 2006). Several studies demonstrated that PKMζ functions by associating with the proteins NSF and PICK1, ultimately releasing synaptic pools of GluR2-containing AMPA receptors to the postsynaptic membrane (Yao et al., 2008; Migues et al., 2010; Sacktor, 2011). However, recent reports questioned the role of PKMζ in LTP and memory by demonstrating that ZIP is not entirely specific for PKMζ in vivo (Wu-Zhang et al., 2012) and that mice lacking PKMζ have normal LTP and memory (Lee et al., 2013; Volk et al., 2013). Although PKCζ mRNA is present at a low level in the adult rat brain, full-length PKCζ protein is almost completely absent (Hernández et al., 2003). PKCλ is a related atypical PKC expressed in brain tissue that contains a catalytic domain similar to PKMζ and the same pseudosubstrate as PKCζ; its activity is also blocked by ZIP (Ren et al., 2013). PKMζ and PKCλ are known to have different functions during neuronal development but have similar substrates and may act redundantly or compensate for each other during LTP maintenance (Parker et al., 2013; Ren et al., 2013).
To give insight into whether each protein could independently sustain long-lasting changes in synaptic strength, we developed a modified TimeSTAMP (TS) reporter to track PKMζ and PKCλ synthesis and stability in response to chemical LTP (cLTP) stimulation. TS reporters were originally designed to characterize the production and lifetime of a specific protein of interest by detection of an epitope tag retained by the protein in a drug-dependent manner (Lin et al., 2008). New versions incorporate the split yellow fluorescent protein (YFP) Venus and the mini singlet oxygen generator (miniSOG) tag for live fluorescence and electron microscopy (EM) readouts and increased temporal and spatial resolution (Butko et al., 2012).
We modified the existing TS:YSOG2 reporter to produce TS:YSOG3, a reporter that can identify both new and old copies of a protein fused to TS via its N terminus. We then used TS:YSOG3 to visualize PKMζ and PKCλ synthesis and degradation in cultured neurons following cLTP to characterize the stability of labeled proteins over time. Whereas both PKMζ and PKCλ are rapidly synthesized and localized to synapses after stimulation, here we demonstrate that only new PKCλ copies remain stable at these synapses during longer time scales of over a day.
Materials and Methods
Reagents.
A plasmid encoding rat PKMζ cDNA was obtained from Alexandra Newton's laboratory (University of California, San Diego). Rat PKMζ untranslated regions (UTRs) and PKCλ cDNA were cloned directly from rat neuron genomic DNA and rat neuron cDNA, respectively. All constructs were inserted into the pCAGGS mammalian expression plasmid, which drives expression via a CMV enhancer fused to a chicken β-actin promoter, and were generated using standard molecular biology techniques: PCR, restriction enzyme digestion, and ligation. The TS:YSOG3-PKMζ version of TS was generated by rearrangement of the original TS:YSOG2 tag followed by testing and determination of optimal modifications. The control 5′UTR sequence derived from the pCAGGS vector is 5′-CTCCAGCCTCGGGGCTGTCCGCGGGGGGACGGCTGCCTTCGGGGGGGACGGGGCAGGGCGGGGTTCGGCTTCTGGCGTGTGACCGGCGGCTCTAGCTAGAGCCTCTGCTAACCATGTTCATGCCTTCTTCTTTTTCCTACAGCTCCTGGGCAACGTGCTGGTTATTGTGCTGTCTCATCATTTTGGCAAAGAATTC-3′ and the control 3′-UTR sequence is 5′ GCGGCCGCTCGA 3′ followed by the rabbit β-globulin poly(A) terminator present in the pCAGGS vector. All subcloned fragments were fully sequenced to confirm successful construction. Forskolin, rolipram, and glycine were used for stimulation (50, 0.1, and 200 μm, respectively; Sigma-Aldrich). Cycloheximide was used at 50 μg/ml to inhibit protein synthesis (Millipore). BILN-2061 was used at 1 μm to inhibit the HCV NS3 protease (ACME Synthetic Chemical). MG132 was used at 5 μm to inhibit proteasome-mediated protein degradation (Millipore). Primary antibodies used were mouse monoclonal anti-PSD95 (Pierce), mouse monoclonal anti-T7 (Millipore), mouse monoclonal anti-FLAG (Invitrogen), rabbit polyclonal anti-PKCζ (Sigma-Aldrich), and rabbit polyclonal anti-miniSOG (custom order from QED Biosciences). For immunoblotting, primary antibodies were used at 0.1–0.4 μg/ml. HRP-conjugated mouse secondary antibody (Cell Signaling Technology) and HRP-conjugated rabbit secondary antibody (Bio-Rad) were used at 0.4 μg/ml. For immunofluorescence, primary antibodies were used at 0.5–1 μg/ml. AlexaFluor 568-conjugated mouse and AlexaFluor 647-conjugated goat secondary antibodies (Invitrogen) were used at 0.5 μg/ml.
Cell culture.
All cell culture reagents were obtained from Invitrogen unless otherwise indicated. HEK-293A cells were obtained (ATCC) and cultured in DMEM supplemented with 10% FBS, 2 mm GlutaMAX, 20 U/ml penicillin, and 50 μg/ml streptomycin. HEK-293A cells were transfected with Lipofectamine 3000. Cortical neurons were dissociated by papain from postnatal day 2 (P2) Sprague Dawley rats of either sex, transfected by Amaxa electroporation (Lonza), then plated on poly-d-lysine-coated MatTek glass-bottom dishes (MatTek Corporation) and cultured in Neurobasal A medium with B27 supplement, 2 mm GlutaMAX, 20 U/ml penicillin, and 50 μg/ml streptomycin as previously described (Butko et al., 2012). All neuron experiments were performed in cultured neurons matured to between 12 and 16 DIV. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Chemical LTP.
Neurons were stimulated as previously described (Kim et al., 2007; Tampellini et al., 2009). Briefly, cultured neurons were incubated for 20 min at room temperature in ACSF (in mm) as follows: 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 22 d-glucose, and 25 HEPES, pH 7.3, then incubated in ACSF lacking MgCl2 and containing 50 μm forskolin and 0.1 μm rolipram for 10 min. Neurons were rinsed in ACSF, then replaced in their original culture medium to which 1 μm BILN had been added. For glycine-dependent chemical LTP, neurons were treated in the same manner but incubated with 200 μm glycine for 15 min instead of forskolin and rolipram.
Immunoblotting.
For analysis of PKMζ synthesis and degradation in neurons by immunoblotting, transfected neurons were plated and maintained in 6-well cell culture plates. After treatment by the desired pharmacological agents, cells were rinsed quickly with ACSF (in mm) as follows: 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 22 d-glucose, and 25 HEPES, pH 7.3, and resuspended in lysis buffer (20 mm Tris-HCl, 1% Triton detergent, 10% glycerol, 2 mm EDTA, 137 mm NaCl, and Roche EDTA-free protease inhibitor, pH 7.4). Cells were homogenized and protein content assessed by Bradford assay. SDS loading buffer was added to lysate; then immunoblotting was performed. Briefly, lysates were run on NuPage 4%–12% Novex Bis-Tris SDS polyacrylamide gels (Invitrogen). Proteins were transferred onto PVDF membranes (Invitrogen) by electroblotting, and membranes were then blocked with 10% nonfat dried milk in Tris-buffered saline with 0.1% Tween 20 (TBST), incubated in primary antibody overnight in 5% BSA in TBST at 4°C, washed in TBST to remove excess primary antibody, incubated in HRP-conjugated secondary antibody in TBST for 1 h at room temperature, and finally rinsed 3 times for 15 min each in TBST. Proteins were visualized by chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and film (Blue Devil, Genesee Scientific). Films were scanned and band densities were quantified using the National Institutes of Health ImageJ program. Band densities were normalized to the GAPDH loading control band in each lane (see Fig. 2E,H) and plotted as the fraction of protein remaining relative to time 0 h (see Fig. 2F, I).
Figure 2.

YFP imaging and TS fusion protein expression do not affect characterization of PKMζ dynamics in neurons. A, Neurons expressing PKMζ-YFP imaged 10 times show no fluorescence bleaching after image acquisition. B, Fluorescence quantification in dendritic spines over the 10 h imaging period indicates that no fluorescence loss occurred. Error bars indicate SEM. C, Neurons were transfected with PKMζ-TS:YSOG2, TS:YSOG3-PKMζ, or left untransfected (Untrans) and immunoblotted for PKMζ to compare levels of endogenous and TS-driven PKMζ production. D, Schematic indicating how transfected neurons were treated with BILN for 24 h and then with 50 μg/ml cycloheximide without removing BILN. This blocked new protein synthesis while preventing protease reactivation. Neurons were lysed at 0, 4, 8, and 12 h after cycloheximide addition. E, Western blots tracking PKMζ-TS:YSOG2 and TS:YSOG3-PKMζ using an antibody specific for miniSOG and PKMζ fused only to the T7 epitope tag using an antibody specific for the T7 epitope, performed as described in D; blots demonstrate that each protein degrades similarly. GAPDH was used as a loading control. F, Quantitative analysis of Western blots in E; band intensities were normalized to GAPDH for each lane and plotted as the fraction of protein remaining over time. G, FLAG-TS:YSOG3-PKMζ fusion protein after BILN addition for labeling, after subsequent BILN washout, and after exposure to denaturing conditions. H, Western blots using antibodies specific for miniSOG and the FLAG epitope tag performed on HEK-293A cells expressing FLAG-TS:YSOG3-PKMζ, lysed at 0, 5, and 10 h after BILN washout and simultaneous MG132 addition to visualize all TS peptides. I, Quantitative analysis of Western blots in H; band intensities were normalized to GAPDH for each lane and plotted as the fraction of protein remaining over time.
Immunofluorescence.
To determine that fusion to TS did not affect PKMζ synaptic localization, cortical neurons were dissociated, electroporated with the plasmid of interest, and plated on MatTek dishes coated with poly-d-lysine. BILN was added to the media 24 h before fixation. Neurons were fixed by addition of one culture volume of 4% PFA and 10% sucrose in PBS for 10 min at room temperature, then permeabilized with 0.1% Triton in PBS, blocked with 5% nonimmune goat serum in PBS with 0.45% BSA, and probed for PSD95 according to standard procedures. Specificity of secondary antibodies was confirmed in control samples without primary antibody. Images were obtained on an Olympus FluoView 1000 Confocal Laser Scanning Microscope using a 40× oil-immersion lens. A stack of optical sections with 1024 × 1024 resolution at 1 μm intervals through each neuron was obtained and then flattened into a maximum intensity projection. Analysis was performed on an Apple Macintosh notebook using the National Institutes of Health ImageJ Program. Immunofluorescence images of related conditions were matched for laser power, gain, and offset.
Time-lapse microscopy.
To follow fluorescence development of TS:YSOG2 and TS:YSOG3 tags in transfected neurons, cells were cultured on MatTek dishes in Neurobasal medium as previously described (Butko et al., 2012). BILN was added to neurons that had been stimulated or left unstimulated. After 24 h of BILN incubation, neurons were washed three times with warmed conditioned media and replaced in HBSS supplemented with B27, 25 mm glucose, 1 mm pyruvate, and 20 mm HEPES. Neurons were imaged on an Olympus FluoView 1000 Confocal Laser Scanning Microscope equipped with a stage-top environmental chamber with temperature set to 37°C and 5% CO2, using a 40× oil objective. For confocal imaging, the following settings were used: excitation with a 488 nm argon-ion laser line at 0.5% power, scan resolution 800 × 800 pixels, scanning speed 2 μs/pixel, photo-multiplier voltage 700 V, digital gain of 1. A stack of optical sections at 1 μm intervals through each neuron was obtained at each position and time point and then flattened in a maximum intensity projection. Analysis was performed on an Apple Macintosh notebook using the National Institutes of Health ImageJ program. Images of related conditions were matched for laser power, gain, and offset.
Stable isotope labeling by amino acids in cell culture.
Neurons were transfected with the PSD95-TS:YSOG2 plasmid and cultured on MatTek dishes in Neurobasal medium as previously described (Butko et al., 2012). On DIV13, neurons were placed in custom-ordered Neurobasal medium lacking arginine and lysine (Invitrogen) and supplemented with 1 μm BILN, 13C6, 15N4 l-arginine-HCl (Arg*), and 13C6, 15N2 l-lysine-2HCl (Lys*) (Cambridge Isotope Laboratories). After 24 h, neurons were either lysed or replaced in normal Neurobasal medium lacking BILN, Arg*, and Lys* for an additional 24 h before lysis. Lysates were analyzed by MS. Peptides were identified using open-source MaxQuant software.
Statistical analysis.
Statistical significance was determined by the unpaired, two-tailed Student's t test; differences were considered to be significant for p values of <0.05.
Results
Development of the TS:YSOG3 reporter to track PKMζ synthesis and degradation
We developed TS:YSOG3 to track populations of PKMζ because we wanted to avoid fusing TS to PKMζ via its C terminus, as PKMζ has a PDZ-binding sequence near the C terminus that interacts with the protein PICK1 (Newton, 2001; Yao et al., 2008). TS:YSOG3 is designed for proteins to be N-terminally fused while the previously developed TS:YSOG2 reporter only allowed proteins to be C-terminally fused (Butko et al., 2012). Like TS:YSOG2, TS:YSOG3 incorporates split YFP, a cis-cleaving Hepatitis C virus (HCV) nonstructured 3 (NS3) protease, and miniSOG, a genetically encoded tag for correlated light and electron microscopy (Shu et al., 2011). In the absence of an inhibitor specific to the protease, the fusion protein is translated, folds, and the protease becomes active. It cleaves the protein of interest away from split YFP and miniSOG, which are then degraded via exposure of a destabilizing histidine at the N terminus of this cleaved peptide. When a small-molecule HCV NS3 protease inhibitor called BILN-2061 (BILN) (Lamarre et al., 2003) is applied, however, the protein of interest remains attached to miniSOG, and the two halves of split YFP fold and complement (Fig. 1A). As a result, new copies of tagged protein become fluorescent while old copies remain unlabeled and invisible.
Figure 1.

Development of TS:YSOG3 and effect of 5′ and 3′ UTRs on PKMζ expression. A, TS:YSOG3 fused to PKMζ, before and after BILN. B, YFP fluorescence quantification in neurons transfected with TS:YSOG3-PKMζ with the PKMζ and control and without 5′ and 3′ UTRs and in untransfected neurons, in the presence and absence of BILN. ***p < 0.001 (unpaired two-tailed Student's t test). n = 23, 19, 22, 18, 15, and 17 neurons per condition, respectively. Error bars indicate SEM. C, YFP complementation and maintenance after BILN addition and washout. D, Schematic indicating treatment with BILN for 24 h, then imaging for 10 h (once per hour) following BILN washout. E, Neurons imaged for 10 h after BILN washout show a loss in TS:YSOG3-PKMζ fluorescence, indicating degradation over time. F, Neurons visualized for 10 h after BILN washout with MG132 added immediately after washout do not show TS:YSOG3-PKMζ loss in fluorescence, indicating that proteasomal inhibition prevented TS:YSOG3-PKMζ from degrading. G, Fluorescence quantification of cell bodies with and without MG132 added at BILN washout. ***p < 0.001 (unpaired two-tailed Student's t test). n = 10 MG132 treated neurons; n = 7 untreated neurons. Error bars indicate SEM. Scale bars, 20 μm. H, TS:YSOG3-PKMζ amino acid sequence.
TS:YSOG3 differs from TS:YSOG2 in the position and type of HCV NS3 protease cleavage sites embedded within the reporter. TS:YSOG2 incorporates cleavage sites flanking the protease and contains the P6-P1 peptide sequence EDVVCC, derived from the NS5A/NS5B cleavage substrate, along with a destabilizing histidine at the P1′ position. In contrast, TS:YSOG3 incorporates two EDVVCC cleavage sites with destabilizing histidines at each P1′ position, both located at the N terminus of the reporter, as well as a cleavage site with the P6-P1 sequence DEMEEC in between the protease and the C-terminal fragment of split YFP (Butko et al., 2012). Two EDVVCC sites were incorporated to ensure complete peptide cleavage. When cleavage occurs at the DEMEEC site in TS:YSOG3, the C terminal peptide is separated from the TS reporter but not targeted for degradation as it would be if fused in the same manner to TS:YSOG2.
The 5′ and 3′ untranslated regions (UTRs) of PKMζ mRNA influence mRNA localization and translation (Muslimov et al., 2004) so we first examined the effect of the PKMζ 5′ and 3′ UTRs on TS:YSOG3-PKMζ translation to ensure that our TS fusion protein was expressed in the same manner as endogenous PKMζ. The 5′ UTR contains a binding site for PIN1, a protein that represses mRNA translation (Hernández et al., 2003), and both UTRs contain motifs that influence PKMζ mRNA transport to dendritic spines and translation initiation (Muslimov et al., 2004; Muslimov et al., 2011; Eom et al., 2014). We produced a TS:YSOG3-PKMζ construct with PKMζ 5′ and 3′ UTRs and a separate TS:YSOG3-PKMζ construct incorporating control 5′ and 3′ UTRs derived from the pCAGGS vector. We expressed each construct in cultured neurons and visualized TS:YSOG3-PKMζ production in neurons that were either treated with BILN or left untreated for 24 h (Fig. 1B). New, marked copies of PKMζ were visible by YFP with somatodendritic localization, consistent with previous observations for PKMζ localization (Hernández et al., 2014). Fluorescence was significantly reduced in neurons transfected with the TS:YSOG3-PKMζ construct incorporating the 5′ and 3′ UTRs, indicating that the PKMζ UTRs strongly affected PKMζ translation. YFP signal was absent in transfected neurons not treated with BILN, indicating that cleaved YFP halves did not complement and cleaved peptides were rapidly degraded. Untransfected neurons treated with BILN were not fluorescent, indicating that BILN treatment alone did not generate any fluorescence signal. While we did not consider PKMζ translocation, we did not expect PKMζ protein to move away from the site of synthesis because atypical PKCs do not translocate from the cytosol to either the membrane or nucleus in response to calcium as do conventional PKCs (Hrabetova and Sacktor, 2001). All following experiments were performed using PKMζ fusions to TS reporters incorporating the PKMζ 5′ and 3′ UTRs.
We next verified that we could track PKMζ degradation by YFP fluorescence over time after BILN washout, at which point old, previously produced, and marked proteins remain visible if stable and new proteins are not visible (Fig. 1C). Neurons transfected with TS:YSOG3-PKMζ were treated with BILN for 24 h. BILN was then washed out, and neurons were imaged every hour for the following 10 h, tracking old, marked proteins until degradation (Fig. 1D). YFP fluorescence in transfected neurons decreased over the 10 h period imaged (Fig. 1E) and was proteasome-dependent because treatment with MG132, a proteasome inhibitor, at the time of BILN washout resulted in no loss of YFP signal (Fig. 1F). Similar results were previously reported for Neuroligin-TS2:YFP and Arc-TS:YFP (De Jaco et al., 2010; Butko et al., 2012). In addition, loss of YFP signal over time was not a result of photobleaching (Fig. 2A,B). Together, these data indicate that complemented YFP remains stable over time after BILN washout and that reduction in YFP fluorescence over time is due to protein degradation, not split YFP dissociation. It was also necessary to characterize the level of TS-mediated PKMζ overexpression in neurons. We used Western blot analysis to compare levels of endogenous PKMζ protein to PKMζ produced from TS reporters. In the absence of BILN, PKMζ copies produced from TS:YSOG2 and TS:YSOG3 remain fused to a fragment of YFP and are larger in molecular weight than endogenous PKMζ. These populations can be differentiated by size. We found that there was considerably less PKMζ produced from each TS reporter compared with endogenous PKMζ, suggesting that TS-mediated overexpression does not dramatically alter PKMζ levels in neurons (Fig. 2C). We additionally used a cycloheximide assay to confirm that fusions to the TS:YSOG2 and TS:YSOG3 reporters do not alter PKMζ turnover (Fig. 2E,F). Finally, we used a FLAG-TS:YSOG3-PKMζ construct expressed in HEK-293A cells to demonstrate that the N terminal EDVVCC cleavage sites in the YSOG3 reporter are not cleaved after BILN washout, indicating that labeled proteins would not be subject to N-terminal histidine-mediated destabilization (Fig. 2G–I).
We further confirmed that complemented YFP holds TS reporters together after BILN washout using stable isotope labeling by amino acids in cell culture (SILAC) combined with mass spectrometry (MS) (Fig. 3A). Cultured neurons were transfected with a PSD95-TS:YSOG2 construct that exhibits elevated expression in neurons compared with PKMζ and is easier to identify by MS (Butko et al., 2012). Neurons were cultured in normal growth media and at DIV 13 were switched to media containing 13C6, 15N2 lysine (Lys*), 13C6, 15N4 arginine (Arg*), and BILN. After 24 h, neurons were either collected and analyzed by MS or replaced in normal growth media lacking Lys*, Arg*, and BILN for an additional 24 h before MS (Fig. 3B). In the sample analyzed immediately after labeling, heavy peptides were identified from regions spanning the full PSD95-TS:YSOG2 fusion protein. In the sample replaced in normal growth media for an additional 24 h, Arg* and Lys* labeled peptides were identified from the N and C termini of PSD95-TS:YSOG2 and not from the NS3 protease (Fig. 3C; Table 1). If complemented YFP dissociated, we would not have detected any peptides from the C terminus of PSD95-TS:YSOG2 in MS. This indicates that the NS3 protease can be reactivated following BILN washout, yet PSD95 remains fused to complemented YFP. This is consistent with previous studies of complemented YFP stability over time (Butko et al., 2012).
Figure 3.
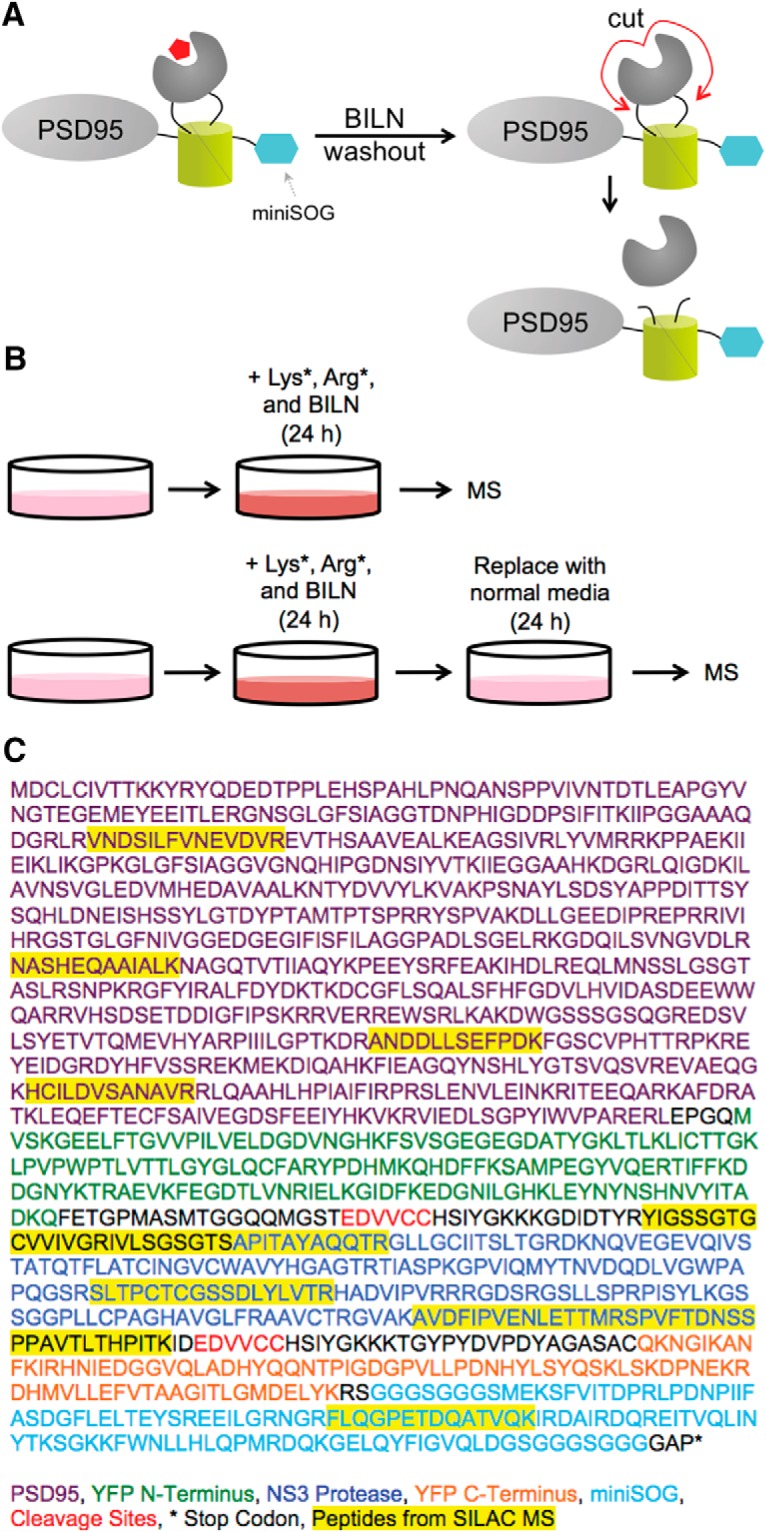
SILAC MS demonstrates that reconstituted YFP does not fall apart when BILN is removed. A, Schematic describing how labeled PSD95 retains complemented YFP after BILN washout despite NS3 protease cleavage. B, SILAC experimental design; the first sample is labeled with Lys*, Arg*, and BILN for 24 h and then immediately analyzed by MS, whereas the second sample undergoes an additional 24 h incubation in media lacking Lys*, Arg*, and BILN. C, PSD95-TS:YSOG2 amino acid sequence and peptides identified from MS analysis.
Table 1.
Peptides identified from SILAC MS analysisa
| Sequence | Index (amino acids) | Mass (Da) | Ratio H/L |
RMSE | |
|---|---|---|---|---|---|
| BILN washout | No BILN washout | ||||
| VNDSILFVNEVDVR | 110–123 | 1617.836 | 0.10 | 7.8 | 0.5 |
| NASHEQAAIALK | 366–377 | 1251.657 | 0.07 | 12.8 | 3.0 |
| ANDDLLSEFPDK | 544–555 | 1362.630 | 0.06 | 4.8 | 0.6 |
| HCILDVSANAVR | 622–633 | 1353.662 | 0.02 | 4.4 | 1.1 |
| YIGSSGTGCVVIVGR | 923–937 | 1523.777 | 0.00 | 7.9 | 0.4 |
| IVLSGSGTSAPITAYAQQTR | 938–957 | 2020.059 | 0.00 | 52.9 | 0.8 |
| SLTPCTCGSSDLYLVTR | 1039–1055 | 1928.897 | 0.00 | 2.6 | 2.2 |
| AVDFIPVENLETTMR | 1112–1126 | 1733.866 | 0.00 | 8.1 | 0.7 |
| SPVFTDNSSPPAVTLTHPITK | 1127–1147 | 2208.143 | 0.00 | 11.3 | 0.6 |
| FLQGPETDQATVQK | 1313–1326 | 1560.778 | 0.02 | 9.3 | 5.7 |
aLys*- and Arg*-containing peptides are identified from the full PSD95-TS:YSOG2 right after a 24 h BILN pulse, although Lys*- and Arg*-containing peptides are identified from only the N- and C-termini of PSD95-TS:YSOG2 and not from the region excised by the NS3 protease when subjected to a 24 h BILN pulse and 24 h BILN washout. This indicates that the TS reporter is held together by complemented YFP despite NS3 protease cleavage upon BILN washout. RMSE, Root mean square error.
Chemical LTP induction results in PKMζ upregulation at synapses
We next aimed to determine whether cLTP increased de novo synthesis of TS:YSOG3-PKMζ the way endogenous PKMζ is upregulated during late LTP (Sacktor et al., 1993; Sacktor, 2011). Cultured neurons were stimulated for 10 min with forskolin and rolipram in ACSF lacking magnesium to induce cLTP, following an established method previously used in both cultured neurons and brain slices (Otmakhov et al., 2004; Kim et al., 2007). We first stimulated neurons expressing PKMζ fused directly and only to YFP and imaged YFP after forskolin and rolipram cLTP at 1 h intervals over a 10 h period. YFP fluorescence increased over time in the cell body and in dendritic spines, indicating that new copies of PKMζ were produced in response to the stimulation. This result was in contrast to the unchanged YFP signal in neurons expressing PKMζ-YFP but left unstimulated (Fig. 4A–C). Because PKMζ is active during late LTP (Hernández et al., 2003), we used TS:YSOG3-PKMζ to specifically visualize new copies of PKMζ produced over the course of several hours after forskolin and rolipram cLTP. Transfected neurons were stimulated and immediately treated with BILN for 24 h. At 24 h after stimulation, neurons were fixed and immunolabeled for the postsynaptic protein PSD95. YFP fluorescence indicated that stimulated neurons had increased PKMζ production and accumulation. New PKMζ copies colocalized with PSD95 in stimulated but not unstimulated neurons, indicating that new PKMζ is preferentially localized to dendritic spines after stimulation (Fig. 4D,E). PKMζ localization to dendritic spines was also observed in neurons transfected with PKMζ C-terminally fused to the TS:YSOG2 reporter (Fig. 5A,B), indicating that PKMζ upregulation following stimulation is not dependent on how PKMζ is fused to TS.
Figure 4.

TS:YSOG3-PKMζ is upregulated and localizes to synapses after forskolin and rolipram cLTP. A, Schematic indicating how neurons expressing PKMζ-YFP were stimulated and visualized over 10 h. B, Stimulation increased PKMζ synthesis and accumulation; unstimulated neurons showed no change in YFP signal. Scale bars, 20 μm. C, Fluorescence quantification of dendritic spines indicates that there was more PKMζ in spines after stimulation. ***p < 0.001 (unpaired two-tailed Student's t test). n = 41 stimulated spines; n = 40 unstimulated spines. Error bars indicate SEM. D, Schematic indicating how neurons transfected with TS:YSOG3-PKMζ were treated with BILN for 24 h and then imaged. E, Transfected neurons stimulated with forskolin and rolipram, left unstimulated, or left without BILN were fixed and immunolabeled for PSD95. TS:YSOG3-PKMζ YFP fluorescence (green pseudocolor in magnified images) and PSD95 immunofluorescence (red pseudocolor) were imaged. Stimulated neurons show increased YFP fluorescence and TS:YSOG3-PKMζ colocalization with PSD95 (yellow pseudocolor). Scale bars: whole cell, 20 μm; magnified pseudocolor, 2 μm.
Figure 5.
PKMζ-TS:YSOG2 is upregulated and localizes to synapses after forskolin and rolipram cLTP. A, PKMζ fusion to TS:YSOG2, before and after BILN addition and subsequent washout. B, Transfected neurons stimulated with forskolin and rolipram or left unstimulated, and untransfected neurons were fixed and immunolabeled for PSD95. PKMζ-TS:YSOG2 YFP fluorescence and PSD95 immunofluorescence were imaged as in Figure 4. Stimulated neurons show increased YFP fluorescence and PKMζ-TS:YSOG2 colocalization with PSD95. Scale bars: whole cell, 20 μm; magnified pseudocolor, 2 μm.
Newly produced PKMζ is degraded rapidly
PKMζ may function by associating with synaptic proteins NSF and PICK1 to release GluR2 AMPA receptors from synaptic pools into the postsynaptic membrane; however, AMPA receptors undergo activity-dependent dephosphorylation and endocytosis (Derkach et al., 2007; Yao et al., 2008; Migues et al., 2010). Thus, to maintain LTP in this manner, PKMζ would need to persist at synapses to maintain sufficient levels of GluR2-containing AMPA receptors at the postsynaptic membrane over long periods of time. To investigate PKMζ stability, we tracked labeled PKMζ copies produced after forskolin and rolipram cLTP to see how long they remained at the synapse after synthesis. We chose to study the total population of PKMζ produced 24 h after stimulation because PKMζ is upregulated during the late, maintenance phase of LTP (Osten et al., 1996). Presumably, memories that persist beyond a short time frame are perpetuated by equally persistent synaptic changes. Thus, the elevated population of PKMζ present some time after forskolin and rolipram cLTP induction would be important in maintaining LTP and is the important population to study. If a small and highly stable population of PKMζ were produced soon after LTP, it would be part of this labeled population of protein. Cultured neurons expressing TS:YSOG3-PKMζ were stimulated and immediately treated with BILN for 24 h to mark PKMζ copies produced after stimulation. BILN was removed after the 24 h period and the sample was transferred to a confocal microscope equipped with a temperature and CO2-controlled chamber. Transfected neurons were identified by YFP fluorescence, and images were captured at 1 h intervals over the following 10 h to track labeled PKMζ degradation over time (Fig. 6A).
Figure 6.

TS:YSOG3-PKMζ degrades rapidly in neuronal cell bodies and dendritic spines after forskolin and rolipram cLTP. A, Schematic indicating how neurons transfected with TS:YSOG3-PKMζ were treated with BILN for 24 h, then imaged once per hour for 10 h after BILN washout. B, Stimulated neuron YFP fluorescence decreased quickly over time, indicating that PKMζ is rapidly degraded. C, Unstimulated neuron YFP fluorescence decreased slowly in the 10 h following BILN washout, indicating that basal PKMζ remains more stable over time. D, YFP quantification in neuron cell bodies (n = 5 stimulated cell bodies; n = 5 unstimulated cell bodies). **p < 0.01 (unpaired two-tailed t test). Error bars indicate SEM. E, YFP quantification in dendritic spines (n = 53 stimulated dendritic spines; n = 51 unstimulated dendritic spines). ***p < 0.001 (unpaired two-tailed t test). Error bars indicate SEM. Scale bars, 20 μm.
As expected, more PKMζ protein was produced in stimulated neurons; however, surprisingly, this population of PKMζ degraded rapidly over the 10 h following BILN washout (Fig. 6B). In contrast, PKMζ in unstimulated neurons was produced at a lower, basal level, but most of this protein remained relatively stable both in the cell body and at dendritic spines (Fig. 4C). The effects of stimulation were quantified by measuring the reduction in fluorescence over time in neuronal cell bodies and dendritic spines (Fig. 4D,E). Thus, PKMζ appeared to turn over more rapidly after cLTP.
Although forskolin and rolipram are widely used for cLTP, we further confirmed that we could also observe elevated PKMζ degradation after stimulation using glycine-dependent cLTP. Glycine is an NMDA coactivator that specifically activates synaptic NMDARs in cultured neurons in conjunction with stochastic glutamate release at synapses (Liao et al., 2001; Tampellini et al., 2009). We found that PKMζ was upregulated in cell bodies and dendritic spines after glycine cLTP as with forskolin and rolipram cLTP, and new PKMζ copies colocalized with the postsynaptic protein PSD95 (Fig. 7A). Following the same experimental scheme as used with forskolin and rolipram, we labeled PKMζ produced during a 24 h period with and without glycine stimulation and then tracked degradation of labeled PKMζ over time after BILN washout. We found that glycine stimulation also resulted in more total PKMζ degradation over the 10 h time period measured, indicating that increased PKMζ degradation following cLTP was not specific to only forskolin and rolipram (Fig. 7B–F). We additionally repeated forskolin and rolipram stimulation on neurons transfected with C-terminally fused PKMζ-TS:YSOG2, which also showed the same increase in PKMζ degradation rate after cLTP (Fig. 8A–E), indicating that this effect is not dependent on how PKMζ is fused to the TS reporter.
Figure 7.

TS:YSOG3-PKMζ degrades rapidly in neuronal cell bodies and dendritic spines after glycine cLTP. A, Transfected neurons stimulated with glycine were fixed and immunolabeled for PSD95. TS:YSOG3-PKMζ YFP fluorescence (green pseudocolor in magnified images) and PSD95 immunofluorescence (red pseudocolor) were imaged. Stimulated neurons show increased YFP fluorescence and TS:YSOG3-PKMζ colocalization with PSD95 (yellow pseudocolor). B, Schematic indicating how transfected neurons were treated with BILN for 24 h and then imaged once per hour for 10 h after BILN washout. C, Stimulated neuron YFP fluorescence decreases quickly over time, indicating that PKMζ is rapidly degraded. D, Unstimulated neuron YFP fluorescence decreases slowly in the 10 h following BILN washout, indicating that basal PKMζ is slowly degraded. n = 52 stimulated dendritic spines; n = 51 unstimulated dendritic spines. PKMζ produced at the basal level remains more stable over time. E, YFP quantification in neuronal cell bodies (n = 5 stimulated cell bodies; n = 5 unstimulated cell bodies) and (F) dendritic spines (n = 55 stimulated dendritic spines; n = 52 unstimulated dendritic spines). *p < 0.05 (unpaired two-tailed t test). **p < 0.01 (unpaired two-tailed t test). ***p < 0.001 (unpaired two-tailed t test). Error bars indicate SEM. NS, Not significant). Scale bars: A, whole cell, 20 μm; magnified pseudocolor, 2 μm; C, D, 20 μm.
Figure 8.

PKMζ-TS:YSOG2 degrades more rapidly in neuronal cell bodies and dendritic spines after forskolin and rolipram cLTP. A, Schematic indicating how neurons transfected with PKMζ-TS:YSOG2 were treated with BILN for 24 h, then imaged once per hour for 10 h following BILN washout. B, YFP fluorescence in neurons stimulated with forskolin and rolipram decreases rapidly over time, indicating that PKMζ is rapidly degraded. C, Unstimulated neuron YFP fluorescence decreases slowly over time, indicating that PKMζ, produced at the basal level, is slowly degraded. D, YFP quantification in neuron cell bodies (n = 5 forskolin and rolipram stimulated cell bodies; n = 5 unstimulated cell bodies). Error bars indicate SEM. E, YFP quantification in dendritic spines (n = 54 forskolin and rolipram-stimulated dendritic spines; n = 55 unstimulated dendritic spines). ***p < 0.001 (unpaired two-tailed t test). Error bars indicate SEM. Scale bar, 20 μm.
PKCλ turnover is not affected by chemical LTP induction
Recent studies suggest that ZIP may not effectively inhibit PKMζ in vivo (Wu-Zhang et al., 2012) or that it may inhibit other synaptic molecules in addition to PKMζ that may be the true mediators of LTP and LTM (Ren et al., 2013; Volk et al., 2013). One alternative target for ZIP is PKCλ, a PKC isoform that is similar to PKMζ and is also inhibited by ZIP (Ren et al., 2013). PKCλ phosphorylates the GluR1 subunit of AMPA receptors and localizes to the postsynaptic membrane via the scaffold protein p62, which is important for AMPA receptor trafficking (Jiang et al., 2006, 2009). PKCλ is active with slightly elevated protein levels during late LTP (Kelly et al., 2007; Ren et al., 2013). To characterize new PKCλ synthesis, we fused TS:YSOG3 to PKCλ and visualized PKCλ production and degradation after forskolin and rolipram cLTP. In contrast to PKMζ, PKCλ strongly localized to synapses both with and without stimulation (Fig. 9A). We marked newly synthesized PKCλ in TS:YSOG3-PKCλ-transfected neurons by treating them with BILN for 24 h and then tracked degradation of labeled PKCλ over a 10 h period after BILN washout to visualize stability of new PKCλ over time (Fig. 9B). PKCλ levels were slightly elevated in dendritic spines after stimulation, as expected. However, unlike PKMζ, PKCλ turnover was unaffected by forskolin and rolipram stimulation, and new copies of PKCλ appeared to stay more stable at synapses over time (Fig. 9C–F). We cannot exclude the possibility that some PKCλ produced in dendritic spines diffuses away from the synapse; here we only examined PKCλ presence in spines regardless of diffusion or degradation. If a population of PKCλ indeed remains stable at the postsynaptic membrane, it could potentially contribute to maintaining long-term postsynaptic changes following LTP induction.
Figure 9.

TS:YSOG3-PKCλ turnover is not altered by forskolin and rolipram cLTP. A, Transfected neurons stimulated with forskolin and rolipram, left unstimulated, or left without BILN were fixed and immunolabeled for PSD95. TS:YSOG3-PKCλ YFP fluorescence and PSD95 immunofluorescence were imaged. Both stimulated and unstimulated neurons show TS:YSOG3-PKMλ colocalization with PSD95 and no YFP fluorescence in untransfected neurons as expected. B, Schematic indicating how transfected neurons were treated with BILN for 24 h and imaged once per hour for 10 h after BILN washout. C, Stimulated neuron YFP fluorescence decreased over time, indicating some PKCλ degradation. D, Unstimulated neuron YFP fluorescence also decreased over time, similar to stimulated neurons. E, YFP quantification in neuronal cell bodies (n = 5 stimulated cell bodies; n = 5 unstimulated cell bodies). NS, Not significant (p > 0.05, unpaired two-tailed t test). Error bars indicate SEM. F, YFP quantification in dendritic spines (n = 51 stimulated dendritic spines; n = 52 unstimulated dendritic spines). *p < 0.05 (unpaired two-tailed t test). Error bars indicate SEM. G, Amino acid sequence of the TS:YSOG3-PKCλ fusion protein. Scale bars: A, whole cell 20 μm; magnified pseudocolor 2 μm; C, D, 20 μm.
Discussion
Here we developed the new TS:YSOG3 reporter and used it along with TS:YSOG2 to characterize PKMζ and PKCλ turnover after cLTP induction in neurons. We found that PKMζ is upregulated following cLTP and that PKMζ copies produced after stimulation localized to synapses. We additionally demonstrated that the turnover of PKMζ copies synthesized after cLTP is more rapid than the turnover of basal PKMζ, both in the neuron cell body and in dendritic spines. These results held for PKMζ fused via its C terminus to TS:YSOG2. Furthermore, we demonstrated that PKCλ remains more stable at synapses than PKMζ and that its turnover is not altered after cLTP. We showed that our TS reporters not only allow identification of new copies of a given protein but also old copies as they degrade over time; this is possible because TS-tagged proteins synthesized during BILN application remain fused to irreversibly complemented YFP that stays stable after BILN washout. Moreover, fusion to TS did not affect PKMζ localization, proteasome-mediated degradation, or rate of turnover. Our experiments were performed in cultured neurons with cLTP induction; although this is only an in vitro model of brain activity, many of the cellular mechanisms are analogous and relevant for understanding processes that occur in the intact brain.
TS reporters confer several advantages over similar techniques for characterizing protein stability. New protein synthesis and turnover can be visualized with photoconvertible fluorescent proteins, such as Dendra2 and mEos2, by tracking protein emission shifts after exposure to violet light. Although these proteins can differentiate between new and old copies of a particular protein, their utility is limited because photoconversion requires transparent or very thin samples and potentially damaging exposure to intense illumination (Chudakov et al., 2007; McKinney et al., 2009). Pulse labeling methods, such as bioorthogonal noncanonical amino acid tagging (BONCAT) and fluorescent noncanonical amino acid tagging (FUNCAT), can also mark all new protein copies, but are not specific to a protein of interest and require cells to incorporate unnatural amino acids before labeling (Dieterich et al., 2006, 2010; Hinz et al., 2013). This can be evaded by pulse labeling proteins expressing a tetracysteine motif with biarsenical dyes FlAsH and ReAsH; however, these dyes can be toxic to live samples and require coadministration of antidotes (Adams et al., 2002; Adams and Tsien, 2008). TS reporters avoid these drawbacks and allow analysis of new or old marked protein copies by Western blotting, MS, and EM in addition to light microscopy.
Our observation that PKMζ is both rapidly synthesized and degraded following cLTP in neurons suggests that a stable amount of PKMζ would be difficult to maintain at synapses and store memories over long time scales. New reports characterizing memory in PKMζ knock-out animals and demonstrating that ZIP is not specific to PKMζ also suggest that PKMζ may not be the primary molecule maintaining LTM (Wu-Zhang et al., 2012; Lee et al., 2013; Volk et al., 2013). Despite the controversy surrounding previous work on PKMζ, the exciting and well-established finding is that ZIP administration into the brain can disrupt fairly long-lasting memories of up to a month (Shema et al., 2007). The protein KIBRA (KIdney/BRAin protein) was newly identified as a synaptic protein that binds to GluR2 AMPA receptors, associates with PKMζ, and is involved in memory (Makuch et al., 2011). A recent study performed in a kidney cell line suggested that KIBRA stabilizes PKMζ (Vogt-Eisele et al., 2014); perhaps this interaction contributes to the elevated levels of PKMζ present after LTP induction. Further studies in neurons are needed to demonstrate whether KIBRA can stabilize PKMζ in the complicated synaptic environment containing proteins, such as PIN1, NSF, PICK1, p62, and GluR2, all of which are implicated in PKMζ-mediated GluR2 transport to the postsynaptic membrane during late LTP.
PKCλ is an atypical PKC with a catalytic domain similar to the catalytic domain in PKMζ (Ren et al., 2013) and is now known to be an additional target of ZIP (Ren et al., 2013). PKCλ could potentially function in retaining AMPA receptors at the postsynaptic membrane in a similar or complementary mechanism as PKMζ. Here we observed that PKCλ turnover was not strongly affected by cLTP and that it remains more stable at synapses than PKMζ. Although these two PKC isoforms have different roles in establishing neuronal polarity during development and differential expression patterns throughout the body, their substrates are similar (Naik et al., 2000; Parker et al., 2013). Thus, it is possible that one could compensate in the absence of the other. PKCλ has a regulatory domain, unlike PKMζ, the latter of which is constitutively active after late LTP. Therefore, the question remains how PKCλ can remain active and functional over long time scales. Perhaps the association of stable PKCλ with scaffolding protein p62 and other structural proteins near the postsynaptic membrane protects a population of the protein from degradation. In our study, we did not account for the likely on-off switches in PKCλ kinase activity as with other PKCs containing a regulatory domain (Newton, 2001), or PKMζ or PKCλ translocation away from the original site of synthesis. There is no evidence for atypical PKC translocation (Hrabetova and Sacktor, 2001); however, the activation state and diffusion of these proteins should be considered in future studies.
Despite extensive work on synaptic proteins, such as PKMζ, the mechanisms behind LTP and LTM maintenance are still elusive. Early theories postulated that memory must be maintained in a highly stable molecule, such as DNA (Crick, 1984), others later attributed memory maintenance to self-perpetuating bistable proteins, such as PKMζ, CPEB, and CaMKII (Sacktor, 2011; Kandel, 2012; Lisman et al., 2012), or permanent alterations in neuronal extracellular structures (Tsien, 2013). For memories to remain stable, the changes made to encode the memory must also perpetuate themselves as the molecules undergo repeated cycles of generational turnover.
In conclusion, our work addressed the stability of synaptic memory-related proteins PKMζ and PKCλ and demonstrated that TS reporters are useful tools for visualizing and tracking specific populations of a given protein over time. These reporters could potentially be used in the intact brain to identify synapses that were modified after a learning experience and that maintain the memory of what was learned. Experiments using confocal brain tissue imaging and live animal brain imaging to visualize TS-tagged proteins synthesized in response to stimulation or a learning paradigm could give further insight into synapse modifications that are long-lasting. Although there is growing evidence against a role for PKMζ in memory, PKCλ has yet to be challenged. Ultimately, the molecular interactions disrupted by ZIP are likely those that are critical in memory maintenance. Future studies on synaptic protein stability using TS reporters could potentially help us understand how synapses are modified to encode and store our memories for as long as a lifetime.
Footnotes
This work was supported by National Institutes of Health Grant NS027177 to R.Y.T. and Grants R01 GM086197 and P41 GM103412 to M.H.E., S.F.P. was supported by the University of California, San Diego Graduate Training Program in Cellular and Molecular Pharmacology (T32 GM007752) and the University of California, San Diego Graduate Training Program in Neuroplasticity of Aging (T32 AG000216). We thank Stephen R. Adams, Hiro Hakozaki, Ranjan Ramachandra, Varda Lev-Ram, John Y. Lin, Alexandra C. Newton, Roberto Malinow, Ali F. Palida, and Mira Sastri for experimental help, advice, and manuscript editing.
The authors declare no competing financial interests.
References
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem. 2008;89:260–268. doi: 10.1016/j.nlm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Adams SR, Tsien RY. Preparation of the membrane-permeant biarsenicals FlAsH-EDT2 and ReAsH-EDT2 for fluorescent labeling of tetracysteine-tagged proteins. Nat Protoc. 2008;3:1527–1534. doi: 10.1038/nprot.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- Butko MT, Yang J, Geng Y, Kim HJ, Jeon NL, Shu X, Mackey MR, Ellisman MH, Tsien RY, Lin MZ. Fluorescent and photo-oxidizing TimeSTAMP tags track protein fates in light and electron microscopy. Nat Neurosci. 2012;15:1742–1751. doi: 10.1038/nn.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Lukyanov S, Lukyanov KA. Using photoactivatable fluorescent protein Dendra2 to track protein movement. Biotechniques. 2007;42:553, 555, 557. doi: 10.2144/000112470. passim. [DOI] [PubMed] [Google Scholar]
- Cohen LD, Zuchman R, Sorokina O, Müller A, Dieterich DC, Armstrong JD, Ziv T, Ziv NE. Metabolic turnover of synaptic proteins: kinetics, interdependencies and implications for synaptic maintenance. PLoS One. 2013;8:e63191. doi: 10.1371/journal.pone.0063191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Memory and molecular turnover. Nature. 1984;312:101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- De Jaco A, Lin MZ, Dubi N, Comoletti D, Miller MT, Camp S, Ellisman M, Butko MT, Tsien RY, Taylor P. Neuroligin trafficking deficiencies arising from mutations in the alpha/beta-hydrolase fold protein family. J Biol Chem. 2010;285:28674–28682. doi: 10.1074/jbc.M110.139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci U S A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom T, Muslimov IA, Tsokas P, Berardi V, Zhong J, Sacktor TC, Tiedge H. Neuronal BC RNAs cooperate with eIF4B to mediate activity-dependent translational control. J Cell Biol. 2014;207:237–252. doi: 10.1083/jcb.201401005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández AI, Blace N, Crary JF, Serrano PA, Leitges M, Libien JM, Weinstein G, Tcherapanov A, Sacktor TC. Protein kinase M zeta synthesis from a brain mRNA encoding an independent protein kinase C zeta catalytic domain: implications for the molecular mechanism of memory. J Biol Chem. 2003;278:40305–40316. doi: 10.1074/jbc.M307065200. [DOI] [PubMed] [Google Scholar]
- Hernández AI, Oxberry WC, Crary JF, Mirra SS, Sacktor TC. Cellular and subcellular localization of PKMzeta. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130140. doi: 10.1098/rstb.2013.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz FI, Dieterich DC, Schuman EM. Teaching old NCATs new tricks: using non-canonical amino acid tagging to study neuronal plasticity. Curr Opin Chem Biol. 2013;17:738–746. doi: 10.1016/j.cbpa.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabetova S, Sacktor TC. Transient translocation of conventional protein kinase C isoforms and persistent downregulation of atypical protein kinase Mzeta in long-term depression. Brain Res Mol Brain Res. 2001;95:146–152. doi: 10.1016/S0169-328X(01)00185-1. [DOI] [PubMed] [Google Scholar]
- Jiang J, Suppiramaniam V, Wooten MW. Posttranslational modifications and receptor-associated proteins in AMPA receptor trafficking and synaptic plasticity. Neurosignals. 2006;15:266–282. doi: 10.1159/000105517. [DOI] [PubMed] [Google Scholar]
- Jiang J, Parameshwaran K, Seibenhener ML, Kang MG, Suppiramaniam V, Huganir RL, Diaz-Meco MT, Wooten MW. AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus. 2009;19:392–406. doi: 10.1002/hipo.20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MT, Crary JF, Sacktor TC. Regulation of protein kinase Mzeta synthesis by multiple kinases in long-term potentiation. J Neurosci. 2007;27:3439–3444. doi: 10.1523/JNEUROSCI.5612-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bös M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagacé L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. Prkcz null mice show normal learning and memory. Nature. 2013;493:416–419. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21:6008–6017. doi: 10.1523/JNEUROSCI.21-16-06008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, Glenn JS, Tsien RY. A drug-controllable tag for visualizing newly synthesized proteins in cells and whole animals. Proc Natl Acad Sci U S A. 2008;105:7744–7749. doi: 10.1073/pnas.0803060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makuch L, Volk L, Anggono V, Johnson RC, Yu Y, Duning K, Kremerskothen J, Xia J, Takamiya K, Huganir RL. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron. 2011;71:1022–1029. doi: 10.1016/j.neuron.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Casadio A, Zhu H, Yaping E, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/S0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Muslimov IA, Nimmrich V, Hernández AI, Tcherepanov A, Sacktor TC, Tiedge H. Dendritic transport and localization of protein kinase Mzeta mRNA: implications for molecular memory consolidation. J Biol Chem. 2004;279:52613–52622. doi: 10.1074/jbc.M409240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslimov IA, Patel MV, Rose A, Tiedge H. Spatial code recognition in neuronal RNA targeting: role of RNA-hnRNP A2 interactions. J Cell Biol. 2011;194:441–457. doi: 10.1083/jcb.201010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Benedikz E, Hernández I, Libien J, Hrabe J, Valsamis M, Dow-Edwards D, Osman M, Sacktor TC. Distribution of protein kinase Mzeta and the complete protein kinase C isoform family in rat brain. J Comp Neurol. 2000;426:243–258. doi: 10.1002/1096-9861(20001016)426:2<243::AID-CNE6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- Osten P, Valsamis L, Harris A, Sacktor TC. Protein synthesis-dependent formation of protein kinase Mzeta in long-term potentiation. J Neurosci. 1996;16:2444–2451. doi: 10.1523/JNEUROSCI.16-08-02444.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, Lisman J. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–1962. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- Parker SS, Mandell EK, Hapak SM, Maskaykina IY, Kusne Y, Kim JY, Moy JK, St John PA, Wilson JM, Gothard KM, Price TJ, Ghosh S. Competing molecular interactions of aPKC isoforms regulate neuronal polarity. Proc Natl Acad Sci U S A. 2013;110:14450–14455. doi: 10.1073/pnas.1301588110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Ren SQ, Yan JZ, Zhang XY, Bu YF, Pan WW, Yao W, Tian T, Lu W. PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. EMBO J. 2013;32:1365–1380. doi: 10.1038/emboj.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. How does PKMzeta maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci U S A. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc Natl Acad Sci U S A. 2013;110:12456–12461. doi: 10.1073/pnas.1310158110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt-Eisele A, Krüger C, Duning K, Weber D, Spoelgen R, Pitzer C, Plaas C, Eisenhardt G, Meyer A, Vogt G, Krieger M, Handwerker E, Wennmann DO, Weide T, Skryabin BV, Klugmann M, Pavenstädt H, Huentelmann MJ, Kremerskothen J, Schneider A. KIBRA (KIdney/BRAin protein) regulates learning and memory and stabilizes protein kinase Mzeta. J Neurochem. 2014;128:686–700. doi: 10.1111/jnc.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu-Zhang AX, Schramm CL, Nabavi S, Malinow R, Newton AC. Cellular pharmacology of protein kinase Mzeta (PKMzeta) contrasts with its in vitro profile: implications for PKMzeta as a mediator of memory. J Biol Chem. 2012;287:12879–12885. doi: 10.1074/jbc.M112.357244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]