

ABSTRACT
Pseudomonas aeruginosa is a Gram-negative bacterium that is ubiquitous in the environment, and it is an opportunistic pathogen that can infect a variety of hosts, including humans. During the process of infection, P. aeruginosa coordinates the expression of numerous virulence factors through the production of multiple cell-to-cell signaling molecules. The production of these signaling molecules is linked through a regulatory network, with the signal N-(3-oxododecanoyl) homoserine lactone and its receptor LasR controlling the induction of a second acyl-homoserine lactone signal and the Pseudomonas quinolone signal (PQS). LasR-mediated control of PQS occurs partly by activating the transcription of pqsR, a gene that encodes the PQS receptor and is necessary for PQS production. We show that LasR interacts with a single binding site in the pqsR promoter region and that it does not influence the transcription of the divergently transcribed gene, nadA. Using DNA affinity chromatography, we identified additional proteins that interact with the pqsR-nadA intergenic region. These include the H-NS family members MvaT and MvaU, and CysB, a transcriptional regulator that controls sulfur uptake and cysteine biosynthesis. We show that CysB interacts with the pqsR promoter and that CysB represses pqsR transcription and PQS production. Additionally, we provide evidence that CysB can interfere with the activation of pqsR transcription by LasR. However, as seen with other CysB-regulated genes, pqsR expression was not differentially regulated in response to cysteine levels. These findings demonstrate a novel role for CysB in influencing cell-to-cell signal production by P. aeruginosa.
IMPORTANCE The production of PQS and other 4-hydroxy-2-alkylquinolone (HAQs) compounds is a key component of the P. aeruginosa cell-to-cell signaling network, impacts multiple physiological functions, and is required for virulence. PqsR directly regulates the genes necessary for HAQ production, but little is known about the regulation of pqsR. We identified CysB as a novel regulator of pqsR and PQS production, but, unlike other CysB-controlled genes, it does not appear to regulate pqsR in response to cysteine. This implies that CysB functions as both a cysteine-responsive and cysteine-unresponsive regulator in P. aeruginosa.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative bacterium that is ubiquitous in the environment, due in part to its metabolic versatility that allows it to obtain the nutrients needed for growth from a wide variety of compounds (1). P. aeruginosa is also an opportunistic pathogen that can infect plants, nematodes, insects, and mammals (2–5). With regard to humans, it can cause a wide range of diseases and is a major cause of health care-associated illnesses and devastating chronic infections in cystic fibrosis patients (6, 7). In order to thrive under such a diverse range of conditions, P. aeruginosa utilizes complex regulatory networks to control gene expression. These networks include global regulatory systems that can direct large-scale changes in gene expression, as well as many systems that allow fine-tuning of gene expression in response to specific environmental stimuli. During infection, P. aeruginosa coordinates the expression of many virulence factors through the production of cell-to-cell signaling molecules. There are three main cell-to-cell signaling systems that together are estimated to control the expression of up to 12% of the genes in the P. aeruginosa genome (2, 8, 9). The las and rhl systems, which, respectively, function through the signaling molecules N-(3-oxododecanoyl) homoserine lactone (3-oxo-C12-HSL) and N-butyryl homoserine lactone, and the regulators LasR and RhlR, are similar to homoserine lactone-based systems found in other Gram-negative bacteria. The third system functions primarily through the signaling molecule 2-heptyl-3-hydroxy-4-quinolone, also referred to as the Pseudomonas quinolone signal (PQS) (10). These three systems are linked in a network, with the las system positively regulating the components of the PQS and rhl systems (10–12). Additionally, the PQS system positively regulates the components of the rhl system, while the rhl system can negatively impact PQS production (13–17). In spite of all this signaling cross talk, each system can also function independently. In the absence of LasR, the rhl system becomes active and can regulate some las-controlled functions (18). PQS is also produced in the absence of a functional las system (14), and, unlike the other two signals, PQS is not produced maximally until the stationary phase of growth (13). Furthermore, each signaling system can also be affected by environmental conditions, such as the availability of nutrients like iron, phosphate, and certain amino acids (16, 19–22). The interplay between these systems allows P. aeruginosa to integrate information from multiple signals and alter gene expression accordingly.
The quinolone signaling system is more complex than either the las or rhl systems. PQS is synthesized by enzymes encoded in the pqsABCDE and pqsH operons from the precursors anthranilate, malonyl coenzyme A (malonyl-CoA), and octanoyl-CoA (23–25). The pqsABCD genes are also important for the synthesis of 55 other quinolone compounds and the metabolites 2,4-dihydroxyquinoline and 2-aminoacetophenone (25–28). Expression of the pqsABCDE operon is controlled by the LysR-type regulator PqsR (also known as MvfR), which is required for virulence (5, 29–31). PQS acts as a coinducer with PqsR to positively regulate pqsABCDE expression, forming a positive autoregulatory feedback loop for quinolone synthesis. The direct precursor of PQS, 2-heptyl-4-quinolone, can also act as a coinducer with PqsR but with 100-fold lower activity (31). Transcription of the pqsABCDE operon leads to the expression of PqsE, which is not required for PQS biosynthesis but instead independently regulates the expression of a majority of the genes controlled by PQS (5, 17, 32, 33). PqsE is required for virulence, and its function is not known; but it appears to work in conjunction with the rhl signaling system to influence gene expression (2, 17, 32, 33). In addition to its role as a signaling molecule, PQS and other 4-hydroxy-2-alkylquinolones produced by P. aeruginosa have been linked to other biological activities. These include roles in iron metabolism, redox cycling, membrane vesicle formation, interspecies interactions, and antimicrobial activity (34–36). These findings show that quinolone production has a major impact on P. aeruginosa physiology that extends beyond the ability of this organism to cause disease.
We became interested in the regulation of pqsR because it directly regulates the pqsABCDE operon. The pqsR promoter sequence is part of a large intergenic region, with 747 bp separating the coding regions of pqsR and the divergently transcribed nadA gene. Two transcriptional start sites were identified for pqsR, located at bp −190 and −278 relative to the pqsR translational start site (TS2 and TS1, respectively) (30). Additionally there is a small open reading frame located upstream from the pqsR coding sequence (uof) (see Fig. 1A). The function of the peptide encoded by uof is not known, but the translation of uof is coupled to the translation of pqsR (37). It has been shown that the transcription of pqsR is positively controlled by LasR and 3-oxo-C12-HSL, which also promotes PQS production by inducing the expression of pqsH (9, 38). Previous studies identified a potential consensus sequence for LasR binding in the pqsR promoter region and showed that this sequence was important for pqsR expression, but they did not fully investigate the interactions of LasR with the pqsR promoter (30, 39). In this study, we directly examined the role of LasR in controlling pqsR transcription and identified an additional factor that impacts pqsR transcription. We report a novel role for CysB, a master regulator of sulfur uptake (40), in controlling pqsR expression and found that it can counteract the positive regulatory influence of LasR.
FIG 1.

Purified LasR interacts only with a 27-bp region in the pqsR promoter centered at −526 relative to the pqsR translational start site. (A) A graphical depiction of the pqsR promoter region and DNA segments used as probes for EMSA. The bent arrows show the relative positions of identified transcriptional start sites. Numbering indicates the position in base pairs relative to the pqsR translational start site. (B) For EMSA, radiolabeled DNA fragments corresponding to either the lasI promoter (p-lasI, positive control), the kynA promoter (p-kynA, negative control), or the pqsR promoter region were incubated in the presence and absence of 7.2 ng of purified LasR as described in Materials and Methods. 3-oxo-C12-HSL (5 μM) was included in each reaction mixture. Samples of binding reaction products were electrophoresed on nondenaturing acrylamide gels, and the position of DNA fragments was visualized by autoradiography. (C) DNase I protection assays with purified LasR were performed as described in Materials and Methods. The results shown were generated using the DNA fragment pqsR-P2 that was end labeled on the pqsR coding strand. Lanes labeled A, C, G, and T contain DNA sequencing ladders that were generated by cycle sequencing and that terminate at the corresponding nucleotide positions. The DNA sequence that was protected from DNase I digestion by LasR is shown in brackets.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Bacterial strains used in this study are listed in Table 1. Bacteria were freshly plated to begin each experiment. Bacteria were cultured in either lysogeny broth (LB; Lennox formulation) or tryptic soy broth (TSB; Becton Dickinson) as indicated in the text. For experiments to investigate the effects of various sulfur sources, bacteria were grown in a modified form of the sulfur-free salts medium (MSFM) described by Kertesz et al. (41). This medium contained 50 mM Tris, pH 7.3, 25 mM sodium succinate, 20 mM NH4Cl, 0.5 mM potassium phosphate, 0.5 mM MgCl2, 2 μM FeCl3, 1 μM MnCl2, 1 μM CaCl2, 1 μM ZnCl2, and 50 μg/ml of each proteinogenic amino acid except for cysteine and methionine. This medium was supplemented with specific sulfur sources as indicated in the text. Unless indicated otherwise, cultures were incubated at 37°C with shaking at 260 to 280 rpm. When necessary for selection or to maintain plasmids, cultures were supplemented with 200 μg/ml carbenicillin and/or 30 μg/ml gentamicin for P. aeruginosa and with 100 μg/ml ampicillin and/or 10 μg/ml tetracycline for Escherichia coli.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotype | Reference or source |
|---|---|---|
| E. coli DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U196 recA1 endA1 hsdR17 (rK− mK−) supE44 thi-1 gyrA relA1 | 66 |
| P. aeruginosa strains | ||
| PAO1 | Wild type | 67 |
| PJF-MT1 | mvaT deletion mutant derived from strain PAO1 | This study |
| PJF-MU1 | mvaU deletion mutant derived from strain PAO1 | This study |
| PJF-CB1 | cysB deletion mutant derived from strain PAO1 | This study |
| PAO1-PpqsRtc | PAO1 harboring chromosomal pqsR′-lacZ | This study |
| PJF-CB1-PpqsRtc | PJF-CB1 harboring chromosomal pqsR′-lacZ | This study |
| Plasmids | ||
| pECP8 | ptac-lasR, Ampr | 30 |
| pLP170 | lacZ transcriptional fusion vector | 68 |
| pJF-NadA2 | nadA′-lacZ transcriptional fusion | This study |
| pJF-NadA2-LasRmut | nadA′-lacZ fusion with mutated LasR binding site | This study |
| pMWC1003 | pqsR′-lacZ transcriptional fusion | 30 |
| pMWC1003-LasRmut | pqsR′-lacZ fusion with mutated LasR binding site | This study |
| pEX18Ap | Suicide vector for P. aeruginosa | 69 |
| pΔmvaT-suc | mvaT deletion suicide vector | This study |
| pΔmvaU-suc | mvaU deletion suicide vector | This study |
| pΔcysB-suc | cysB deletion suicide vector | This study |
| pHERD20T | E. coli-P. aeruginosa expression vector | 57 |
| pJF60 | PBAD-cysB on pHERD20T | This study |
| pJF-Tn7T-pqsR′Tc | pqsR′-lacZ transcriptional fusion plasmid for chromosomal integration | 70 |
| pJF-Tn7T-cysH′Tc | cysH′-lacZ transcriptional fusion plasmid for chromosomal integration | This study |
| pJF1003 | pqsR′-lacZ tacp-lasR | This study |
| pACYC184 | Cloning vector for E. coli | 71 |
| pACYC-PBAD | araC, PBAD | This study |
| pJF52 | araC, PBAD-cysB | This study |
Purification of LasR.
LasR was purified using the procedure described by Schuster et al. (42). E. coli strain DH5α carrying LasR expression plasmid pECP8 was grown overnight in LB medium with ampicillin. Cells were subcultured in 1 liter of prewarmed LB containing 50 mM morpholinepropanesulfonic acid (MOPS), pH 7, 100 μg/ml ampicillin, and 5 μM 3-oxo-C12-HSL to an optical density at 600 nm (OD600) of 0.08, and the culture was incubated at 37°C. When the OD600 reached 0.05, the culture was chilled to 20°C, supplemented with isopropyl-β-d-thiogalactoside (IPTG) to 0.4 mM, and incubated at 17°C overnight. Cells were then harvested by centrifugation and stored at −70°C. For protein purification, cells from 500 ml of culture were thawed and suspended in 8.5 ml of LasR purification buffer (25 mM Tris-HCl, 150 mM NaCl, 1 mM dithiothreitol [DTT], 1 mM EDTA, 10% glycerol, and 0.05% Tween 20). Cells were disrupted by passing the suspension through a French pressure cell at 16,000 lb/in2. The remaining steps in the purification procedure were carried out exactly as described by Schuster et al. (42). Fractions containing LasR were >95% pure as judged by SDS-PAGE. Protein concentrations were determined by the Bradford assay using reagents from Bio-Rad.
EMSA and DNase I protection assays with LasR.
Electrophoretic mobility shift assays (EMSA) and DNase I protection assays were performed essentially as described by Schuster et al. (42). DNA probes for EMSA were amplified by PCR using primers listed in Table 2. DNA fragments were purified from an agarose gel and labeled on both ends using [γ-32P]ATP (PerkinElmer) and T4 polynucleotide kinase. DNA binding reactions were carried out in a buffer containing 20 mM Tris-HCl, pH 7.6, 50 mM KCl, 1 mM EDTA, 1 mM DTT, 5% glycerol, 100 μg/ml bovine serum albumin (BSA), 15 μg/ml sheared salmon sperm DNA, and 5 μM 3-oxo-C12-HSL. For binding reactions, approximately 0.06 to 0.12 fmol of radiolabeled DNA probe was incubated either with or without the addition of 0.27 pmol of purified LasR in a total volume of 20 μl for 20 min at room temperature. Aliquots of binding reaction mixtures were then electrophoresed on nondenaturing 5% acrylamide gels in 0.5× Tris-borate-EDTA (TBE) buffer. Gels were visualized by autoradiography.
TABLE 2.
Primers and probes used in this study
| Primer or probe function and name | Sequence (5′−3′)a |
|---|---|
| EMSA and DNase I protection | |
| PQSR-PRR1 | AAGCGCAAGGGCTGGCGT |
| PQSR-PRF2 | CCGTTCCTGGCTCGGCTC |
| PQSR-PRR4 | ACCAAGGGCCTAATACCAAA |
| PQSR-PRF4 | ACTGACGATTGCAGGTTTCG |
| PQSR-F52 | CCGGTAAATATCCGGATT |
| P cysH F | GCGGGAATGGTAGCAAAGG |
| P cysH R | CATCAGCACGTCGTAGTCCG |
| lasIP-F | TTTTGGGGCTGTGTTCTCTC |
| lasIP-R | CACTTGAGCACGCAACTTGT |
| kynA promoter fwd | GAGTGAGGGCAAGGACACAT |
| kynA promoter rev | CGCGAGTGATCCGAAATTCG |
| Primer extension analysis | |
| NadA P EXT2 | TCTCCTGCTCGCTCAACACACGG |
| PQSR-PRF4 | ACTGACGATTGCAGGTTTCG |
| Cloning lacZ fusions and mutational analysis | |
| NadA-F2 | AAAAAAGCTTTAGATAGCGCCCGAGTGTTT |
| NadA-R1 | AAAAATCTAGACTCCTGCTCGCTCAACAC |
| PQSRP mutLasR FWD | AGAACAAAAGTCATTAGTTTCGGTCGTTAGACCTACGCGAC |
| PQSRP mutLasR REV | CGTAGCCACCGGCCAGGCC |
| LasR tac F | AAAAGAATTCTAAATCACTGCATAATTCGTGTCG |
| LasR tac R | GCGATCTGTCTATTTCGTTCATCC |
| CYSH UP | AAAACTGCAGAGAGAATCACCACCTGGAAGCG |
| CYSH DOWN | AAAAAAGCTTGCGGGAATGGTAGCAAAGG |
| DNA affinity chromatography | |
| PQSR PRR10B | GTGCGAGGGTGGAATCGG |
| PQSR PRF1 | ATCCCGGCATCCGTACCG |
| Generation of mutant strains | |
| MVAT P1.2 | AAAACTGCAGTCGGTCACGTAATGGCTGGTCGC |
| MVAT P2 | GGAACTGACTGGTTTAGCCGAGCAGGCGATATTCGTTGATCAGGGACATGTCAGG |
| MVAT P3 | CCTGACATGTCCCTGATCAACGAATATCGCCTGCTCGGCTAAACCAGTCAGTTCC |
| MVAT P4 | AAAACTGCAGCCGCTGGTGCATGTC |
| MVAT SEQ1 | CGCGGTTTACTTACAGTTTCGC |
| MVAT SEQ2 | CTATTCGCTGGAGACTTGA |
| MVAU P1 | AAAACTGCAGAACGACCGGTCACTCGTTGCG |
| MVAU P2 | GGCTTAGCGTTGCAGCCAGGATTCGAACTCGGCAAGTTTGGACATTCG |
| MVAU P3 | CGAATGTCCAAACTTGCCGAGTTCGAATCCTGGCTGCAACGCTAAGCC |
| MVAU P4 | AAAACTGCAGTTCGAGAAGCAAGGGACGATCGC |
| MVAU SEQ1 | ACTTATCCTTTGCCGCGGACAC |
| MVAU SEQ2 | GTCGCAGAATTGATCTGGCGAGG |
| CYSB P1 | TTTTGGATCCCTATGCGGACCAACTGCTG |
| CYSB P2 | GATCAGTAGACCGGCAGTTCGATAACCTCCCAGATATAGCGC |
| CYSB P3 | GCGCTATATCTGGGAGGTTATCGAACTGCCGGTCTACTGATC |
| CYSB P4 | TTTTGGATCCCTATCGTTTCATCGACCAGGTCC |
| CYSB SEQ2 | CAATCTCGACATGCTCTGAGGG |
| CYSB SEQ3 | TATACCTTCGGTTCTTGATCGC |
| Quantitative real-time PCR | |
| PQSR RT FWD2 | ATTGCGCGGACCCTTGTTGA |
| PQSR RT REV3 | GGACGGCTACAAGGTCGAAC |
| NADA RT FWD3 | GTTTCGGCAACCAGCATCCG |
| NADA RT REV3 | GGCATCAGCACACGCTTCTC |
| CLPX RT1 | CCTGTGCAATGACATCATCC |
| CLPX RT2 | AGGATGGTGCGGATCTCTTT |
| RPLU RT1 | GGTGGCAAGCAGCACAAAGTCACCG |
| RPLU RT2 | GCGGACCTTGTCGTGACGGCCGTGG |
| Cloning CysB expression plasmids and controls | |
| CYSB EX FWD | AAGCTTCAGCAATTGCGCTATATCTG |
| CYSB EX REV | AAAACTGCAGACACCGTGTTCATGAGCTTCC |
| HERD Pbad F | CTACTTCACCTATCCTGC |
| HERD Pbad R | CCCAGTCACGACGTTG |
Underlined sequence denotes a restriction site utilized for cloning. Nucleotides shown in bold are altered compared to the original template sequence.
To generate probes for DNase I protection assays, oligonucleotide primers PQSR-PRF4 and PQSR-PRR4 (Table 2) were labeled on the 5′ end using [γ-32P]ATP and T4 polynucleotide kinase. A single radiolabeled primer was used in PCRs to generate DNA fragments that were labeled on a single strand, and the resulting fragments were gel purified. Approximately 0.2 pmol of each fragment was incubated with 10 pmol of LasR and 5 μM 3-oxo-C12-HSL in the same buffer used for EMSA but in a final volume of 120 μl. Reaction mixtures were incubated for 20 min at room temperature; then 0.24 U of RQ1 DNase (diluted to 0.04 U/μl in 25 mM Tris-HCl, pH 7.6, 30 mM MgCl2, 30 mM CaCl2; Promega) was added, and reaction mixtures were incubated for another 8 min. Next, 25 μl of stop solution (3 M ammonium acetate, 0.25 M EDTA, 15 μg/ml sheared salmon sperm DNA) was added to each reaction mixture, and then DNA was precipitated by adding 370 μl of ethanol. DNA sequencing ladders were generated with radiolabeled primers PQSR-PRF4 and PQSR-PRR4 using a Thermo Sequenase cycle sequencing kit (USB/Affymetrix). Cycle sequencing reactions were performed using dideoxynucleoside triphosphate (ddNTP) termination mixes that contained dGTP instead of 7-deaza-dGTP. DNase I digestion products and sequencing ladders were separated on denaturing 8% polyacrylamide gels and visualized by autoradiography.
Primer extension analysis.
To isolate RNA for primer extension analyses, P. aeruginosa strain PAO1 was subcultured in LB medium to an OD660 of 0.05 and incubated at 37°C for 3 h. Cells were harvested by centrifugation and treated with RNAprotect bacteria reagent (Qiagen). Cells were then incubated in lysis buffer (30 mM Tris, pH 8, 1 mM EDTA, 10 mg/ml lysozyme, 2 mg/ml proteinase K) for 5 min at room temperature, and RNA was isolated using TRIzol reagent (Life Technologies). For extension reactions, oligonucleotide primers were labeled on the 5′ end using [γ-32P]ATP and T4 polynucleotide kinase. Approximately 160 fmol of radiolabeled primer was mixed with 80 μg of RNA in 20 μl of a buffer containing 167 mM HEPES, pH 7.5, 1 M NaCl, and 0.33 mM EDTA. Mixtures were heated to 95°C for 1 min and then incubated at 50°C for 30 min. Next, nucleic acids were collected by ethanol precipitation and suspended in diethyl pyrocarbonate (DEPC)-treated water. The entire volume was used for reverse transcription reactions using SuperScript III reverse transcriptase (Life Technologies). Each reaction mixture contained 1× first-strand buffer, 5 mM DTT, 5% dimethyl sulfoxide (DMSO), a 0.5 mM concentration of each deoxynucleoside triphosphate (dNTP), 2 U/μl RNase OUT (Life Technologies), and 125 U of SuperScript III reverse transcriptase in a total volume of 25 μl. Reaction mixtures were incubated at 55°C for 90 min. Then, nucleic acids were isolated by extraction with 1:1 phenol-chloroform, followed by ethanol precipitation. DNA sequencing ladders were generated with the same radiolabeled oligonucleotides used for extension reactions, using a Thermo Sequenase cycle sequencing kit as described for DNase I protection assays. Extension products and sequencing ladders were separated on denaturing 8% polyacrylamide gels and visualized by autoradiography.
Construction of nadA′-lacZ fusions and β-galactosidase (β-Gal) assays.
To generate an nadA′-lacZ reporter plasmid, a 738-bp fragment from +78 to −661 relative to the nadA translational start site was amplified by PCR. For this amplification, the oligonucleotide primers contained XbaI and HindIII restriction sites, and strain PAO1 chromosomal DNA was used as a template. The amplified fragment was digested with XbaI and HindIII, purified from an agarose gel, and ligated into pLP170 digested with the same enzymes to produce reporter plasmid pJF-NadA2. To construct a reporter plasmid that contained mutations in the LasR binding site, plasmids pJF-NadA2 and pMWC1003 were each amplified by PCR with oligonucleotide primers PQSRP mutLasR FWD and PQSRP mutLasR REV. These primers contained alterations from the original nucleotide sequence to generate the desired mutations. The amplified fragments were purified from an agarose gel and ligated to produce reporter plasmids pJF-NadA2-LasRmut and pMWC1003-LasRmut. Plasmids were transformed into P. aeruginosa by electroporation.
To assay for β-Gal activity, washed cells from overnight cultures were used to inoculate 10-ml subcultures of LB supplemented with carbenicillin to an OD660 of 0.05. Cultures were incubated at 37°C for 6 h, and then aliquots of cultures were diluted 1:10 in fresh LB medium and assayed for β-Gal activity in duplicate (43).
DNA affinity chromatography.
DNA affinity chromatography experiments were based on the procedure of Imperi et al. (44) with some modifications. A DNA fragment corresponding to −177 to −570 relative to the pqsR transcriptional start site was amplified by PCR using oligonucleotide primers PQSR PRR10B, which was labeled with biotin at the 5′ end, and PQSR PRF1. The amplified fragment was purified using a QIAquick PCR purification kit (Qiagen). The purified, biotin-labeled fragment was coupled to Dynabeads M-280 Streptavidin (Life Technologies) as directed by the manufacturer. To prepare cell lysates, washed cells from an overnight culture of P. aeruginosa strain PAO1 grown in LB medium were used to inoculate cultures of fresh LB medium to an OD660 of 0.05. Cultures were incubated at 37°C for either 2.5 h or 8 h, and then cells from 200 to 300 ml of culture were harvested by centrifugation and stored at −70°C. Next, cell pellets were thawed, suspended in 10 ml of lysis buffer (50 mM Tris, pH 8, 100 mM NaCl, 0.5 mM EDTA, 2 mg/ml lysozyme), and incubated at room temperature for 15 min. Cells were then disrupted by sonication, and the resulting cell lysates were cleared by centrifugation at 20,000 × g for 30 min at 4°C. The total protein concentration in the cleared lysates was estimated using the Bradford assay.
For DNA affinity chromatography experiments, aliquots of either uncoupled or DNA-coupled Dynabeads were equilibrated in binding buffer (50 mM Tris, pH 8, 100 mM NaCl, 0.5 mM EDTA, 10% glycerol, 0.05% Triton X-100). The Dynabeads were then incubated for 1 h at room temperature, with gentle mixing, in binding buffer with 1 mM DTT, 15 μg/ml sheared chromosomal DNA from P. aeruginosa strain PAO1, and cleared cell lysates (0.6 mg of total protein for every 10 μl of Dynabead suspension used). Next, the Dynabeads were collected by magnetic separation and washed three times with 10 volumes of binding buffer. Bound proteins were then eluted by washing the Dynabeads three times with two volumes of binding buffer containing 2 M NaCl. The eluted proteins were concentrated and exchanged into a buffer containing 50 mM Tris, pH 8, 50 mM NaCl, and 0.5 mM EDTA using Amicon Ultra centrifugal filters (3,000-Da-molecular-mass cutoff). The proteins were then separated by SDS-PAGE on a 15% polyacrylamide gel. Individual protein bands were excised from the gel and identified by mass spectrometry (performed by the Proteomics Core Facility at the University of California, Davis).
Generation of mutant strains.
Mutant alleles for mvaT, mvaU, and cysB were generated using PCR as described elsewhere (21). Alleles were constructed to contain in-frame deletions in the coding DNA sequence corresponding to amino acids 9 to 121 for mvaT (91% of protein sequence), 8 to 111 for mvaU (90% of protein sequence), and 13 to 318 for cysB (94% of protein sequence). The mutated fragments were also designed to contain the following at each end: PstI sites for mvaT and mvaU and BamHI sites for cysB. Each fragment and the vector plasmid pEX18Ap were digested with the appropriate restriction enzyme, purified from an agarose gel, and ligated to produce suicide plasmids pΔmvaT-suc, pΔmvaU-suc, and pΔcysB-suc. These plasmids were used to transfer the mutant alleles onto the chromosome of P. aeruginosa strain PAO1 as described elsewhere (21). Potential mutants were screened by PCR using appropriate flanking primers, and mutants were further confirmed by determining the DNA sequence of the PCR products.
qRT-PCR analysis.
To isolate RNA for quantitative real-time PCR (qRT-PCR) analyses, washed cells from overnight cultures of P. aeruginosa were used to inoculate 10-ml cultures of LB medium to an OD660 of 0.05. Cultures were incubated at 37°C for 6 h. Then, 400-μl samples of each culture were treated with RNAprotect bacteria reagent (Qiagen), and RNA was isolated from each sample using an RNeasy Midi kit according to the manufacturer's protocol (Qiagen). Isolated RNA samples were extracted twice with 1:1 phenol-chloroform and once with chloroform and then collected by ethanol precipitation. Next, the RNA was suspended in nuclease-free water and treated with RQ1 DNase (Promega) according to the manufacturer's protocol. Following DNase treatment, samples were extracted once with 1:1 phenol-chloroform and once with chloroform and then collected by ethanol precipitation. The purified RNA was suspended in nuclease-free water and stored at −70°C. The concentration of RNA in each sample was determined using a NanoDrop ND-1000 spectrophotometer. The integrity of the RNA in each sample was assessed by electrophoresis on an agarose gel.
To generate cDNA for qRT-PCR analysis, 2 μg of total RNA was primed with random hexamers (approximately 60% GC content). Superscript III reverse transcriptase (Life Technologies) was used to synthesize cDNA according to the manufacturer's protocol, including the addition of 40 U of RNaseOUT RNase inhibitor (Life Technologies) to each reaction mixture. Reaction mixtures were incubated at 55°C for 1 h as directed by the manufacturer. For real-time PCR analysis, cDNA preparations were diluted either 1:50 or 1:60 in nuclease-free water. PCRs were performed on a Bio-Rad CFX96 real-time system using FastStart SYBR green master mix (Roche) and the following program: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 58°C for 20 s, and 72°C for 20 s, with a final step at 95°C for 10 s, followed by a melt curve analysis. The oligonucleotide primers used to detect expression of each gene of interest are listed in Table 2. The efficiency and limits of detection for each primer set were determined by performing real-time PCR with serial dilutions of strain PAO1 chromosomal DNA in 100 μg/ml Saccharomyces cerevisiae tRNA (Ambion) as a template (45). Relative gene expression was calculated using the method of Pfaffl (46), with rplU as a reference gene for comparison. At least two technical replicates were performed for each cDNA sample analyzed.
PQS extraction and quantification.
To analyze PQS production, 10-ml cultures of freshly prepared medium were inoculated with several colonies of freshly grown P. aeruginosa cells and incubated at 37°C. Washed cells from starter cultures were used to inoculate 10-ml subcultures to an OD660 of 0.05, which were incubated at 37°C for either 6 h or 18 h as indicated in the text. Next, 0.3-ml samples of each culture were extracted with 0.9 ml of acidified ethyl acetate as described elsewhere (47). One half of the resulting organic phase was evaporated to dryness at 37°C and reconstituted in 50 μl of 1:1 acidified ethyl acetate-acetonitrile. Samples of each extract were analyzed by thin-layer chromatography (TLC) as described elsewhere (10). TLC plates were photographed under long-wave UV light, and the amount of PQS present was determined by comparison to known amounts of synthetic PQS using densitometry (47).
Construction of a CysB expression plasmid.
In order to generate a plasmid for controlled expression of CysB, a 1,156-bp DNA fragment, which began at the second codon of cysB and ended 175 bp past the stop codon, was amplified by PCR. The oligonucleotide primers for this amplification were designed to contain a single PstI site downstream from the stop codon. Vector plasmid pHERD20T was digested with NcoI and then treated with Pfu Turbo DNA polymerase (Agilent Technologies) to generate blunt ends. The plasmid was then digested with PstI. The prepared plasmid DNA was ligated with the cysB-containing fragment, which had also been digested with PstI, to produce the CysB expression plasmid pJF60. This construct placed the expression of cysB under the control of the inducible PBAD promoter.
Chromosomal lacZ fusion strains and β-Gal assays.
To construct a cysH promoter-lacZ fusion, a 448-bp DNA fragment corresponding to the cysH-thrH intergenic region (from −403 to +26 relative to the cysH translational start site) was amplified by PCR. The oligonucleotide primers for this amplification contained an HindIII restriction site downstream and a PstI restriction site upstream from the cysH translational start site. The amplified fragment was digested with HindIII and PstI, purified from an agarose gel, and ligated into pUC18T-mini-Tn7T-lacZ-Gm, which was digested with the same enzymes, to produce plasmid pJF-Tn7T-cysH′tc. To generate P. aeruginosa strains carrying either a pqsR′-lacZ fusion or a cysH′-lacZ fusion integrated on the bacterial chromosome at the attTn7 site, cells were transformed with plasmid pJF-Tn7T-pqsR′tc or pJF-Tn7T-cysH′tc as described elsewhere (48). The gentamicin resistance gene carried by the plasmid was also removed from the chromosome as described previously (48).
For assays to assess the effects of CysB overexpression, washed cells from overnight cultures were used to inoculate 10-ml subcultures of TSB, supplemented with either water or 0.1% arabinose, to an OD660 of 0.05. Cultures were incubated at 37°C for 6 h, and then aliquots of cultures were diluted 1:10 in fresh TSB medium and assayed for β-Gal activity in duplicate as described elsewhere (43). To test the effects of cysteine or its precursors on gene expression in rich medium, washed cells from overnight cultures were used to inoculate fresh TSB to an OD660 of 0.05, and 980-μl aliquots of inoculated medium was transferred to tubes and supplemented with either water, 5 mM l-cysteine, 5 mM Na2SO4, or 5 mM O-acetyl-l-serine. Cultures in tubes were incubated at 37°C with agitation as indicated in the text and assayed for β-Gal activity in duplicate. For β-Gal assays from bacteria cultured in defined medium, cells from overnight cultures of MSFM supplemented with 100 μM l-cysteine and 100 μM l-methionine were collected by centrifugation and suspended in unsupplemented MSFM. The washed cells were used to inoculate 10-ml cultures of MSFM supplemented with a 200 μM concentration of sulfur sources (as indicated in the legend to Fig. 6) to an OD660 of 0.05. Then, cultures were incubated at 37°C for 6 h and assayed for β-Gal activity. For some experiments washed cells were used to inoculate MSFM supplemented with 400 μM l-cysteine to an OD660 of 0.05, and then aliquots of inoculated medium were transferred to tubes and supplemented with potential CysB ligands. Cultures in tubes were incubated at 37°C as indicated and then assayed for β-Gal activity.
FIG 6.

Expression of both pqsR and cysH is controlled by CysB, but only cysH expression is altered in response to available sulfur sources. (A and B) P. aeruginosa strains carrying either a cysH′-lacZ or a pqsR′-lacZ fusion integrated on the bacterial chromosome were grown for 3 h or 6 h, respectively, in TSB medium supplemented with either water, 5 mM l-cysteine, 5 mM Na2SO4, or 5 mM O-acetyl-l-serine, and then cultures were assayed for β-Gal activity. (C and D) P. aeruginosa strains carrying either a cysH′-lacZ or a pqsR′-lacZ fusion integrated on the bacterial chromosome were grown for 6 h in MSFM supplemented with either l-cysteine, Na2SO4, or 1-hexyl sulfate as the sole sulfur source, and then cultures were assayed for β-Gal activity. (E) P. aeruginosa strains carrying either a cysH′-lacZ or a pqsR′-lacZ fusion integrated on the bacterial chromosome were grown for 6 h in MSFM supplemented with 400 μM l-cysteine as a sulfur source and additionally either water, 5 mM O-acetyl-l-serine, or 5 mM N-acetyl-dl-serine, and then cultures were assayed for β-Gal activity. Data for all β-Gal assays are presented in Miller units as the means ± standard deviations of results from duplicate assays from at least three separate experiments. (F) PQS production by strain PAO1 was analyzed after 10 h of growth in MSFM supplemented with either l-cysteine, Na2SO4, or 1-hexyl sulfate as the sole sulfur source. Data are presented as the means ± standard deviations of results from four independent experiments.
Purification of CysB.
To purify CysB, we overexpressed the native P. aeruginosa CysB protein in E. coli. This approach has also been used to purify the CysB proteins for Salmonella enterica serovar Typhimurium and Klebsiella aerogenes (49, 50). E. coli strain DH5α(pJF60) was grown overnight in LB medium with ampicillin. Cells were subcultured in 250 ml of LB with ampicillin to an OD600 of 0.08, and the culture was incubated at 37°C. When the OD600 reached 0.6, arabinose was added to the culture at 0.1% to induce cysB expression, and the culture was incubated at 28°C for 4 h. Then the cells were harvested by centrifugation, washed with sterile phosphate-buffered saline, and stored at −70°C.
To prepare a cell lysate, cells were thawed and suspended in 5 ml of ice-cold buffer A [50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 M (NH4)2SO4] containing 10 μl/ml HALT protease inhibitor cocktail (Thermo Scientific). Cells were disrupted by sonication, and the resulting lysate was centrifuged at 10,000 × g for 30 min at 4°C. The supernatant was collected, 291 mg/ml (NH4)2SO4 was then added, and the solution was incubated on ice with stirring for 30 min. Next, the sample was centrifuged at 10,000 × g for 30 min at 4°C to collect the precipitated material. The precipitate was suspended in 5 ml of 50 mM Tris-HCl, pH 7.5, and 1 mM EDTA and stored at −70°C.
For protein purification, the sample was thawed on ice and diluted 1:1 with 50 mM Tris-HCl, pH 7.5, 1 mM EDTA, and 2 M (NH4)2SO4. A 1-ml HiTrap Phenyl FF (low sub) column (GE Healthcare) was equilibrated with buffer A at room temperature, and then the sample was applied at a flow rate of 0.5 ml/min. Next, the column was washed with approximately 20 volumes of buffer A to remove unbound material. Bound proteins were eluted with a step gradient of 1 M to 0 M (NH4)2SO4 in 50 mM Tris-HCl (pH 8.0)–1 mM EDTA. Each step was eluted with a volume of 1 ml, with the (NH4)2SO4 concentration decreasing in 0.1 M increments. CysB eluted in fractions containing 0.3 to 0.1 M (NH4)2SO4, as determined by SDS-PAGE. These fractions were pooled, and glycerol was added to a final concentration of 10% before storage at −70°C. To complete the purification procedure, the sample was thawed and diluted 1:1 with buffer B (20 mM Tris-HCl, pH 7.0, 1 mM EDTA, 1 mM DTT, 10% glycerol). The sample was then applied to a 5-ml HiTrap Heparin column (GE Healthcare), which had been equilibrated with buffer B, at a flow rate of 0.5 ml/min. The column was washed with approximately 3 volumes of buffer B to remove unbound material. Bound proteins were eluted with a 50-ml linear gradient of 0 to 1 M NaCl in buffer B at a flow rate of 1 ml/min, and 3-ml fractions were collected and placed on ice. The CysB protein eluted at approximately 0.7 to 0.8 M NaCl and appeared as a single band on Coomassie blue-stained SDS-PAGE gels (see Fig. S1 in the supplemental material). Protein concentrations in each fraction were determined by the Bradford assay using reagents from Bio-Rad.
EMSA with CysB.
DNA fragments for EMSA were amplified by PCR using primers listed in Table 2, and fragments were prepared and labeled with 32P as described for EMSA with LasR. DNA binding reactions were carried out in a buffer containing 40 mM Tris-HCl, pH 8.0, 90 mM NaCl, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml bovine serum albumin, and 11.25 μg/ml sheared salmon sperm DNA. Each binding reaction mixture contained approximately 1.75 to 3 pmol of the appropriate radiolabeled DNA fragment and amounts of CysB protein as indicated in Fig. 7 and 8 in a final volume of 20 μl. Reaction mixtures were incubated for 10 min at room temperature. Aliquots of binding reactions were then electrophoresed on nondenaturing 5% acrylamide gels in 0.5× TBE buffer. Gels were visualized by autoradiography. For EMSA to test the DNA binding activity of both CysB and LasR simultaneously, experiments were performed as described for LasR except that reaction mixtures contained 18 μg/ml sheared salmon sperm DNA and the addition of 25 mM NaCl.
FIG 7.
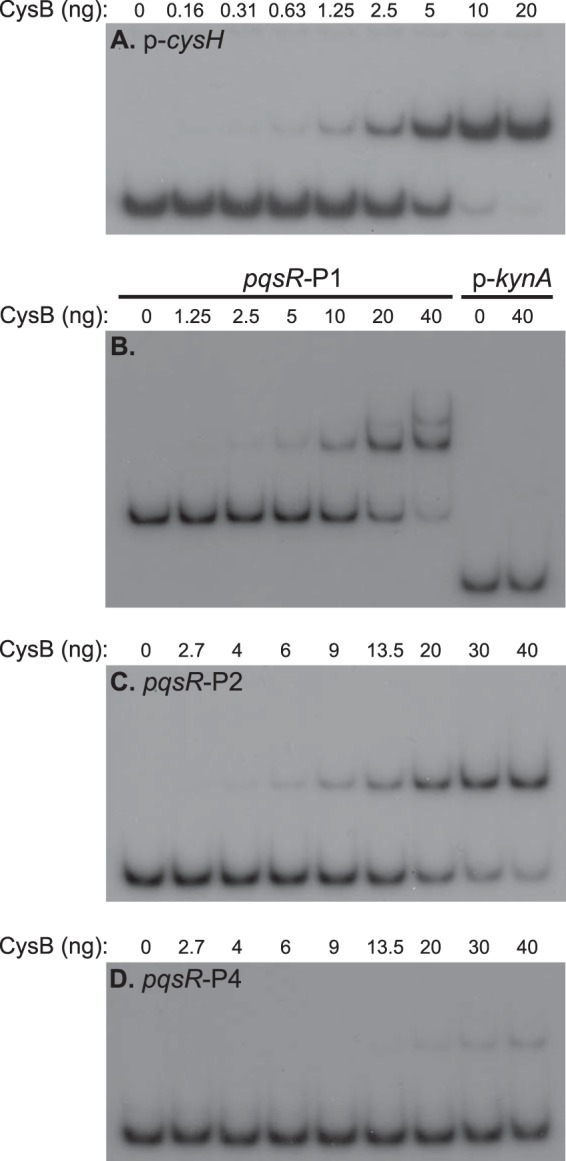
Purified CysB interacts with the pqsR and cysH promoter regions. Radiolabeled DNA fragments were incubated with purified CysB as described in Materials and Methods. Figure 1A shows the relative location of the pqsR promoter fragments. Binding reaction mixtures contained between 0.5 and 1 fmol of labeled DNA for each fragment tested. Equal aliquots of binding reaction mixtures were electrophoresed on nondenaturing 5% acrylamide gels, and the position of DNA fragments was visualized by autoradiography. The data presented are representative of at least three independent experiments.
FIG 8.

Purified CysB competes with LasR for binding to the pqsR promoter during in vitro DNA binding assays. (A and C) Radiolabeled DNA fragments were incubated with purified CysB and LasR as described in Materials and Methods. Binding reaction mixtures contained between 0.02 and 0.05 fmol of labeled DNA for each fragment tested. Equal aliquots of binding reaction mixtures were electrophoresed on nondenaturing 5% acrylamide gels, and the position of DNA fragments was visualized by autoradiography. The amount of each protein included in each binding reaction mixture and the molar ratio of CysB monomers to LasR monomers are shown above each lane. Black arrowheads show the position of CysB-DNA complexes, and white arrowheads show the position of LasR-DNA complexes. (B and D) The results from panels A and C were analyzed by densitometry using Bio-Rad ImageQuant software.
Two-plasmid system to analyze pqsR′-lacZ control by LasR and CysB in E. coli.
To generate a plasmid that carried both a pqsR′-lacZ fusion and tacp-lasR, a DNA fragment containing tacp-lasR was amplified by PCR using pECP8 as a template. The oligonucleotide primers used for this amplification added an EcoRI site upstream from the tac promoter. The amplified fragment was digested with EcoRI and ScaI and then ligated into pMWC1003, which had been digested with the same enzymes, to produce pJF1003. A second set of plasmids was generated for CysB expression and as a control. For these constructs, DNA fragments containing either araC and PBAD or araC and PBAD-cysB were amplified by PCR using either pHERD20T or pJF60 as a template. Vector plasmid pACYC184 was digested with DraI, treated with calf intestinal phosphatase, and purified from an agarose gel. The araC-PBAD or the araC-PBAD-cysB fragment was ligated into the prepared vector to produce control plasmid pACYC-PBAD or CysB expression plasmid pJF52, respectively. These plasmids were transformed into E. coli strain DH5α.
For β-Gal assays, cells from overnight cultures were used to inoculate 10-ml subcultures of LB, supplemented with ampicillin and tetracycline, to an OD600 of 0.08. Cultures were incubated at 37°C until the OD600 reached 0.2. Then IPTG was added to 1 mM, and 1-ml aliquots of cultures were transferred to tubes in the presence or absence of 20 nM 3-oxo-C12-HSL. Cultures in tubes were additionally supplemented with either water or 1% arabinose and incubated at 37°C with mixing for 2 h. Then samples were taken from each culture and assayed for β-Gal activity in duplicate (43).
RESULTS
LasR controls pqsR transcription from a single binding site but does not regulate nadA.
Previous studies showed that LasR positively regulates the transcription of pqsR and that LasR and 3-oxo-C12-HSL could induce pqsR expression in E. coli (9, 30). In addition, mutation of a consensus sequence for LasR binding, centered at 248 bp upstream from the pqsR transcriptional start site 1 (TS1) (Fig. 1A), caused a large decrease in pqsR transcription (39). With this in mind, we set out to directly determine the location of LasR binding sites in the pqsR promoter region. To investigate this, we expressed LasR in E. coli in the presence of 3-oxo-C12-HSL and purified the protein using the method of Schuster et al. (42). We then performed in vitro DNA binding experiments to localize LasR binding sites in the pqsR promoter. As a positive control, we tested the ability of our purified LasR to bind to the lasI promoter. LasR caused a shift in mobility of more than 90% of the labeled lasI promoter fragment at the concentration tested (Fig. 1B). There was also a small amount of a higher-molecular-weight complex visible, possibly due to the formation of LasR multimers. LasR did not bind to the promoter region of kynA, an unrelated gene (Fig. 1B). When we tested a 516-bp fragment of the pqsR-nadA intergenic region that included both of the identified pqsR transcriptional start sites and extended beyond the las-rhl box (Fig. 1A, pqsR-P1), we found that LasR did, indeed, interact directly with this fragment (Fig. 1B). Next, we divided this segment into two smaller overlapping DNA fragments (Fig. 1A, pqsR-P2 and pqsR-P3) and tested the ability of LasR to bind to each. LasR caused a shift in the mobility of fragment pqsR-P2, which included the predicted LasR binding consensus sequence but did not affect the mobility of fragment pqsR-P3, showing that LasR only binds beyond −471 bp relative to the pqsR translational start site (Fig. 1B). Finally, we performed DNase I protection assays to determine the exact site of the interaction between LasR and fragment pqsR-P2. This experiment showed that LasR protected a single 27-bp region from DNase I digestion, which completely overlapped the previously identified putative las-rhl box (Fig. 1C). When we repeated the experiment with a DNA fragment that was radiolabeled on the opposite strand, we observed a nearly identical region of protection (data not shown). Together, these experiments indicated that LasR interacts with a single binding site in the pqsR-nadA intergenic region.
This binding site placed bound LasR in closer proximity to the divergently transcribed nadA gene (centered at bp −221 relative to the nadA translational start site) than to the pqsR transcriptional start site TS1. The nadA gene is predicted to encode a protein involved in the de novo biosynthesis of NADH, and it has not been linked to PQS synthesis. We decided to investigate whether LasR binding has any effect on nadA transcription since it is known that LasR can regulate divergently transcribed genes from a single binding site (51). First, we mapped the transcriptional start site of nadA by primer extension analysis. We identified a single start site that was verified by analyses with two separate primers in independent experiments. This site corresponded to a C residue located at bp −141 relative to the nadA translational start site (Fig. 2A). Relative to this transcriptional start site, the las-rhl box was centered at bp −81. To analyze the effects of LasR on both nadA and pqsR transcription, we constructed lacZ fusions to the promoter of each gene and then made five base pair substitutions in the las-rhl box on each fusion (Table 2 gives specific mutations). These mutations altered residues that were identified as highly conserved in LasR binding sequences (38), and the binding of purified LasR-3-oxo-C12-HSL to fragment pqsR-P2 was completely abolished when these mutations were present (Fig. 2B). Expression from the nadA′-lacZ fusion was almost identical with either the wild-type or mutated las-rhl box present, indicating that this regulatory element does not control nadA (Fig. 2C). In contrast, there was a large decrease in expression from the pqsR promoter when the las-rhl box was mutated (Fig. 2D). These results showed that despite the fact that LasR binds in closer proximity to the nadA transcriptional start site, it does not regulate the transcription of nadA under these conditions. Instead, the primary function of LasR binding to the pqsR-nadA intergenic region appears to be to control pqsR transcription.
FIG 2.
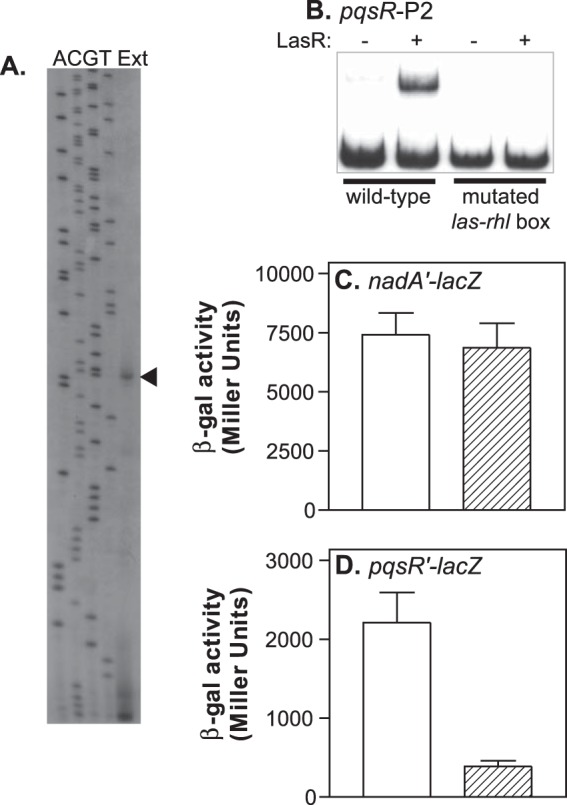
LasR does not affect the transcription of nadA. (A) Primer extension analysis of the nadA transcript (lane Ext). Lanes labeled A, C, G, and T contain DNA sequencing ladders that were generated by cycle sequencing and that terminate at the corresponding nucleotide positions. Figure 1A shows the relative location of the primer extension product identified by the arrowhead. (B) Radiolabeled DNA fragments corresponding to pqsR promoter fragment pqsR-P2 or pqsR-P2 with a mutated LasR binding site were incubated in the presence or absence of purified LasR and 5 μM 3-oxo-C12-HSL. Samples of binding reaction products were analyzed on nondenaturing acrylamide gels, and the position of DNA fragments was visualized by autoradiography. (C) P. aeruginosa strain PAO1 carrying either a plasmid containing the wild-type nadA promoter fused to lacZ (open bars) or a plasmid containing an nadA promoter with a mutated LasR binding site fused to lacZ (striped bars) was grown in LB medium for 6 h, and then cultures were assayed for β-Gal activity. (D) P. aeruginosa strain PAO1 carrying either a plasmid containing the wild-type pqsR promoter fused to lacZ (open bars) or a plasmid containing a pqsR promoter with a mutated LasR binding site fused to lacZ (striped bars) was grown in LB medium for 6 h, and then cultures were assayed for β-Gal activity. β-Gal data are presented in Miller units as the means ± standard deviations of results from duplicate assays from at least three separate experiments.
Identification of other proteins that bind to the pqsR-nadA intergenic region.
Once we had established that LasR interacted with a single site 250 bp upstream from the pqsR TS1, we questioned how LasR could affect transcription from this distal binding site. Also, data from other studies showed that pqsR expression and PQS production are delayed but still occur in an lasR mutant strain (14, 18, 39). Therefore, we hypothesized that additional factors which affect pqsR transcription could be binding to the pqsR-nadA intergenic region. In an attempt to identify these factors, we utilized DNA affinity chromatography to isolate proteins that interact with this region. In these experiments, a 393-bp DNA fragment, spanning from −177 to −570 relative to the pqsR translational start site, was amplified by PCR using a biotinylated oligonucleotide primer and conjugated to streptavidin-coated magnetic beads. These were incubated with P. aeruginosa strain PAO1 cell lysates from either the logarithmic or stationary phase of growth. Next, the unbound proteins were washed away, and bound proteins were eluted, separated by SDS-PAGE, and identified by mass spectroscopy. We identified three proteins that were consistently isolated in separate experiments using this technique (Fig. 3). We did not isolate these proteins when we performed the experiment with unconjugated magnetic beads or when we used a different DNA fragment (pqsL promoter) (K. A. Tipton and E. C. Pesci, unpublished data). The first of these proteins, CysB, is an LysR-type transcriptional regulator that controls sulfur uptake and cysteine biosynthesis (40). The second and third proteins, MvaT and MvaU, are considered to be members of the H-NS family of nucleoid-associated proteins. These two proteins are similar to one another, bind to the same regions of the chromosome, and appear to have similar or overlapping functions (52). They can also affect gene expression and primarily act to silence transcription (52). In a previous study, data from experiments using chromatin immunoprecipitation coupled with microarray technology (ChIP-on-chip) suggested that MvaT and MvaU associate with the pqsR-nadA intergenic region in vivo (52). Based on their established regulatory functions, each of the proteins we identified could potentially influence pqsR or nadA expression.
FIG 3.

The proteins CysB, MvaT, and MvaU interact with the pqsR-nadA promoter region. A DNA fragment corresponding to −177 to −570 relative to the pqsR translational start site was used to capture proteins by DNA affinity chromatography as described in Materials and Methods. Cleared cell lysates from strain PAO1 grown in LB medium for either 2.5 h or 8 h were used in the procedure. Isolated proteins were separated by SDS-PAGE, and the gel was stained with SYPRO Ruby and visualized on a UV transilluminator. The image shown is a representative of four independent experiments.
CysB is a repressor of pqsR and PQS production.
To investigate the role of each of these proteins in the regulation of pqsR and nadA, we constructed isogenic, in-frame deletion mutants for the mvaT, mvaU, and cysB genes. We also tried to construct an mvaT mvaU double mutant strain but were unable to after multiple attempts. This result agrees with previous reports, which established that the presence of either MvaT or MvaU is necessary for cell viability in order to silence the expression of prophage genes encoded in the P. aeruginosa genome (52, 53). The phenotypes displayed by each mutant strain were similar to what has been described in previous studies (14, 54, 55). The mvaT and mvaU mutant strains both overproduced the blue-green pigment pyocyanin, and this occurred at a much greater degree in the mvaT mutant strain. The cysB mutant strain was able to grow on the defined medium MSFM supplemented with cysteine as the sole sulfur source but was unable to grow when supplied with either sulfate or 1-hexyl sulfate as a sulfur source (see Fig. S1 in the supplemental material). All of the mutant strains grew at a rate similar to that of the wild-type strain when cultured on rich medium.
Next, we isolated RNA from each P. aeruginosa strain grown to the early stationary phase of growth in LB medium and analyzed pqsR and nadA expression by qRT-PCR. We observed no changes in the relative expression level of nadA in the mvaT, mvaU, and cysB mutant strains compared to the level in the wild-type strain (Fig. 4A). This result indicated that nadA transcription is not affected by the removal of each of the respective proteins individually. We also did not observe any significant change in the relative expression of pqsR in either the mvaT or mvaU mutant strain (Fig. 4A). However, there was approximately a 2.5-fold increase in pqsR expression in the cysB mutant strain (Fig. 4A). We also analyzed PQS production by the cysB mutant strain. Somewhat surprisingly, we did not find any significant difference in the levels of PQS produced by the wild-type and cysB mutant strains when each was cultured in LB medium (data not shown). However, when we examined PQS levels during growth in TSB medium, we found that the cysB mutant produced almost 2-fold more PQS than the wild-type strain (Fig. 4B). We also observed a similar increase in PQS production by the cysB mutant in a third growth medium, peptone-tryptic soy broth (56) (data not shown). It is unclear why this phenotype was not observed during growth in LB medium, but perhaps the availability of PQS precursors or differential regulation of PQS biosynthetic genes by factors other than PqsR was able to make the regulatory effects of CysB less apparent. Overall, these data showed that CysB can act as a repressor of both pqsR expression and PQS production.
FIG 4.
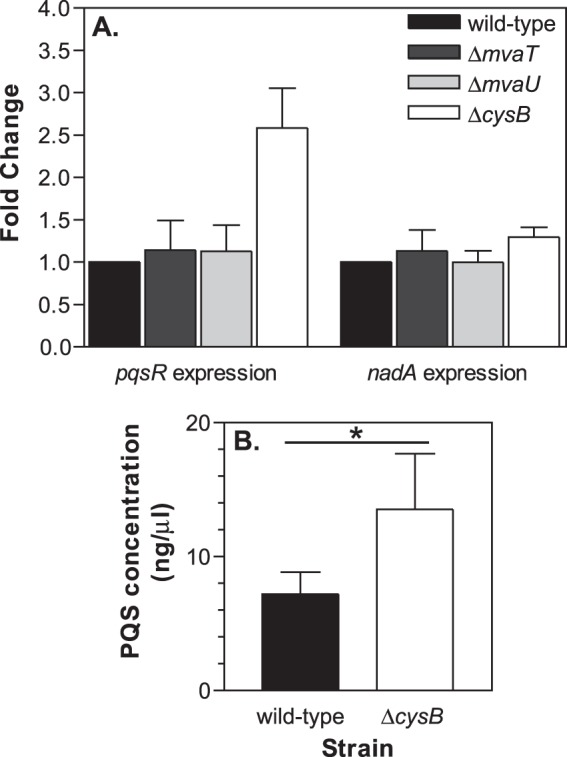
CysB can repress pqsR expression and PQS production. (A) The expression of pqsR and nadA was analyzed by qRT-PCR using total RNA isolated from P. aeruginosa strains PAO1 (wild type), PJF-MT1 (ΔmvaT), PJF-MU1 (ΔmvaU), and PJF-CB1 (ΔcysB). RNA was isolated from each strain after 6 h of growth in LB medium. Results are shown as the average fold change, relative to the expression in strain PAO1 ± standard deviations from at least three separate experiments. (B) PQS production by strains PAO1 (black bar) and PJF-CB1 (white bar) was analyzed after 18 h of growth in TSB medium. Data are presented as the averages ± standard deviations of results from six independent experiments. *, P < 0.01, as determined by Student's t test.
We obtained more support for this conclusion when we complemented the cysB mutant strain by supplying an intact cysB gene on a multicopy plasmid. We cloned cysB onto the E. coli-P. aeruginosa expression vector pHERD20T (57). This placed cysB under the control of the PBAD promoter, allowing cysB expression to be induced by supplementing cultures with l-arabinose. To examine the effects on pqsR expression, we transformed the vector and cysB expression plasmid pJF60 into strains PAO1-PpqsRtc (wild type) and PJF-CB1-PpqsRtc (ΔcysB), that each harbor a pqsR′-lacZ transcriptional fusion integrated onto the chromosome at the attTn7 site. In the strains carrying the vector plasmid, the cysB mutant strain produced approximately 2.3-fold more β-Gal activity than the wild-type strain [1,148 ± 55 units from strain PJF-CB1-PpqsRtc(pHERD20T) versus 491 ± 17 units from strain PAO1-PpqsRtc(pHERD20T)] (Fig. 5A). When cysB was supplied on pJF60, the β-Gal activity produced by both the wild-type and cysB mutant strains decreased (Fig. 5A). Each strain produced approximately an equivalent amount of β-Gal activity in the absence of arabinose (Fig. 5A), and this level of activity was slightly lower than the level of activity produced by the wild-type strain carrying the vector plasmid. This result showed that the cysB mutant was fully complemented by cysB expression from pJF60 even in the absence of inducer, apparently due to a basal level of expression from the PBAD promoter. When arabinose was added to cultures of strains carrying pJF60, the β-Gal activity decreased further, to approximately 20% of the activity produced by the wild-type strain carrying the vector plasmid (Fig. 5A). These data demonstrated that CysB can repress pqsR transcription.
FIG 5.
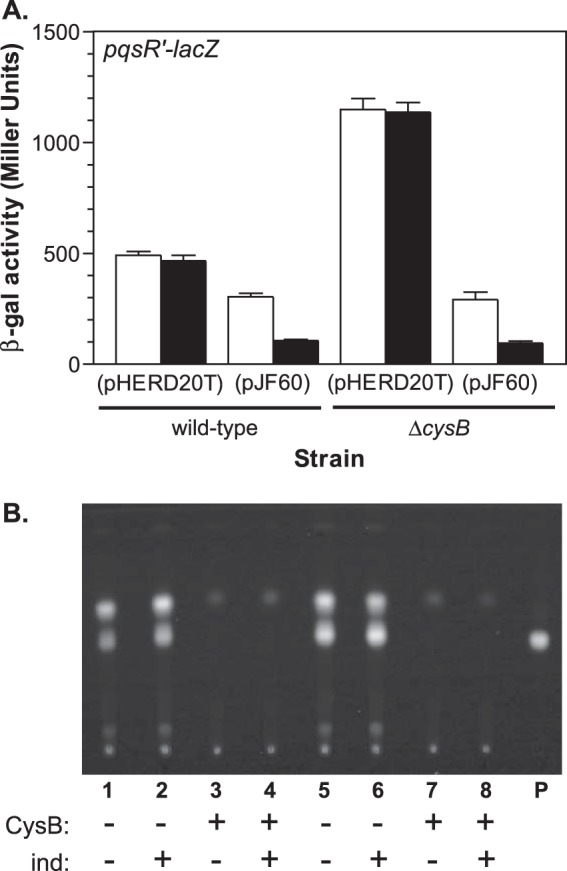
Overexpression of CysB represses pqsR expression and inhibits PQS production. (A) Strains PAO1-PpqsRtc (wild type) and PJF-CB1-PpqsRtc (ΔcysB), that each contain a pqsR′-lacZ fusion integrated on the bacterial chromosome, were transformed with either the vector control plasmid pHERD20T or CysB expression plasmid pJF60. Each strain was cultured for 6 h in TSB medium with the addition of either water (open bars) or 0.1% arabinose (black bars) to induce CysB expression, and then cultures were assayed for β-Gal activity. Data are presented in Miller units as the means ± standard deviations of results from duplicate assays from at least three separate experiments. (B) Organic extracts from P. aeruginosa strains carrying either the vector plasmid pHERD20T or CysB expression plasmid pJF60 and grown for 6 h in TSB medium in either the presence or absence of 0.1% arabinose (ind) were analyzed by TLC to detect PQS production. Lanes show results of extracts from the following strains: lanes 1 and 2, PAO1(pHERD20T); lanes 3 and 4, PAO1(pJF60); lanes 5 and 6, PJF-CB1(pHERD20T); lanes 7 and 8, PJF-CB1(pJF60); lane P, 50 ng of synthetic PQS.
We observed a similar but more striking trend when we analyzed PQS production in the presence of the CysB expression plasmid. The cysB mutant PJF-CB1(pHERD20T) appeared to produce a slightly increased amount of PQS (Fig. 5B, lanes 5 and 6) compared to the wild-type PAO1(pHERD20T) (Fig. 5B, lanes 1 and 2), and this was not affected when cultures were supplemented with arabinose. However, when we transformed either strain with the CysB expression plasmid, PQS production was completely abolished, even in the absence of inducer (Fig. 5B, wild type, lanes 3 and 4, and cysB mutant, lanes 7 and 8). These results showed that even the basal level of CysB expression from pJF60 could significantly impact PQS production. We should also note that we obtained nearly identical results when we analyzed pqsR′-lacZ activity and PQS production in either LB medium (data not shown) or TSB medium (Fig. 5). Taken together, these data provide clear evidence that CysB can repress both pqsR expression and PQS production.
Expression of pqsR is not affected by cysteine availability.
Since CysB has an established regulatory role in controlling cysteine biosynthesis, we next investigated the role of cysteine and sulfur source availability on pqsR expression and PQS production. Studies on CysB in several enteric bacteria showed that the primary role of CysB was to activate the expression of genes necessary for sulfur uptake in response to cysteine starvation (40). This regulatory activity occurs in response to the ligand N-acetylserine (NAS). NAS is derived from the cysteine precursor O-acetylserine (OAS), which accumulates when there is not enough sulfur available for cysteine biosynthesis, and acts as a coinducer with CysB to stimulate sulfate uptake and cysteine production (40, 50). Similarly, in P. aeruginosa and Pseudomonas putida, CysB appears to act as a master regulator of sulfur assimilation pathways although its exact regulatory functions are not clearly defined (58, 59). So to examine the influence of cysteine availability, we compared the expression of pqsR′-lacZ and cysH′-lacZ fusions in response to various sulfur sources as well as OAS and NAS. The cysH gene encodes a 5′-adenylylsulfate reductase, an enzyme that is involved in sulfate assimilation and cysteine biosynthesis (60). It is expected to be part of the CysB regulon based on studies in other Gram-negative bacteria (40, 60). In addition, cysH is located downstream from the cysB gene in P. aeruginosa but on the opposite strand.
First, we analyzed pqsR and cysH expression in TSB medium supplemented with either excess cysteine or cysteine precursors. We observed that the activity produced by the cysH′-lacZ fusion in the wild-type strain decreased when cultures were supplemented with cysteine but not when the cultures were supplemented with sulfate or OAS (Fig. 6A). Additionally, cysH′-lacZ expression was much lower in the cysB mutant strain, indicating that CysB regulates cysH, and the β-Gal activity in this strain remained the same in the presence of excess cysteine. In contrast, expression of the pqsR′-lacZ fusion was not affected by the addition of cysteine to cultures of either the wild-type or cysB mutant strain (Fig. 6B). We also examined cysH and pqsR expression in the wild-type strain during growth on a chemically defined growth medium with various sulfur sources. There was a 1.5-fold increase in β-Gal activity produced by the cysH′-lacZ fusion during growth on sulfate compared to the activity produced during growth on cysteine and a 2-fold increase in activity during growth on an alternative sulfur source, the organic sulfur compound 1-hexyl sulfate (Fig. 6C). However, we observed only minor changes in pqsR′-lacZ expression and PQS production during growth on different sulfur sources (Fig. 6D and F). These experiments showed that CysB positively regulates cysH, and in keeping with its predicted role in cysteine biosynthesis, cysH was expressed at lower levels in the presence of excess cysteine and at higher levels in the absence of cysteine. Unlike what has been described in enteric bacteria, these regulatory effects do not appear to depend on the cysteine precursor OAS or the potential CysB ligand NAS since neither compound was able to stimulate increased cysH′-lacZ expression (Fig. 6A and E). Somewhat surprisingly, pqsR expression and PQS production did not change in response to cysteine availability, implying that CysB does not regulate pqsR expression in the same manner as sulfur acquisition and cysteine biosynthetic genes.
CysB directly interacts with the pqsR promoter region.
Having established that CysB can regulate both pqsR and cysH, we wanted to confirm that CysB could directly interact with the pqsR and cysH promoter regions. To do this, we overexpressed the native P. aeruginosa CysB in E. coli and purified the protein as described in Materials and Methods (see also Fig. S2 in the supplemental material). We then tested the ability of the purified P. aeruginosa CysB protein to interact with promoter DNA fragments using EMSA. First, we examined the ability of CysB to bind to the cysH promoter region. As expected, CysB caused a shift in the mobility of the cysH promoter fragment, with 20 ng of CysB causing a shift of at least 95% of the labeled probe (Fig. 7A). As a control, we found that 40 ng of CysB did not cause any change in the mobility of the kynA promoter (Fig. 7B). This indicated that CysB was interacting specifically with the cysH promoter fragment. Most interestingly, the pqsR promoter fragment pqsR-P1 (Fig. 7A) also exhibited a shift in mobility when CysB was added (Fig. 7B). In this case, 20 ng of CysB caused a shift of only approximately 50% of the labeled pqsR promoter fragment, suggesting that CysB has a lower affinity for the pqsR promoter than the cysH promoter. Additionally, we observed a second, higher-molecular-weight complex formed when larger amounts of CysB were incubated with the pqsR promoter (Fig. 7B). To begin to more clearly define the site or sites where CysB interacts with the pqsR promoter, we tested two smaller, nonoverlapping fragments of the pqsR promoter region (Fig. 1A shows relative positions). CysB interacted with pqsR promoter fragment pqsR-P2, causing the formation of a single, higher-molecular-weight complex (Fig. 7C). CysB also formed a single complex with the pqsR-P4 fragment, but this occurred only when more than 10 ng of CysB was included in the binding reaction mixtures (Fig. 7D). These results indicated that CysB can interact with two separate sites in the pqsR promoter region in vitro and provided an explanation for the two distinct CysB-DNA complexes formed with the pqsR-P1 fragment (Fig. 7B). However, because CysB appeared to bind to the pqsR-P4 fragment with low affinity, it is unclear whether this interaction is relevant in vivo or is simply due to nonspecific interactions between CysB and DNA sequences close to the pqsR transcriptional start sites. Aside from a preference for A-T-rich DNA sequences, CysB binding sites lack a strong consensus sequence (59, 61), and we were unable to clearly identify specific sequences in the pqsR or cysH promoter that might be recognized by CysB. Nevertheless, these data provide evidence that both cysH and pqsR are directly regulated by CysB in P. aeruginosa.
Activation of pqsR can be blocked by CysB.
Since part of our original goal was to investigate LasR-mediated control of pqsR and since we found that LasR and CysB interact with similar regions of the pqsR promoter, we wondered if CysB could interfere with the activation of pqsR transcription by LasR and 3-oxo-C12-HSL. First, we tested whether both proteins could interact with the pqsR promoter region using EMSAs. We incubated the pqsR-P2 DNA fragment with either LasR and 3-oxo-C12-HSL, CysB, or both proteins and analyzed the DNA-protein complexes formed in each reaction. We observed a single DNA-protein complex formed when the pqsR-P2 fragment was incubated with only LasR and a single complex of even higher molecular weight when the fragment was incubated with only CysB (Fig. 8A). In reaction mixtures that contained both proteins, we observed complexes that migrated at the same rate as each of the individual LasR- or CysB-DNA complexes, but we did not detect the formation of any larger-molecular-weight complexes (Fig. 8A) even after X-ray film was exposed to the gels for extended periods of time, suggesting that the protein-DNA complexes we observed contained either LasR or CysB exclusively. Furthermore, as an increasing ratio of CysB to LasR was included in each binding reaction mixture, the amount of the pqsR-P2 fragment bound by LasR decreased from 36% to 16% (Fig. 8B). We also performed these experiments using a DNA fragment corresponding to the lasI promoter. CysB formed only a small amount of a complex when incubated with the lasI promoter alone, most likely due to nonspecific interactions with this DNA fragment, and this complex could not be detected when LasR was present (Fig. 8C). Instead, we observed a single complex formed in all reactions when the reaction mixture contained LasR, and the addition of CysB had little effect on the formation of this complex (Fig. 8C and D). This result showed that CysB does not adversely affect the ability of LasR to bind DNA, and together these experiments suggest that LasR and CysB compete for similar or overlapping binding sites at the pqsR promoter.
In order to test the effects of these two proteins in vivo, but in the absence of other P. aeruginosa-encoded factors, we expressed each protein in E. coli and analyzed the effects on the activation of a pqsR′-lacZ transcriptional fusion. To do this, we transformed E. coli strain DH5α with a plasmid carrying either a pqsR′-lacZ fusion or a pqsR′-lacZ fusion and tacp-lasR. Then, we added a second plasmid carrying either araC and PBAD-cysB so that we could control CysB expression or araC and PBAD without any coding sequence cloned downstream from the promoter as a control. In the absence of LasR, CysB expression caused no significant change in the basal level of β-Gal activity produced by the pqsR′-lacZ fusion in E. coli (Fig. 9). With the expression of LasR and only the control plasmid present, we found that 354 ± 14 units of β-Gal activity was produced, and this activity increased to 837 ± 21 units when 3-oxo-C12-HSL was added to cultures (Fig. 9). This showed that LasR and 3-oxo-C12-HSL can activate pqsR in E. coli. However, when CysB was expressed along with LasR, the addition of 3-oxo-C12-HSL caused only a minor increase in β-Gal activity (Fig. 9), indicating that CysB blocked the induction of pqsR′-lacZ by LasR and 3-oxo-C12-HSL. This also implies that CysB may modulate pqsR expression in P. aeruginosa by interfering with the LasR-mediated activation of pqsR transcription.
FIG 9.
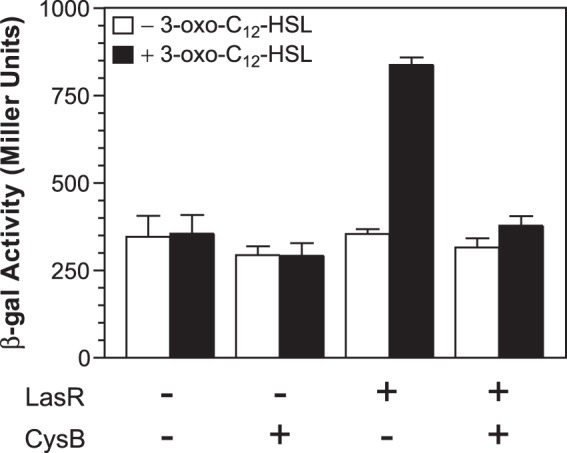
CysB can block the activation of pqsR′-lacZ transcription by LasR and 3-oxo-C12-HSL in E. coli. E. coli strain DH5α was transformed with two plasmids: one carrying either a pqsR′-lacZ fusion (pMWC1003) or a pqsR′-lacZ fusion and tacp-lasR (pJF1003) and a second one carrying either a vector control (pACYC-PBAD) or PBAD-cysB (pJF52). Bacteria were grown for 2 h in LB medium supplemented with 1 mM IPTG and 1% arabinose either in the absence or presence of 20 nM 3-oxo-C12-HSL. The β-Gal activity produced in each culture was assayed, and data are presented in Miller units as the means ± standard deviations of results from duplicate assays from at least three separate experiments.
DISCUSSION
In P. aeruginosa, PqsR plays a key role in the induction of 4-quinolone biosynthesis. We set out to investigate the regulation of pqsR transcription since it seemed likely to involve the integration of both cell-to-cell signaling systems and environmental signals. We began by focusing on LasR, the only factor that has been shown to positively regulate pqsR directly. Our experiments showed that LasR interacts with a single binding site in the pqsR-nadA intergenic region (Fig. 1), and this interaction only affects pqsR transcription (Fig. 2). The LasR binding site is centered 248 bp upstream from the pqsR TS1, and it is unclear how LasR activates the transcription of pqsR from this distal binding site. Based on the spacing, it seems unlikely that LasR could make direct contact with RNA polymerase without additional DNA binding proteins present to facilitate bending in the pqsR promoter region. LasR may instead act indirectly, possibly through recruiting additional factors to the pqsR promoter or by affecting DNA supercoiling in the region (62). A genome-wide identification of LasR binding sites showed that the spacing between these sites and established transcriptional start sites varied widely (38), suggesting that LasR can use diverse mechanisms to affect gene expression.
To identify additional factors that could bind to the pqsR promoter region and affect pqsR transcription, we used DNA affinity chromatography and found that the MvaT, MvaU, and CysB proteins could associate with the pqsR-nadA intergenic region. This experiment did not isolate either LasR or PmpR, a regulatory protein reported to bind the pqsR promoter and repress transcription (63). This could have been due to low abundance of these proteins, poor solubility, poor DNA binding under the conditions tested, or a combination of these issues, which illustrates some of the limitations of this technique. Our observation that MvaT and MvaU associated with the pqsR-nadA intergenic region was supported by a previous genome-wide identification of MvaT and MvaU binding sites (52). We did not observe any effect on either pqsR or nadA expression in mvaT or mvaU single mutant strains, and we were unable to construct an mvaT mvaU double mutant. However, Li et al. reported the successful construction of an mvaT mvaU mutant and additionally reported that PQS production was greatly reduced in this strain (54). While this finding might suggest a potential role for MvaT and MvaU in positively regulating pqsR, it is inconsistent with the established functions of MvaT and MvaU, which like other H-NS family members appear to oligomerize along A-T-rich regions of DNA to silence gene expression (64). Additionally, depletion of both MvaT and MvaU from P. aeruginosa cells appears to cause wide-scale changes in gene expression (52), so it is difficult to tell whether the phenotype reported by Li et al. was due to direct or indirect regulatory effects. Therefore, the role of MvaT and MvaU in controlling pqsR expression, if any, remains unclear.
With all of this in mind, we decided to focus on CysB since no previous studies had provided any connection between CysB and PQS biosynthesis. CysB was first characterized in enteric bacteria as a master regulator of sulfur assimilation through the cysteine biosynthetic pathway, which is the predominant route of sulfur acquisition in bacteria (40). CysB also appears to have this role in P. aeruginosa based on the growth phenotypes displayed by cysB mutants. Additionally, we found evidence that CysB directly regulates cysH in P. aeruginosa (Fig. 6 and 7), and others showed that CysB regulates cysP, which encodes the periplasmic sulfate binding protein (44). In enteric bacteria the genes for sulfate uptake and reduction to sulfide are activated by CysB when it binds the ligand NAS, which is derived from the direct cysteine precursor OAS. However, although cysH was differentially regulated in response to cysteine levels, we found that neither NAS nor OAS was able to stimulate the expression of cysH (Fig. 6). Furthermore, previous studies showed that the in vitro DNA binding activity of recombinant CysB proteins from P. aeruginosa and P. putida was not affected by NAS or OAS (44, 59). So while CysB seems to fulfill the same regulatory functions in P. aeruginosa, it appears to respond to different signals than those in enteric bacteria. Instead, CysB may interact with other intermediates in the cysteine and methionine biosynthetic pathway, or it may regulate in response to sulfide, which binds to CysB and acts as an anti-inducer in Salmonella Typhimurium (65).
We confirmed that CysB can directly interact with the pqsR promoter region (Fig. 6), and found that CysB can repress pqsR transcription and PQS production (Fig. 4 and 5). We also provided evidence that CysB can compete for binding with LasR at the pqsR promoter (Fig. 8) and showed that CysB can block LasR and 3-oxo-C12-HSL from activating a pqsR′-lacZ fusion in E. coli (Fig. 9), providing a potential mechanism for these regulatory effects. But unexpectedly and in contrast to cysH expression, pqsR expression was not altered in response to cysteine levels (Fig. 6). This finding suggests that CysB may regulate pqsR in response to other signals. Alternatively, CysB may interact with the pqsR promoter independent from ligands, and the regulatory effects may be due instead to the amounts of CysB that are present to compete with LasR for binding at the pqsR promoter. In enteric bacteria CysB appears to be autoregulatory (40), but cysB expression has not been investigated in P. aeruginosa. Interestingly, CysB also regulates other genes in P. aeruginosa that are not directly linked to sulfur metabolism or cysteine biosynthesis. CysB positively regulates the expression of algD, which encodes an enzyme that is essential for synthesis of the polysaccharide alginate (55). It also positively regulates the pvdS gene, which encodes an extracytoplasmic function sigma factor that controls numerous genes involved in iron metabolism and virulence, including genes for the biosynthesis of the siderophore pyoverdine (44). Since CysB can modulate the expression of several genes that affect host colonization and virulence factor production, it is interesting to speculate that perhaps CysB could play a role in the transition from a free-living to host-associated growth environment, which would also include changes in available sulfur sources. Overall, CysB appears to have multiple functions in P. aeruginosa that extend beyond sulfur uptake and cysteine biosynthesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a research grant from the National Institute of Allergy and Infectious Diseases (grant R01-AI076272).
We thank K. Tipton and M. Ellison for helpful discussions and thoughtful insights.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00246-15.
REFERENCES
- 1.Ramos J-L. 2004. Pseudomonas. Kluwer Academic/Plenum, Boston, MA. [Google Scholar]
- 2.Déziel E, Gopalan S, Tampakaki AP, Lépine F, Padfield KE, Saucier M, Xiao G, Rahme LG. 2005. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-l-homoserine lactones. Mol Microbiol 55:998–1014. doi: 10.1111/j.1365-2958.2004.04448.x. [DOI] [PubMed] [Google Scholar]
- 3.Rahme LG, Tan MW, Le L, Wong SM, Tompkins RG, Calderwood SB, Ausubel FM. 1997. Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc Natl Acad Sci U S A 94:13245–13250. doi: 10.1073/pnas.94.24.13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chugani SA, Whiteley M, Lee KM, D'Argenio D, Manoil C, Greenberg EP. 2001. QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98:2752–2757. doi: 10.1073/pnas.051624298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J Bacteriol 184:6472–6480. doi: 10.1128/JB.184.23.6472-6480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 7.Döring G, Flume P, Heijerman H, Elborn JS. 2012. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros 11:461–479. doi: 10.1016/j.jcf.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. 2003. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol 185:2080–2095. doi: 10.1128/JB.185.7.2080-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuster M, Lostroh CP, Ogi T, Greenberg EP. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol 185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pesci EC, Milbank JB, Pearson JP, McKnight S, Kende AS, Greenberg EP, Iglewski BH. 1999. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96:11229–11234. doi: 10.1073/pnas.96.20.11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. 1996. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol Microbiol 21:1137–1146. doi: 10.1046/j.1365-2958.1996.00063.x. [DOI] [PubMed] [Google Scholar]
- 12.Pesci EC, Pearson JP, Seed PC, Iglewski BH. 1997. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol 179:3127–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKnight SL, Iglewski BH, Pesci EC. 2000. The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol 182:2702–2708. doi: 10.1128/JB.182.10.2702-2708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diggle SP, Winzer K, Chhabra SR, Worrall KE, Cámara M, Williams P. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol Microbiol 50:29–43. doi: 10.1046/j.1365-2958.2003.03672.x. [DOI] [PubMed] [Google Scholar]
- 15.McGrath S, Wade DS, Pesci EC. 2004. Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS). FEMS Microbiol Lett 230:27–34. doi: 10.1016/S0378-1097(03)00849-8. [DOI] [PubMed] [Google Scholar]
- 16.Jensen V, Löns D, Zaoui C, Bredenbruch F, Meissner A, Dieterich G, Münch R, Häussler S. 2006. RhlR expression in Pseudomonas aeruginosa is modulated by the Pseudomonas quinolone signal via PhoB-dependent and -independent pathways. J Bacteriol 188:8601–8606. doi: 10.1128/JB.01378-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hazan R, He J, Xiao G, Dekimpe V, Apidianakis Y, Lesic B, Astrakas C, Déziel E, Lépine F, Rahme LG. 2010. Homeostatic interplay between bacterial cell-cell signaling and iron in virulence. PLoS Pathog 6:e1000810. doi: 10.1371/journal.ppat.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dekimpe V, Déziel E. 2009. Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: the transcriptional regulator RhlR regulates LasR-specific factors. Microbiology 155:712–723. doi: 10.1099/mic.0.022764-0. [DOI] [PubMed] [Google Scholar]
- 19.Bazire A, Dheilly A, Diab F, Morin D, Jebbar M, Haras D, Dufour A. 2005. Osmotic stress and phosphate limitation alter production of cell-to-cell signal molecules and rhamnolipid biosurfactant by Pseudomonas aeruginosa. FEMS Microbiol Lett 253:125–131. doi: 10.1016/j.femsle.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 20.Kim EJ, Wang W, Deckwer WD, Zeng AP. 2005. Expression of the quorum-sensing regulatory protein LasR is strongly affected by iron and oxygen concentrations in cultures of Pseudomonas aeruginosa irrespective of cell density. Microbiology 151:1127–1138. doi: 10.1099/mic.0.27566-0. [DOI] [PubMed] [Google Scholar]
- 21.Farrow JM III, Pesci EC. 2007. Two distinct pathways supply anthranilate as a precursor of the Pseudomonas quinolone signal. J Bacteriol 189:3425–3433. doi: 10.1128/JB.00209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown SA, Palmer KL, Whiteley M. 2008. Revisiting the host as a growth medium. Nat Rev Microbiol 6:657–666. doi: 10.1038/nrmicro1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calfee MW, Coleman JP, Pesci EC. 2001. Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98:11633–11637. doi: 10.1073/pnas.201328498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bredenbruch F, Nimtz M, Wray V, Morr M, Muller R, Häussler S. 2005. Biosynthetic pathway of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines. J Bacteriol 187:3630–3635. doi: 10.1128/JB.187.11.3630-3635.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dulcey CE, Dekimpe V, Fauvelle DA, Milot S, Groleau MC, Doucet N, Rahme LG, Lépine F, Déziel E. 2013. The end of an old hypothesis: the Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem Biol 20:1481–1491. doi: 10.1016/j.chembiol.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Déziel E, Lépine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG. 2004. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A 101:1339–1344. doi: 10.1073/pnas.0307694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lépine F, Milot S, Déziel E, He J, Rahme LG. 2004. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J Am Soc Mass Spectrom 15:862–869. doi: 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 28.Zhang YM, Frank MW, Zhu K, Mayasundari A, Rock CO. 2008. PqsD is responsible for the synthesis of 2,4-dihydroxyquinoline, an extracellular metabolite produced by Pseudomonas aeruginosa. J Biol Chem 283:28788–28794. doi: 10.1074/jbc.M804555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao H, Krishnan G, Goumnerov B, Tsongalis J, Tompkins R, Rahme LG. 2001. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc Natl Acad Sci U S A 98:14613–14618. doi: 10.1073/pnas.251465298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wade DS, Calfee MW, Rocha ER, Ling EA, Engstrom E, Coleman JP, Pesci EC. 2005. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J Bacteriol 187:4372–4380. doi: 10.1128/JB.187.13.4372-4380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao G, Déziel E, He J, Lépine F, Lesic B, Castonguay MH, Milot S, Tampakaki AP, Stachel SE, Rahme LG. 2006. MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol Microbiol 62:1689–1699. doi: 10.1111/j.1365-2958.2006.05462.x. [DOI] [PubMed] [Google Scholar]
- 32.Farrow JM III, Sund ZM, Ellison ML, Wade DS, Coleman JP, Pesci EC. 2008. PqsE functions independently of PqsR-Pseudomonas quinolone signal and enhances the rhl quorum-sensing system. J Bacteriol 190:7043–7051. doi: 10.1128/JB.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rampioni G, Pustelny C, Fletcher MP, Wright VJ, Bruce M, Rumbaugh KP, Heeb S, Cámara M, Williams P. 2010. Transcriptomic analysis reveals a global alkyl-quinolone-independent regulatory role for PqsE in facilitating the environmental adaptation of Pseudomonas aeruginosa to plant and animal hosts. Environ Microbiol 12:1659–1673. doi: 10.1111/j.1462-2920.2010.02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Cámara M. 2011. Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev 35:247–274. doi: 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huse H, Whiteley M. 2011. 4-Quinolones: smart phones of the microbial world. Chem Rev 111:152–159. doi: 10.1021/cr100063u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tashiro Y, Yawata Y, Toyofuku M, Uchiyama H, Nomura N. 2013. Interspecies interaction between Pseudomonas aeruginosa and other microorganisms. Microbes Environ 28:13–24. doi: 10.1264/jsme2.ME12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonnleitner E, Gonzalez N, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. 2011. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol Microbiol 80:868–885. doi: 10.1111/j.1365-2958.2011.07620.x. [DOI] [PubMed] [Google Scholar]
- 38.Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M. 2009. Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol Microbiol 73:1072–1085. doi: 10.1111/j.1365-2958.2009.06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao G, He J, Rahme LG. 2006. Mutation analysis of the Pseudomonas aeruginosa mvfR and pqsABCDE gene promoters demonstrates complex quorum-sensing circuitry. Microbiology 152:1679–1686. doi: 10.1099/mic.0.28605-0. [DOI] [PubMed] [Google Scholar]
- 40.Kredich NM. 2008. Biosynthesis of cysteine. EcoSal Plus doi: 10.1128/ecosalplus.3.6.1.11. [DOI] [PubMed] [Google Scholar]
- 41.Kertesz MA, Leisinger T, Cook AM. 1993. Proteins induced by sulfate limitation in Escherichia coli, Pseudomonas putida, or Staphylococcus aureus. J Bacteriol 175:1187–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuster M, Urbanowski ML, Greenberg EP. 2004. Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc Natl Acad Sci U S A 101:15833–15839. doi: 10.1073/pnas.0407229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 44.Imperi F, Tiburzi F, Fimia GM, Visca P. 2010. Transcriptional control of the pvdS iron starvation sigma factor gene by the master regulator of sulfur metabolism CysB in Pseudomonas aeruginosa. Environ Microbiol 12:1630–1642. doi: 10.1111/j.1462-2920.2010.02210.x. [DOI] [PubMed] [Google Scholar]
- 45.Nolan T, Hands RE, Bustin SA. 2006. Quantification of mRNA using real-time RT-PCR. Nat Protoc 1:1559–1582. doi: 10.1038/nprot.2006.236. [DOI] [PubMed] [Google Scholar]
- 46.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carty NL, Layland N, Colmer-Hamood JA, Calfee MW, Pesci EC, Hamood AN. 2006. PtxR modulates the expression of QS-controlled virulence factors in the Pseudomonas aeruginosa strain PAO1. Mol Microbiol 61:782–794. doi: 10.1111/j.1365-2958.2006.05269.x. [DOI] [PubMed] [Google Scholar]
- 48.Choi KH, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 49.Miller BE, Kredich NM. 1987. Purification of the cysB protein from Salmonella typhimurium. J Biol Chem 262:6006–6009. [PubMed] [Google Scholar]
- 50.Lynch AS, Tyrrell R, Smerdon SJ, Briggs GS, Wilkinson AJ. 1994. Characterization of the CysB protein of Klebsiella aerogenes: direct evidence that N-acetylserine rather than O-acetylserine serves as the inducer of the cysteine regulon. Biochem J 299:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whiteley M, Greenberg EP. 2001. Promoter specificity elements in Pseudomonas aeruginosa quorum-sensing-controlled genes. J Bacteriol 183:5529–5534. doi: 10.1128/JB.183.19.5529-5534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castang S, McManus HR, Turner KH, Dove SL. 2008. H-NS family members function coordinately in an opportunistic pathogen. Proc Natl Acad Sci U S A 105:18947–18952. doi: 10.1073/pnas.0808215105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castang S, Dove SL. 2012. Basis for the essentiality of H-NS family members in Pseudomonas aeruginosa. J Bacteriol 194:5101–5109. doi: 10.1128/JB.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li C, Wally H, Miller SJ, Lu CD. 2009. The multifaceted proteins MvaT and MvaU, members of the H-NS family, control arginine metabolism, pyocyanin synthesis, and prophage activation in Pseudomonas aeruginosa PAO1. J Bacteriol 191:6211–6218. doi: 10.1128/JB.00888-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delic-Attree I, Toussaint B, Garin J, Vignais PM. 1997. Cloning, sequence and mutagenesis of the structural gene of Pseudomonas aeruginosa CysB, which can activate algD transcription. Mol Microbiol 24:1275–1284. doi: 10.1046/j.1365-2958.1997.4121799.x. [DOI] [PubMed] [Google Scholar]
- 56.Ohman DE, Cryz SJ, Iglewski BH. 1980. Isolation and characterization of Pseudomonas aeruginosa PAO mutant that produces altered elastase. J Bacteriol 142:836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74:7422–7426. doi: 10.1128/AEM.01369-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kertesz MA, Schmidt-Larbig K, Wüest T. 1999. A novel reduced flavin mononucleotide-dependent methanesulfonate sulfonatase encoded by the sulfur-regulated msu operon of Pseudomonas aeruginosa. J Bacteriol 181:1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kouzuma A, Endoh T, Omori T, Nojiri H, Yamane H, Habe H. 2008. Transcription factors CysB and SfnR constitute the hierarchical regulatory system for the sulfate starvation response in Pseudomonas putida. J Bacteriol 190:4521–4531. doi: 10.1128/JB.00217-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bick JA, Dennis JJ, Zylstra GJ, Nowack J, Leustek T. 2000. Identification of a new class of 5′-adenylylsulfate (APS) reductases from sulfate-assimilating bacteria. J Bacteriol 182:135–142. doi: 10.1128/JB.182.1.135-142.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hryniewicz MM, Kredich NM. 1991. The cysP promoter of Salmonella typhimurium: characterization of two binding sites for CysB protein, studies of in vivo transcription initiation, and demonstration of the anti-inducer effects of thiosulfate. J Bacteriol 173:5876–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheridan SD, Benham CJ, Hatfield GW. 1998. Activation of gene expression by a novel DNA structural transmission mechanism that requires supercoiling-induced DNA duplex destabilization in an upstream activating sequence. J Biol Chem 273:21298–21308. doi: 10.1074/jbc.273.33.21298. [DOI] [PubMed] [Google Scholar]
- 63.Liang H, Li L, Dong Z, Surette MG, Duan K. 2008. The YebC family protein PA0964 negatively regulates the Pseudomonas aeruginosa quinolone signal system and pyocyanin production. J Bacteriol 190:6217–6227. doi: 10.1128/JB.00428-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castang S, Dove SL. 2010. High-order oligomerization is required for the function of the H-NS family member MvaT in Pseudomonas aeruginosa. Mol Microbiol 78:916–931. doi: 10.1111/j.1365-2958.2010.07378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ostrowski J, Kredich NM. 1990. In vitro interactions of CysB protein with the cysJIH promoter of Salmonella typhimurium: inhibitory effects of sulfide. J Bacteriol 172:779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith SS, Michael MZ, Graham MW. 1989. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res 17:3469–3478. doi: 10.1093/nar/17.9.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holloway BW, Krishnapillai V, Morgan AF. 1979. Chromosomal genetics of Pseudomonas. Microbiol Rev 43:73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Preston MJ, Seed PC, Toder DS, Iglewski BH, Ohman DE, Gustin JK, Goldberg JB, Pier GB. 1997. Contribution of proteases and LasR to the virulence of Pseudomonas aeruginosa during corneal infections. Infect Immun 65:3086–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 70.Tipton KA, Coleman JP, Pesci EC. 2013. QapR (PA5506) represses an operon that negatively affects the Pseudomonas quinolone signal in Pseudomonas aeruginosa. J Bacteriol 195:3433–3441. doi: 10.1128/JB.00448-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.