

ABSTRACT
The opportunistic pathogen Pseudomonas aeruginosa utilizes an injectisome-type III secretion system (injectisome-T3SS) to elicit cytotoxicity toward epithelial cells and macrophages. Macrophage killing results from the cytotoxic properties of the translocated effector proteins (ExoS, ExoT, ExoU, and ExoY) and inflammasome-mediated induction of pyroptosis. Inflammasome activation can occur following Nlrc4-mediated recognition of cytosolic translocated flagellin (FliC). In the present study, we demonstrate that FliC is a secretion substrate of both the injectisome- and flagellum-associated T3SSs. Molecular analyses indicate that the first 20 amino-terminal residues of FliC are sufficient for secretion by the injectisome-T3SS and that the first 100 residues are sufficient for translocation of FliC into host cells. Although maximal inflammasome activation requires FliC, activation can also occur in the absence of FliC. This prompted us to examine whether other flagellar components might also be translocated into cells to elicit inflammasome activation. Indeed, we find that the flagellar cap (FliD), hook-associated (FlgK and FlgL), hook (FlgE), and rod (FlgE) proteins are secretion substrates of the injectisome-T3SS. None of these proteins, however, result in increased inflammasome activation when they are overexpressed in a fliC mutant and appear to be translocated into host cells. While a role in inflammasome activation has been excluded, these data raise the possibility that flagellar components, which are highly conserved between different bacterial species, trigger other specific host responses from the extracellular milieu or contribute to the pathogenesis of P. aeruginosa.
IMPORTANCE The inflammasome is a host defense mechanism that recognizes invading bacteria and triggers an inflammatory immune response. The opportunistic pathogen P. aeruginosa produces both inflammasome agonists and antagonists. In this study, we demonstrate that overexpression of an agonist suppresses the activity of an antagonist, thereby resulting in inflammasome activation. Since the relative expression levels of agonists and antagonists likely vary between strains, these differences could be important predictors of whether a particular P. aeruginosa strain elicits inflammasome activation.
INTRODUCTION
Innate immunity constitutes the first line of defense against invading microorganisms and functions, in part, by detecting pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively) and activating host defense mechanisms (1, 2). PAMPs and DAMPs are detected through a diverse array of pattern recognition receptors (PRRs) that may be secreted, surface exposed, or localized to various intracellular compartments. The nucleotide-binding domain and leucine-rich repeat-containing proteins (NLRs) represent a large family of intracellular PRRs (3). NLR receptors detect a variety of endogenous and exogenous signals that include double-stranded DNA (dsDNA), extracellular ATP, uric acid crystals, peptidoglycan, and bacterial flagellin (1, 4). Upon signal recognition, the NLRs trigger NF-κB-dependent signaling and/or inflammasome activation, both of which are important for innate and adaptive immunity (2).
The inflammasome is a general term for a complex of proteins that can activate caspase-1. Activated caspase-1 cleaves prointerleukin 1β (pro-IL-1β) and pro-IL-18 into mature cytokines, thereby promoting their release from cells, where they act to recruit phagocytes to infection sites. Caspase-1 can also activate a form of programmed cell death in macrophages, termed pyroptosis. Inflammasomes are named for the specific NLR found within the complex (5). For instance, the Nlrc4 inflammasome contains Nlrc4, the adaptor protein ASC, caspase-1, and a Naip (NLR family apoptosis inhibitory protein) family member which provides ligand specificity (6). The Nlrc4 receptor consists of an amino-terminal caspase-1 recruitment domain (CARD) (7), an internal nucleotide binding (NACHT) domain, and a carboxy-terminal leucine-rich repeat (LRR) region thought to participate in signal recognition. Nlrc4 has been implicated in detection of Salmonella enterica serovar Typhimurium, Legionella pneumophila, Shigella flexneri, and Pseudomonas aeruginosa (7–13).
The signal to which Nlrc4 responds is not entirely clear. Studies suggest that flagellin translocated by the S. Typhimurium injectisome-type III secretion system (injectisome-T3SS) or the L. pneumophila type IV secretion system elicits Nlrc4-dependent activation of caspase-1 (14–16). In support of this, purified flagellin delivered to host cells by protein transfection also activates caspase-1, suggesting that flagellin is directly or indirectly recognized by Nlrc4 (7, 15, 16). S. flexneri, which naturally lacks flagella, and nonflagellated mutants of P. aeruginosa, however, can also activate caspase-1 through Nlrc4 in a manner requiring a functional injectisome-T3SS (10, 17). These findings indicate that Nlrc4 responds to additional injectisome-delivered signals. Importantly, Nlrc4 utilizes the Naip subfamily to mediate specificity for ligand recognition. Murine Naip1 recognizes the needle protein of the T3SS and in conjunction with Nlrc4 induces inflammasome activation (18, 19). Naip2 detects the basal rod structure of the T3SS, and cytosolic flagellin binds to Naip5 and Naip6 (19, 20). Humans have one NAIP homolog which binds to the needle protein of the T3SS (20).
The bacterial injectisome system is highly related to the bacterial flagellum. Both consist of ring-like structures embedded in the inner and outer membranes and a hollow rod-like structure that spans the periplasmic space (21). Assembly of both the injectisome and flagellum is dependent upon a T3SS located in the inner membrane. Biosynthesis of the flagellum is a highly ordered process dependent upon sequential secretion and assembly of the rod, hook, hook-associated, cap, and flagellin proteins (22). The injectisome is thought to assemble in a similar manner (23). Given the conserved nature of the injectisome and flagellum assembly processes and the common requirement for a T3SS, we hypothesized that additional flagellar components may also be substrates of the injectisome-T3SS. In the present study, we use P. aeruginosa as a model system to demonstrate that the first 20 amino-terminal residues of flagellin are sufficient for injectisome-dependent secretion and that flagellin is translocated into host cells through the injectisome. In addition, we find that the flagellar rod, hook, hook-associated, and capping proteins are also secreted by the P. aeruginosa injectisome system. These additional flagellar components, however, do not contribute to inflammasome activation.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are listed in Table S1 in the supplemental material. Escherichia coli strain DH5α was used for cloning, and E. coli strain Sm10(λpir) was used to conjugate plasmids into P. aeruginosa. E. coli strains were maintained on LB-agar plates containing antibiotics (ampicillin, 100 μg/ml; gentamicin, 15 μg/ml) as required. P. aeruginosa strains were grown on LB-agar with antibiotics as required (gentamicin, 75 μg/ml; carbenicillin, 300 μg/ml). Expression of the injectisome-T3SS was induced by growing strains in Trypticase soy broth (TSB) supplemented with 1% glycerol, 100 mM monosodium glutamate, and 1 mM EGTA at 30°C.
Plasmid and mutant construction.
The primer sequences used to generate PCR products, the cloning vectors, and cloning details are provided in Tables S2 to S4 in the supplemental material. DNA fragments flanking the upstream (1,100 bp) and downstream (960 bp) regions of flhB were amplified by PCR and sequentially ligated into pEX18Gm (24), resulting in an in-frame deletion of codons 4 to 376. The resulting plasmid (pEX18GmΔflhB) was mobilized by conjugation from E. coli SM10(λpir) to wild-type PAK, PAK ΔexoSTY (ΔSTY strain) and PAK ΔexsA as previously described (25). Chromosomal DNA from the PAK ΔfliC mutant was used to amplify the in-frame fliC deletion allele using the primer described in Table S4 and fliC sequences from GenBank accession numbers AF332547 and L81176. The fliC deletion allele was cloned into pEX18Gm and mobilized to PAK ΔexoSTY as described above. All mutants were confirmed by PCR using primers outside the cloned regions, and all PCR products were verified by sequencing.
The previously described pUY30 expression plasmid carries an exsE-cyaA translational fusion under the transcriptional control of the PBAD promoter (26) and was used as a backbone to construct plasmids expressing the flagellar components. The pFliC1–20-CyaA and pFliC1–394-CyaA plasmids were constructed by cloning the FliC coding region for residues 1 to 20 and 1 to 394 into the NdeI-XbaI restriction sites of pUY30. A vesicular stomatitis virus (VSV) epitope tag was added to the 3′ end of fliC via PCR primers as follows. First, the coding sequence for the amino-terminal 100 residues of FliC was cloned into the NdeI-XbaI sites in pUY30. Next, a VSV tag was added via PCR primers to the 5′ end of the coding sequence for residues 101 to 394 and cloned into the XbaI-SacI sites of the vector carrying the first 100 residues of FliC, resulting in plasmid pFliC1–100-VSV-101–394. To construct pFliC1–100-VSV-IAD, the coding sequence for the carboxy-terminal 40 residues of FliC, termed the inflammasome activating domain (IAD), was amplified with primers incorporating a VSV epitope tag and cloned into the XbaI-SacI sites of pFliC1–100-VSV-101–394. pFliC1–120-VSV-IAD was constructed by replacing the NdeI-XbaI fragment in pFliC1–100-VSV-101–394 with a fragment encoding the first 20 amino-terminal residues of FliC.
Secretion assays.
Cells were grown under inducing conditions for T3SS expression at 30°C to an optical density of 1 at 540 nm. Aliquots of cell cultures were centrifuged, and the cell-associated and trichloroacetic acid-precipitated supernatant fractions were subjected to immunoblotting as previously described (25). Rabbit anti-VSV glycoprotein antibody was purchased from Sigma-Aldrich and used at a 1:2,000 dilution. Polyclonal antiflagellin antibody, kindly provided by R. Ramphal, and polyclonal PcrV antibody, a gift from D. Frank, were both used at 1:10,000 dilutions.
Coculture experiments with J774A.1 macrophages.
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin G, and 100 μg/ml streptomycin was used to maintain J774A.1 macrophages. For coculture experiments the macrophages were grown in 24-well plates (3.0 × 105 cells/well) and activated with 50 ng/ml lipopolysaccharide (LPS) from E. coli serotype O111:B4 (Invivogen) for 12 to 16 h. The macrophages were washed twice with DMEM, and bacteria suspended in antibiotic-free DMEM supplemented with 10% FCS were added to the macrophages at a multiplicity of infection (MOI) of 20. Plates were centrifuged (350 × g) for 5 min at room temperature and incubated at 37°C with 5% CO2. Supernatants were collected postinfection at time points specified in the figure legends. Macrophage cell death was determined as previously described (27) using a cytotoxicity (lactate dehydrogenase [LDH]) detection kit (CytoTox 96 nonradioactive cytotoxicity assay; Promega). An IL-1β enzyme-linked immunosorbent assay (ELISA) was performed using antibody pairs (anti-mouse IL-1β and biotinylated anti-mouse IL-1β) and recombinant mouse IL-1β purchased from R&D Systems.
Statistical analyses.
One-way analysis of variance (ANOVA) and Tukey's posttest analyses were performed using Prism, version 5.0c (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Requirements for inflammasome activation.
Activation of the Nlrc4 inflammasome by P. aeruginosa requires a functional injectisome-T3SS and is maximal in strains that express FliC, the major flagellin subunit (7–10). Based on these data, it has been proposed that FliC is translocated into host cells by the injectisome-T3SS. To determine whether FliC is indeed secreted through the injectisome system, we used allelic exchange to construct a nonpolar flhB deletion mutant (ΔflhB strain) in P. aeruginosa strain PAK. FlhB is required for flagellar assembly (28), and, as expected, all of the ΔflhB mutants constructed for this study were nonmotile (data not shown). The ΔflhB allele was also introduced into an ΔSTY mutant lacking the known effectors of the injectisome-T3SS (ExoS, ExoT, and ExoY; exoU is absent from the PAK chromosome) and into an exsA mutant which is defective for expression of the entire injectisome-T3SS (29–31). An ΔexoSTY-fliC mutant was also constructed to serve as a negative control. The resulting panel of strains was grown to mid-log phase under nonpermissive (high Ca2+, no EGTA) and permissive (low Ca2+, with EGTA) conditions for expression of the injectisome-T3SS. Cells were then harvested, separated into cell-free culture supernatant and whole-cell fractions, and immunoblotted for PcrV or FliC. PcrV is a secreted substrate of the injectisome-T3SS and served as a positive control. Consistent with previous findings (32), PcrV was detected in the supernatant fraction when strains with a functional injectisome-T3SS (i.e., exsA+ strain) were grown under low-Ca2+ conditions (Fig. 1A, lanes 2, 4, 8, 12, and 14). PcrV secretion was unaffected by the flhB mutation (lanes 8 and 12). Immunoblotting the same fractions for FliC revealed two distinct expression patterns. High levels of FliC were detected in the cell-associated and supernatant fractions of the fhlB+ strains (PAK, ΔSTY, and exsA strains) irrespective of the growth conditions (Fig. 1A, lanes 1 to 6). In contrast, FliC expression was significantly reduced in the cell-associated fractions from the flhB mutant strains and was undetectable in the supernatant fractions (lanes 7 to 12). Reduced FliC expression was expected because transcription of the late flagellar genes (which include fliC) is inhibited by a feedback regulatory mechanism in the flhB mutants (33).
FIG 1.
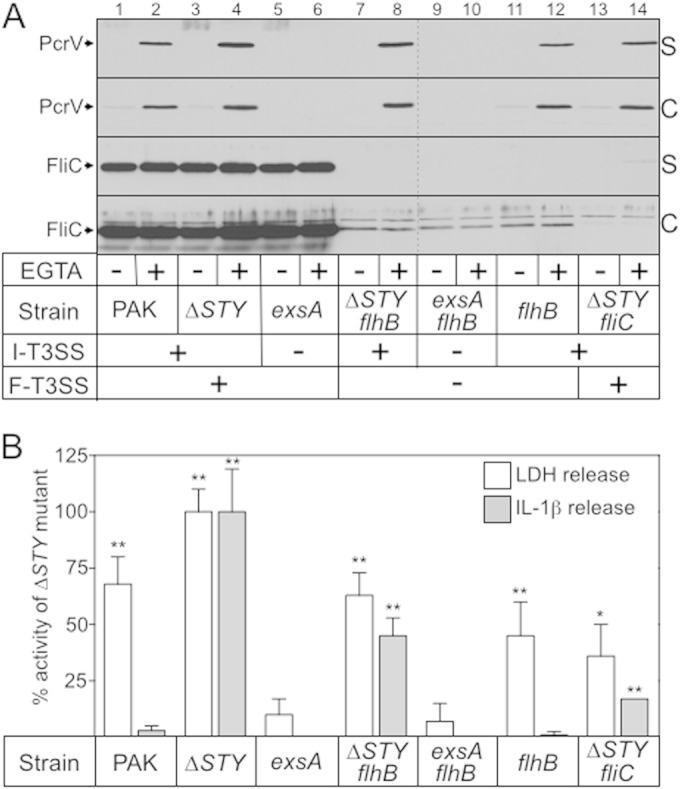
Role of the injectisome-T3SS and flagellum-T3SS in flagellin secretion and inflammasome activation. (A) The indicated strains were grown in TSB under noninducing (−EGTA) or inducing (+EGTA) conditions for expression of the injectisome-T3SS. Cultures were harvested at mid-log phase (A600 of ∼1.0), fractionated into cell-free supernatant (S) and whole-cell lysate (C) samples (normalized by cell number), separated by SDS-PAGE, and subjected to immunoblotting with rabbit polyclonal serum raised against PcrV or FliC. Strains that possess a functional injectisome-T3SS (I-T3SS) and/or flagellum-T3SS (F-T3SS) are indicated. (B) J774A.1 cells were LPS primed for 12 to 16 h and then cocultured with the indicated P. aeruginosa strains (20:1 MOI) for 4 h at 37°C. Cell-free culture supernatants were assayed for lactate dehydrogenase (LDH) activity as a measure of T3SS-dependent cytotoxicity and for IL-1β as a marker for inflammasome activation. Percent LDH and IL-1β release values were calculated relative to those of uninfected J774A.1 cells and J774A.1 cells cocultured with the ΔSTY strain (0 and 100% release, respectively). Coculture with the ΔSTY strain released 65% of the total LDH presented relative to J774A.1 cells that were completely lysed with Triton X-100 and 663 pg/ml IL-1β. The reported values represent the averages from at least three independent experiments, and error bars indicate the standard errors of the means (*, P < 0.01; **, P < 0.001, compared to the value for the exsA mutant). ΔSTY, exoΔSTY.
To determine whether FliC secretion correlated with inflammasome activation, the above panel of strains was cocultured with LPS-primed J774A.1 cells, a murine macrophage cell line, at an MOI of 20 for 3 h. Coculture supernatants were then assayed for the release of host-derived lactate dehydrogenase (LDH) to measure J774A.1 cell lysis and for IL-1β as a marker for inflammasome activation. Significant levels of LDH release were observed following coculture of the J774A.1 cells with wild-type PAK and the ΔSTY, ΔexoSTY-flhB, flhB, and ΔexoSTY-fliC mutants. Strains lacking the injectisome-T3SS (i.e., exsA mutant), however, were highly attenuated for cytotoxicity. The corresponding IL-1β ELISA data indicate that two distinct mechanisms are involved in eliciting cytotoxicity. The first mechanism, seen upon coculture with the ΔSTY, ΔexoSTY-flhB, and ΔexoSTY-fliC mutants, coincided with a significant increase in IL-1β levels compared to the level of the exsA mutant and is consistent with inflammasome activation and pyroptotic death. The observation of IL-1β release with the ΔexoSTY-fliC mutant is supportive of previous reports showing that inflammasome activation can also occur in an FliC-independent manner (9, 10). The increased IL-1β release seen with the ΔexoSTY-flhB mutant relative to the ΔexoSTY-fliC strain may result from low, undetectable levels of secreted FliC (Fig. 1A) or from an enhancement of the FliC-independent activation mechanism.
The second killing mechanism, observed upon coculture with wild-type PAK and the flhB mutant, was not associated with IL-1β release and likely results from the cytotoxic properties of the translocated ExoS, ExoT, and ExoY effectors (34). The lack of IL-1β release following coculture with wild-type PAK (compared to that with the ΔSTY effectorless mutant) was previously attributed to the activity of the ExoS effector protein, which interferes with inflammasome activation (9). Translocated ExoS likely accounts for the lack of IL-1β release in the flhB mutant as well (Fig. 1B, ΔexoSTY-flhB strain versus flhB strains).
P. aeruginosa flagellin is secreted by the injectisome-T3SS.
Because FliC is poorly expressed in flhB mutants (Fig. 1A, lanes 7 to 12), we were unable to determine whether FliC is secreted in the absence of a functional flagellum-T3SS. To address this question, FliC was cloned into an expression vector under the control of an arabinose-inducible promoter. Under these conditions, FliC expression was detected in the cell-associated fractions of each of the tested strains (Fig. 2A). The FliC steady-state expression level was somewhat elevated in wild-type PAK and in the ΔSTY and exsA mutants relative to the levels of the remaining strains (Fig. 2A, lanes 1 to 6 versus lanes 7 to 14). This increase likely reflects the combined expression from the plasmid and chromosomal copies of fliC. Comparison of the supernatant fractions from the various strains revealed three distinct FliC secretion patterns. (i) In wild-type PAK and the ΔSTY, exsA, and ΔexoSTY-fliC mutants, FliC secretion was largely Ca2+ independent although there was a modest increase in secretion under low-Ca2+ conditions (Fig. 2A, lanes 1 to 6, 13, and 14). This pattern is consistent with most of the FliC being secreted by the flagellum-T3SS and a smaller amount being secreted by the injectisome-T3SS in a low-Ca2+-dependent manner. (ii) In the ΔexoSTY-flhB and flhB mutants, FliC secretion was entirely dependent upon low-Ca2+ conditions, a pattern indicative of secretion through the injectisome-T3SS only (Fig. 2A, lanes 7 to 8, 11, and 12). Interestingly, the amount of secreted FliC in the flhB mutant was consistently lower than the levels seen in the ΔexoSTY-flhB strain, possibly reflecting a competition between ExoS, ExoT, ExoY, and FliC for access to the injectisome-T3SS. (iii) FliC was undetectable in the supernatant fraction of the exsA-flhB mutant, which is deficient in both the injectisome- and flagellum-T3SSs (Fig. 2A, lanes 9 and 10). These combined data demonstrate that FliC is a secreted substrate of both the injectisome- and flagellum-T3SSs.
FIG 2.
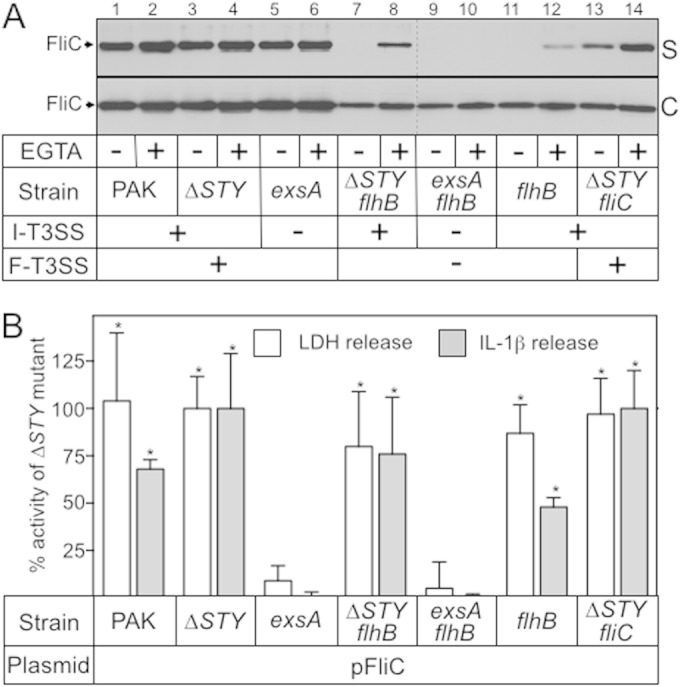
FliC expression suppresses ExoS-dependent inhibition of inflammasome activation. (A) The indicated strains carrying the FliC expression plasmid were grown in TSB in the absence or presence of 1 mM EGTA. Expression of plasmid-encoded FliC was induced by the addition of 0.5% arabinose (0.5%) when the culture A600 reached 0.4. Cells were harvested at mid-log phase (A600 of ∼1.0) and separated into cell-free supernatant (S) and whole-cell lysate (C) fractions (normalized by cell number). Fractions were separated by SDS-PAGE and subjected to immunoblotting with FliC rabbit polyclonal serum. (B) J774A.1 cells were LPS primed for 12 to 16 h, cocultured with the indicated P. aeruginosa strains, and assayed for LDH activity as a measure of T3SS-dependent cytotoxicity and for IL-1β as a marker for inflammasome activation as described in the legend of Fig. 1B. Percent LDH release was calculated relative to the level of an uninfected control (0% cytotoxicity) and by the amount of LDH released by coculture with the ΔSTY strain (100% cytotoxicity). Under these conditions the ΔSTY strain released 82% of the LDH compared to the level in J774A.1 cells completely lysed with Triton X-100 and 450 pg/ml IL-1β. The reported values represent the average from at least three independent experiments, and error bars indicate the standard errors of the means (*, P < 0.001, compared to the value for the exsA mutant).
The development of a system permissive for FliC expression in flhB mutant strains allowed us to further evaluate the requirements for inflammasome activation. J774A.1 cells were cocultured with strains expressing plasmid-encoded FliC and then assayed for LDH release and secreted IL-1β as described above. The overall findings were similar to those reported in Fig. 1B with one notable exception: both wild-type PAK and the flhB mutant expressing plasmid-encoded FliC now triggered significant IL-1β release (Fig. 2B). We speculated that strains carrying the FliC expression plasmid might translocate more FliC into host cells, thereby suppressing ExoS-mediated inhibition of inflammasome activation. Alternatively, increased FliC expression might compete with and reduce the amount of ExoS that is translocated into host cells. To distinguish between these two possibilities the ΔexoSTY-flhB and flhB mutants were transformed with a vector control (pJN105) or the pFliC expression plasmid and then examined for FliC or ExoS secretion. Immunoblotting the culture supernatant fractions confirmed that strains expressing plasmid-encoded FliC have a significant increase in the amount of secreted and, presumably, translocated FliC relative to levels of strains carrying the vector control (Fig. 3, lanes 2 and 6 versus lanes 4 and 8). Increased FliC secretion by the injectisome-T3SS is also likely occurring in wild-type PAK carrying the pFliC expression plasmid although this could not be confirmed due to the large amount of FliC secreted by the flagellum-T3SS (Fig. 2A). We next addressed whether FliC and ExoS compete for access to the injectisome-T3SS. Comparison of the supernatant samples from the ΔexoSTY-flhB and flhB strains revealed no difference in the total amounts of secreted FliC (Fig. 3, lane 4 versus 8). There was a modest but reproducible decrease in the amounts of secreted ExoS and ExoT in the flhB mutant expressing plasmid-encoded FliC (Fig. 3, lane 6 versus lane 8). The finding that the FliC expression plasmid results in a significant increase in secreted FliC and only a modest decrease in ExoS/ExoT secretion suggests that increased delivery of FliC to host cells can circumvent ExoS-mediated inhibition of inflammasome activation (Fig. 1).
FIG 3.
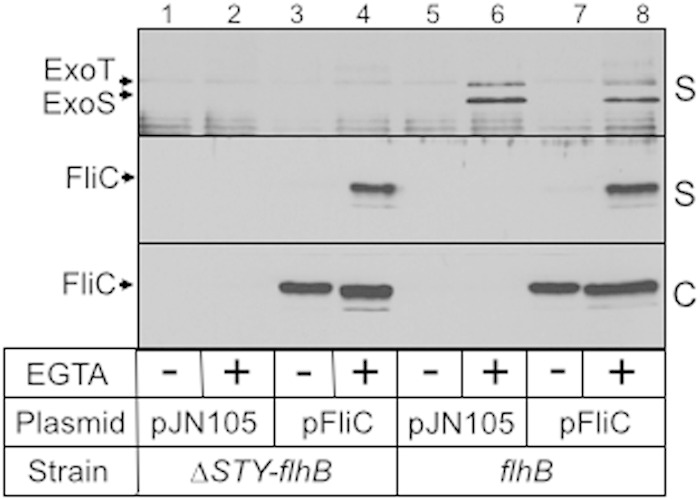
FliC overexpression reduces secretion of ExoS. The ΔexoSTY-flhB and flhB mutants carrying either a vector control (pJN105) or the pFliC expression plasmid were cultured as described in the legend of Fig. 1, separated into cell-free supernatant (S) and whole-cell lysate (C) fractions, and immunoblotted for FliC.
The first 20 amino-terminal residues of FliC are sufficient for secretion.
The secretion signal for substrates of the injectisome- and flagellum-T3SSs is typically located within the first 15 to 20 amino-terminal residues (35–37). To examine the requirements for FliC secretion, we fused full-length FliC (FliC1–394-CyaA) or the first 20 amino-terminal residues of FliC (FliC1–20-CyaA) to a truncated form of the calmodulin-dependent adenylate cyclase domain (∼50 kDa) from Bordetella pertussis CyaA. The truncated form of CyaA lacks its native secretion signal and has been used previously to map signals required for secretion and translocation of effector proteins into host cells (38). Strains carrying the FliC1–394-CyaA and FliC1–20-CyaA plasmids were cultured under noninducing and inducing conditions for expression of the injectisome-T3SS. The expression levels of the FliC1–394-CyaA and FliC1–20-CyaA fusions were similar in the cell-associated fraction for each of the genetic backgrounds tested (Fig. 4, lanes 1 to 10). As expected the FliC1–394-CyaA fusion was secreted by wild-type PAK and the mutants lacking either the injectisome-T3SS or flagellum-T3SS (Fig. 4, lanes 2, 4, 6, and 10). FliC1–394-CyaA secretion was not detected in the double mutant (exsA flhB strain) defective for both T3SSs (Fig. 4, lanes 7 and 8). Curiously, both injectisome- and flagellum-dependent secretion of the FliC1–394-CyaA fusion was dependent upon growth in the presence of EGTA. While secretion under these conditions is a well-documented property of the P. aeruginosa injectisome-T3SS (39), it is unclear why secretion through the flagellum-T3SS would also demonstrate EGTA dependence.
FIG 4.
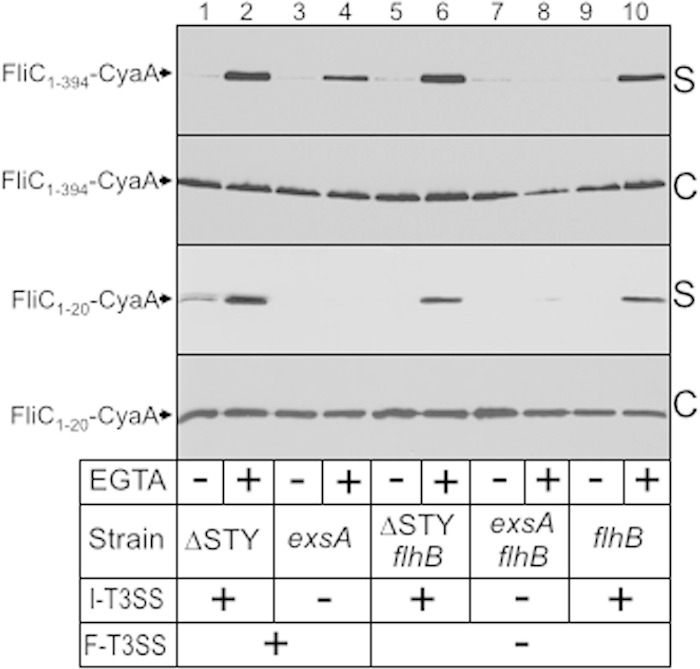
The first 20 amino-terminal residues of FliC are sufficient for secretion of a heterologous protein by the injectisome-T3SS. The indicated strains expressing full-length FliC (FliC1–394-CyaA) or the first 20 amino-terminal residues of FliC (FliC1–20-CyaA) fused to a truncated form of the calmodulin-activated adenylate cyclase (CyaA) from Bordetella pertussis were grown in TSB in the absence or presence of EGTA to mid-log phase, separated into culture supernatant (S) and cell-associated (C) fractions, and subjected to immunoblotting using monoclonal antibody to CyaA.
In contrast to our findings with full-length FliC1–394-CyaA, secretion of the FliC1–20-CyaA fusion was detected only in strains with a functional injectisome-T3SS (Fig. 4, lanes 2, 6, and 10). While these data demonstrate that the amino-terminal 20 residues of FliC are sufficient for secretion by the injectisome-T3SS, they also indicate that secretion by the flagellum-T3SS requires more than the first amino-terminal 20 residues. Finally, we attempted to address the question of whether the first 20 residues were necessary for secretion by generating an FliC21–394-CyaA fusion but found that the fusion protein was not stably expressed in P. aeruginosa (data not shown).
The first 100 amino-terminal residues of FliC are sufficient for translocation into host cells.
To the best of our knowledge, most secretion substrates of injectisome-T3SSs are also translocated into host cells and rely upon a translocation signal located within the first ∼100 amino-terminal residues. The challenge in detecting translocation lies in differentiating translocated substrate from substrate in the coculture supernatant, bound to the surface of the host cell, and/or associated with bacteria adhering to the host cell. Several assays have been developed for this purpose, and they generally involve expression of a translocation substrate fused to an enzymatic reporter that has detectable activity only when translocated. Our initial attempts to determine whether FliC is translocated into host cells made use of the previously described translocation reporters CyaA or Bla (beta-lactamase) fused to the carboxy terminus of FliC (38, 40). Whereas expression of FliC in the ΔexoSTY-fliC mutant results in the lysis of J774A.1 cells and IL-1β secretion (Fig. 2B), neither the FliC-CyaA nor the FliC-Bla fusion retained those activities (data not shown). This result suggested that FliC proteins carrying carboxy-terminal tags are not translocated and/or interfere with inflammasome activation. The latter possibility is consistent with the previous finding that the carboxy-terminal 35 residues of FliC are necessary and sufficient for inflammasome activation (41).
As an alternative to the CyaA and Bla fusion approaches we used the C-terminal 40 residues of FliC (designated here as the IAD for inflammasome activation domain) as a reporter for inflammasome activation. We first confirmed that the IAD of P. aeruginosa FliC is sufficient for inflammasome activation by fusing the IAD to the carboxy terminus of ExsE (Fig. 5A). ExsE is a regulator of the injectisome-T3SS that is translocated into host cells but has no known activity within the host (26). Whereas PAK ΔexoSTY-fliC expressing VSV-tagged ExsE had no effect on IL-1β secretion, the ExsE-VSV-IAD fusion resulted in significant stimulation of IL-1β secretion (Fig. 5B). The FliC IAD, therefore, is sufficient for inflammasome activation when fused to a partner that can promote injectisome-dependent translocation into host cells.
FIG 5.
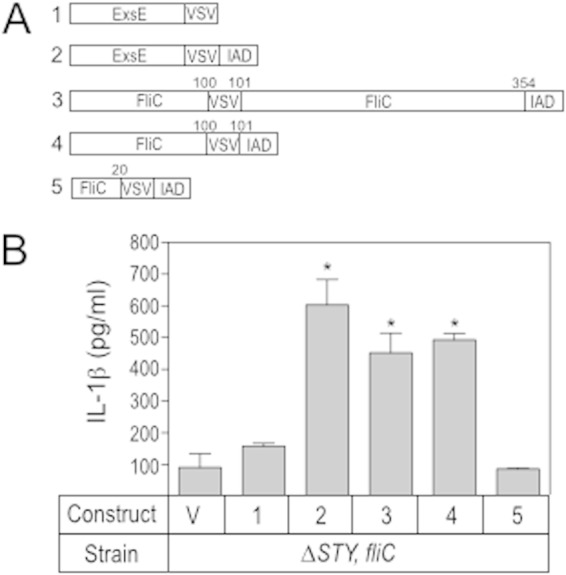
The first 100 residues of FliC are required for injectisome-T3SS-dependent delivery into host cells. (A) Diagram of the constructs used in the experiment shown in panel B. IAD, inflammasome activation domain; VSV, vesicular stomatitis virus epitope tag. (B) J774A.1 cells were LPS primed for 12 to 16 h, cocultured with the P. aeruginosa ΔexoSTY-fliC mutant carrying either a vector control (V) or constructs 1 to 5 as shown in panel A and assayed for secreted IL-1β. The reported values represent the averages from at least three independent experiments, and error bars indicate the standard errors of the means (*, P < 0.05, compared to the value for the ΔexoSTY-fliC mutant carrying the vector control).
We next used the IAD to determine which portion of FliC is required for delivery via the injectisome-T3SS by making deletion constructs lacking residues 101 to 359 or 21 to 359 (Fig. 5A). Both full-length FliC and the deletion construct lacking residues 101 to 359 stimulated IL-1β secretion, indicating that the first 100 residues are sufficient for both FliC secretion and translocation. The fusion consisting of only the first 20 residues of FliC fused to the IAD did not elicit an IL-1β response, indicating that they are not sufficient for translocation.
Secretion of additional flagellar components.
The observation that FliC is translocated into host cells raised the possibility that additional flagellar components, including the cap (FliD), hook-associated (FlgK and FlgL), hook (FlgE), and rod (FlgB) proteins, which are secreted by the flagellum-T3SS, also are substrates of the injectisome-T3SS. To address this question, each component was tagged on the carboxy terminus with the VSV epitope tag and assayed for secretion using the same panel of strains described for Fig. 1. Similar to results for FliC, secretion of each of the flagellar components required that cells possess either a functional injectisome- or flagellum-T3SS (Fig. 6). The mutant lacking both T3SSs (exsA fhlB strain) was completely deficient in secretion. Curiously secretion of FlgL, FlgE, and FlgB by the flagellum-T3SS demonstrated EGTA dependence while FliD and FlgK were secreted in an EGTA-independent manner (Fig. 6, lane 3 versus lane 4). This former finding is similar to our data with the FliC1–394-CyaA fusion (Fig. 4) and indicates that the property is substrate specific.
FIG 6.
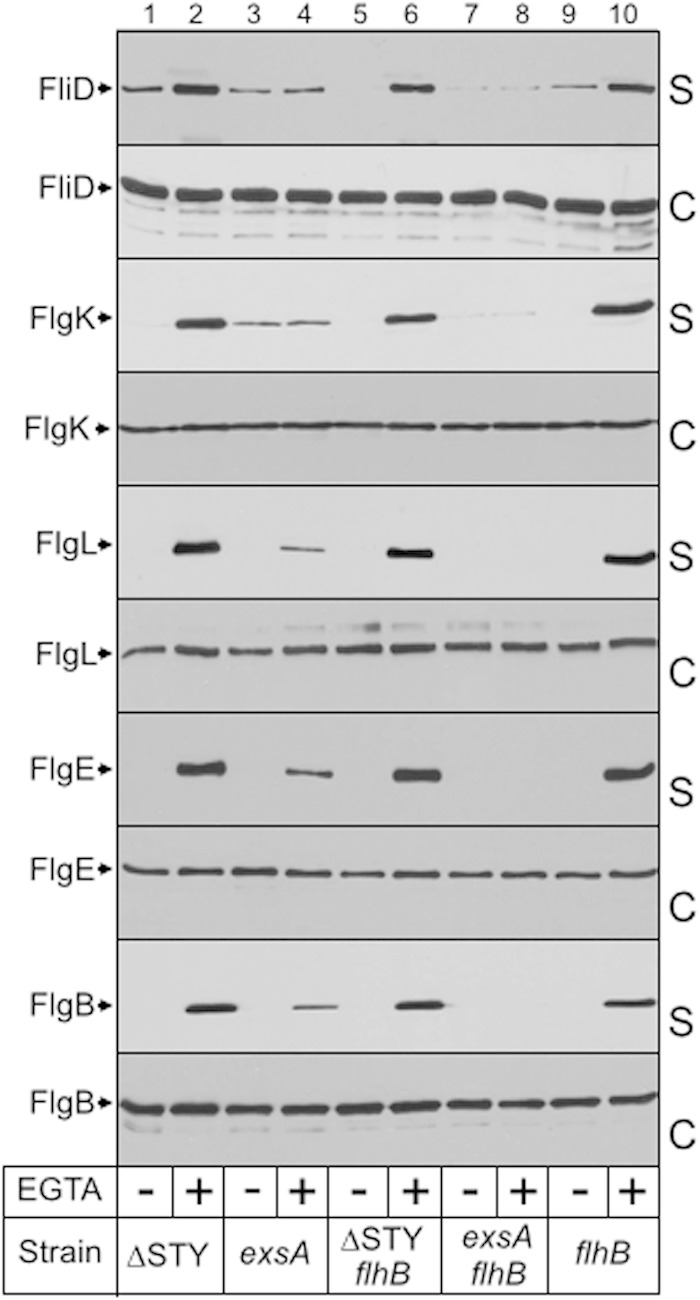
The flagellar cap, hook-associated, hook, and rod proteins are secreted by the injectisome-T3SS. The indicated strains were transformed with plasmids expressing FliD, FlgK, FlgL, FlgE, or FlgB tagged at the carboxy terminus with the VSV epitope. The resulting strains were grown in TSB in the absence or presence of EGTA to mid-log phase, fractionated into supernatant (S) and whole-cell (C) fractions, and immunoblotted using polyclonal antibody to the VSV epitope tag.
The flagellar cap, hook-associated, hook, and rod proteins do not trigger IL-1β release.
Like FliC, the other flagellar components are highly conserved between Gram-negative organisms and could serve as PAMPs that are detected by host cells to trigger defensive responses. Data presented in Fig. 1B and those from another study (10) demonstrate that inflammasome activation can occur in the absence of FliC but still requires a functional injectisome-T3SS. To determine whether the rod (FlgB), hook (FlgE), hook-associated (FlgK and FlgL), and cap (FliD) proteins trigger inflammasome activation, the PAK ΔexoSTY-fliC mutant was transformed with plasmids expressing untagged FlgB, FlgE, FlgL, FlgK, or FliD. The resulting strains were cocultured with LPS-primed J774A.1 macrophages at an MOI of 20:1, and IL-1β release into the culture supernatants was measured by ELISA. None of the additional flagellar components, however, resulted in a detectable increase in IL-1β secretion (Fig. 7A). This result could mean that the flagellar components are not translocated or, if they are, that they do not activate the inflammasome to cause IL-1β secretion. To test the former possibility, each of the flagellar components was expressed in the PAK ΔexoSTY-fliC background as a fusion protein tagged with the IAD at the carboxy-terminal end. Strains were cocultured with J774A.1 macrophages as described above and assayed for IL-1β release in the coculture supernatants. None of the other flagellar components, however, resulted in a significant increase in IL-1β. These findings suggest that the other flagellar components are not translocated by the injectisome-T3SS.
FIG 7.
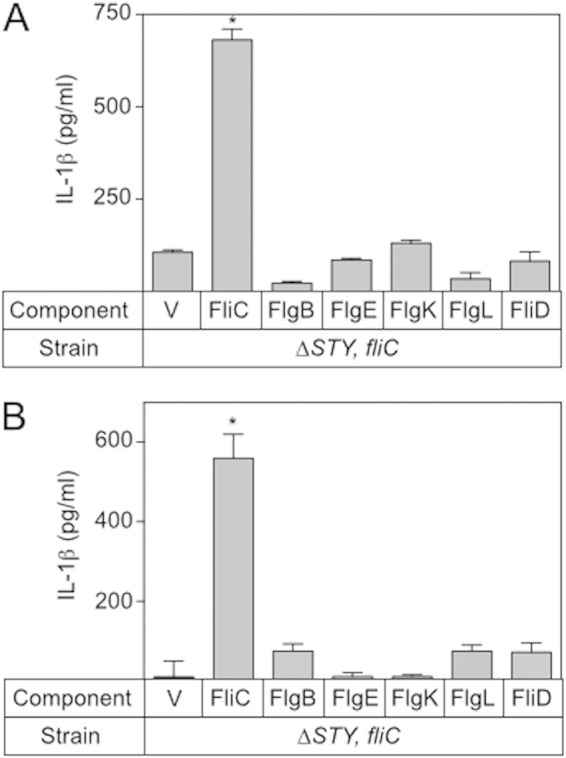
The flagellar cap, hook-associated, hook, and rod proteins do not contribute to FliC-independent inflammasome activation. The ΔexoSTY-fliC mutant was transformed with a vector control (V) or a plasmid expressing FliC, FlhB, FlgE, FlgK, FlgL, or FliD (A) or with a plasmid expressing the same protein fused to the IAD from FliC (B). The resulting strains were cocultured with LPS-stimulated J774A.1 cells as described in the legend to Fig. 1B and assayed for inflammasome activation by measuring IL-1β release. The reported values represent the total IL-1β release for each strain and represent the averages from at least three independent experiments with error bars indicative of the standard errors of the means (*,P < 0.001, compared to the value for the ΔexoSTY-fliC mutant carrying the vector control).
Injectisome substrates are not secreted through the flagellum-associated T3SS.
Our finding that flagellar components are secreted through the injectisome-T3SS prompted us to ask whether injectisome substrates could also be secreted through the flagellum-T3SS. To address this question we used a mutant lacking PscC, a structural component of the injectisome-T3SS. Because T3SS gene expression is coupled to secretory activity, pscC mutants are defective for T3SS gene expression (42). To circumvent this issue, we introduced an ExsA expression vector (the master regulator of T3SS gene expression) to induce expression of the injectisome substrates (43). Wild-type PAK served as the positive control, and PAK pscC with a vector control served as the negative control. Secretion assays were performed as previously described, and cell-free supernatants and whole-cell fractions were blotted for ExoS, ExoT, PopD, PopN, and PcrV. As expected, whole-cell fractions showed that wild-type PAK expressed the substrates only under inducing conditions (low Ca2+) and that the PAK pscC mutant lacked expression under both noninducing and inducing conditions. When the pscC mutant was provided with plasmid-expressed ExsA, expression of each secretion substrate was detected under noninducing and inducing conditions (Fig. 8, lanes 5 and 6, cell-associated fractions). Only PcrV was detected in the cell-free supernatants, suggesting possible secretion through the flagellum-T3SS. We also blotted for an intracellular protein (ExsA) as a control for lysis and found no contamination of the supernatant fraction with ExsA. Taking these results together, we conclude that effector proteins are poor substrates of the flagellum-T3SS.
FIG 8.
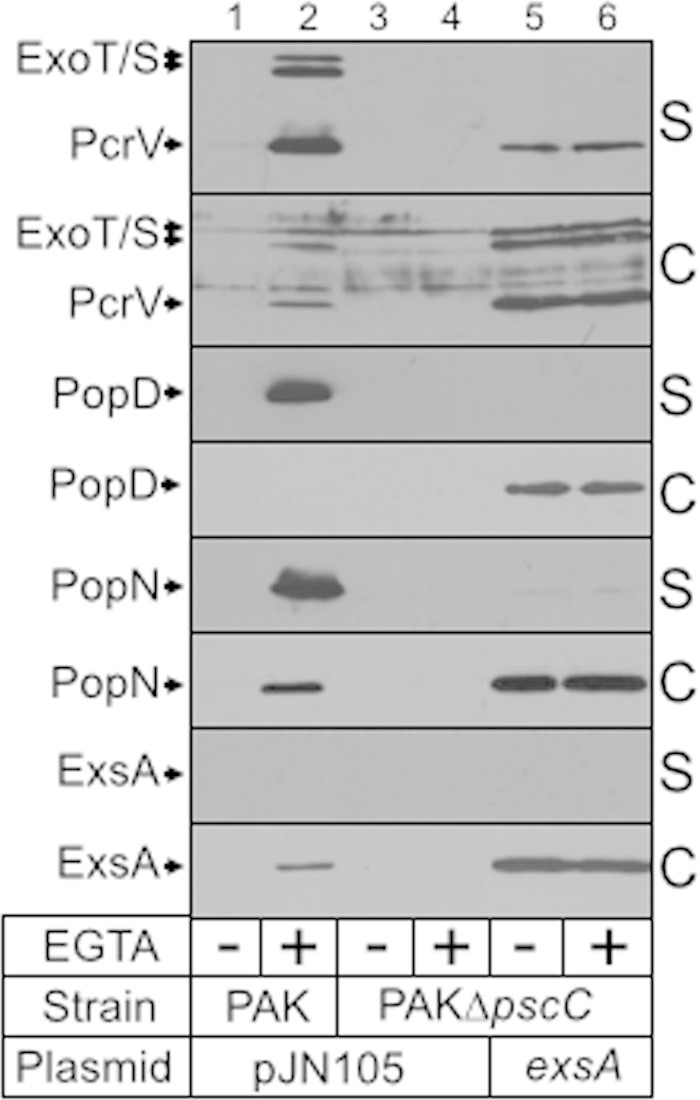
ExoT, ExoS, PcrV, PopD, and PopN are not secreted by the flagellum-associated T3SS. Wild-type PAK and an isogenic pscC mutant carrying either a vector control (V) or an ExsA expression plasmid were cultured in TSB in the absence or presence of EGTA to mid-log phase, fractionated into supernatant (S) and whole-cell (C) fractions, and immunoblotted using polyclonal antibody to the indicated proteins. ExsA is a cytoplasmic protein that served as a control for cellular lysis.
DISCUSSION
An important question relevant to potential therapeutic intervention is whether activation of the Nlrc4 inflammasome is beneficial or detrimental to the host in the context of P. aeruginosa infection. A host-centric view would suggest that inflammasome activation benefits the host by recruiting neutrophils to promote clearance of the invading microorganisms. Conversely, a pathogen-centric view might argue that inflammasome activation benefits P. aeruginosa; otherwise inflammasome-activating traits would be selected against. Indeed, there have been conflicting reports as to whether inflammasome activation is beneficial or detrimental in the response to wild-type P. aeruginosa. Whereas several studies indicate that Nlrc4−/− mice have a defect in the clearance of P. aeruginosa (8, 10), others show that Nlrc4−/− mice have enhanced clearance, survival, and reduced lung injury (44, 45). Likewise, reduced expression of IL-18 or IL-1β, each of which is a downstream effector of Nlrc4 inflammasome activation, has been linked to increased clearance of P. aeruginosa (44, 46), again suggesting that inflammasome activation might be detrimental to the host. These discrepancies may reflect variability in the experimental model systems. Perhaps the variable response to wild-type P. aeruginosa is not that unexpected. Multiple P. aeruginosa proteins trigger inflammasome activation, including pilin (PilA), RhsT, flagellin (FliC), and the injectisome-T3SS needle protein and rod proteins (PscF and PscI) (8, 10, 20, 47–49). While it remains unclear whether PilA and RhsT are secreted via the injectisome-T3SS, we now provide definitive evidence that flagellin is a substrate of both the flagellum- and injectisome-T3SSs. Although the flagellum-T3SS can secrete FliC, it is not sufficient for translocation of FliC and activation of the inflammasome. The injectisome-T3SS, however, delivers both agonists (FliC and PscF) and antagonists (ExoS and ExoU) of inflammasome activation. What then dictates whether a particular P. aeruginosa strain stimulates or inhibits inflammasome activation? Our finding that FliC overexpression suppresses the inhibitory activity of ExoS (Fig. 2) demonstrates that the relative expression level of the agonists and antagonists could be an important variable. It is worth noting that a survey of 74 clinical strains detected significant variation in the expression of the ExoS and ExoU antagonists (50). Whether there is a correlation between ExoS or ExoU expression level and inflammasome activation would be an interesting area for future studies.
While the relative expression of agonists and antagonists is one potential variable, differential timing in their expression is another. One study found that flagellum- and injectisome-T3SS gene expression are inversely controlled in P. aeruginosa (51). This suggests that inflammasome activation might be favored when flagellar genes are preferentially expressed and inhibited when expression of the injectisome-T3SS is favored. A final possibility is hierarchal expression and/or secretion of agonists and antagonists, resulting in a more complex scenario in which both inflammasome activation and inhibition are dynamically controlled during specific phases of the infection cycle. Our finding that overexpressed FliC suppresses ExoS-mediated inhibition of inflammasome activation (Fig. 2) suggests that competition between substrates for the injectisome-T3SS could influence the timing and/or degree of inflammasome activation.
Although significantly reduced, inflammasome activation still occurs in the absence of FliC. The residual activity is dependent upon the presence of a functional injectisome-T3SS, suggesting that an additional secreted protein contributes to inflammasome activation. In addition to FliC, we found that the flagellar rod (FlgB), hook (FlgE), hook-associated (FlgK and FlgL), and capping (FliD) proteins are secreted by the injectisome-T3SS (Fig. 6). In the absence of FliC, however, none of the other flagellar proteins led to an appreciable increase in inflammasome activation when overexpressed alone or as an IAD fusion protein (Fig. 7). A likely candidate remains the pore-forming activity of the translocase itself, a possibility that is consistent with previous observations that several pore-forming toxins trigger inflammasome activation (52).
The flagellar rod, hook, hook-associated, and cap proteins are highly conserved in bacteria and, like FliC, may represent PAMPS. A final observation is that FliD, FlgK, FlgL, FlgE, and FlgB are secreted more efficiently by the injectisome-T3SS than by the flagellum-T3SS (Fig. 6, lane 4 versus lane 6). While these proteins do not appear to be translocated, elevated secretion by the injectisome-T3SS raises a possibility that they exert antihost activity in the extracellular milieu or be recognized by yet to be described host receptors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dara Frank, Erik Hewlett, Matthew Wolfgang, Arne Rietsch, and Reuben Ramphal for providing strains and reagents, Nedim Ince for assistance with the tissue culture, and Mark Urbanowski and Dongwong Wang for technical assistance.
This work was supported by NIH grants N01 AI30040 (T.L.Y.) and R01 AI087630 (F.S.S.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00030-15.
REFERENCES
- 1.Areschoug T, Gordon S. 2008. Pattern recognition receptors and their role in innate immunity: focus on microbial protein ligands. Contrib Microbiol 15:45–60. doi: 10.1159/000135685. [DOI] [PubMed] [Google Scholar]
- 2.Geddes K, Magalhaes JG, Girardin SE. 2009. Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Discov 8:465–479. doi: 10.1038/nrd2783. [DOI] [PubMed] [Google Scholar]
- 3.Shaw MH, Reimer T, Kim YG, Nunez G. 2008. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol 20:377–382. doi: 10.1016/j.coi.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benko S, Philpott DJ, Girardin SE. 2008. The microbial and danger signals that activate Nod-like receptors. Cytokine 43:368–373. doi: 10.1016/j.cyto.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot JP, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V, Ward PA. 2008. The NLR gene family: a standard nomenclature. Immunity 28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sutterwala FS, Flavell RA. 2009. NLRC4/IPAF: a CARD carrying member of the NLR family. Clin Immunol 130:2–6. doi: 10.1016/j.clim.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. 2008. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci U S A 105:2562–2567. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. 2007. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol 37:3030–3039. doi: 10.1002/eji.200737532. [DOI] [PubMed] [Google Scholar]
- 9.Galle M, Schotte P, Haegman M, Wullaert A, Yang HJ, Jin S, Beyaert R. 2007. The Pseudomonas aeruginosa type III secretion system plays a dual role in the regulation of caspase-1 mediated IL-1β maturation. J Cell Mol Med 12:1767–1776. doi: 10.1111/j.1582-4934.2007.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 12.Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, Kuida K, Mariathasan S, Dixit VM, Flavell RA, Dietrich WF, Roy CR. 2006. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol 7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G. 2007. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog 2:e18. doi: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat Immunol 7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 16.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol 7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 17.Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Zhao Y, Shi J, Shao F. 2013. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A 110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kofoed EM, Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 21.Macnab RM. 2004. Type III flagellar protein export and flagellar assembly. Biochim Biophys Acta 1694:207–217. doi: 10.1016/j.bbamcr.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Minamino T, Imada K, Namba K. 2008. Mechanisms of type III protein export for bacterial flagellar assembly. Mol Biosyst 4:1105–1115. doi: 10.1039/b808065h. [DOI] [PubMed] [Google Scholar]
- 23.Wagner S, Konigsmaier L, Lara-Tejero M, Lefebre M, Marlovits TC, Galan JE. 2010. Organization and coordinated assembly of the type III secretion export apparatus. Proc Natl Acad Sci U S A 107:17745–17750. doi: 10.1073/pnas.1008053107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 25.McCaw ML, Lykken GL, Singh PK, Yahr TL. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol Microbiol 46:1123–1133. doi: 10.1046/j.1365-2958.2002.03228.x. [DOI] [PubMed] [Google Scholar]
- 26.Urbanowski ML, Brutinel ED, Yahr TL. 2007. Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect Immun 75:4432–4439. doi: 10.1128/IAI.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dasgupta N, Ashare A, Hunninghake GW, Yahr TL. 2006. Transcriptional induction of the Pseudomonas aeruginosa type III secretion system by low Ca2+ and host cell contact proceeds through two distinct signaling pathways. Infect Immun 74:3334–3341. doi: 10.1128/IAI.00090-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apel D, Surette MG. 2008. Bringing order to a complex molecular machine: the assembly of the bacterial flagella. Biochim Biophys Acta 1778:1851–1858. doi: 10.1016/j.bbamem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Frank DW, Nair G, Schweizer HP. 1994. Construction and characterization of chromosomal insertional mutations of the Pseudomonas aeruginosa exoenzyme S trans-regulatory locus. Infect Immun 62:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee VT, Smith RS, Tummler B, Lory S. 2005. Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infect Immun 73:1695–1705. doi: 10.1128/IAI.73.3.1695-1705.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfgang MC, Lee VT, Gilmore ME, Lory S. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4:253–263. doi: 10.1016/S1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 32.Sawa T, Yahr TL, Ohara M, Kurahashi K, Gropper MA, Wiener-Kronish JP, Frank DW. 1999. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat Med 5:392–398. doi: 10.1038/7391. [DOI] [PubMed] [Google Scholar]
- 33.Gillen KL, Hughes KT. 1991. Negative regulatory loci coupling flagellin synthesis to flagellar assembly in Salmonella typhimurium. J Bacteriol 173:2301–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vegh BM, Gal P, Dobo J, Zavodszky P, Vonderviszt F. 2006. Localization of the flagellum-specific secretion signal in Salmonella flagellin. Biochem Biophys Res Commun 345:93–98. doi: 10.1016/j.bbrc.2006.04.055. [DOI] [PubMed] [Google Scholar]
- 36.Neal-McKinney JM, Christensen JE, Konkel ME. 2010. Amino-terminal residues dictate the export efficiency of the Campylobacter jejuni filament proteins via the flagellum. Mol Microbiol 76:918–931. doi: 10.1111/j.1365-2958.2010.07144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Badea L, Beatson SA, Kaparakis M, Ferrero RL, Hartland EL. 2009. Secretion of flagellin by the LEE-encoded type III secretion system of enteropathogenic Escherichia coli. BMC Microbiol 9:30. doi: 10.1186/1471-2180-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sory MP, Boland A, Lambermont I, Cornelis GR. 1995. Identification of the YopE and YopH domains required for secretion and internalization into the cytosol of macrophages, using the cyaA gene fusion approach. Proc Natl Acad Sci U S A 92:11998–12002. doi: 10.1073/pnas.92.26.11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank DW. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol Microbiol 26:621–629. doi: 10.1046/j.1365-2958.1997.6251991.x. [DOI] [PubMed] [Google Scholar]
- 40.Sun YH, Rolan HG, Tsolis RM. 2007. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J Biol Chem 282:33897–33901. doi: 10.1074/jbc.C700181200. [DOI] [PubMed] [Google Scholar]
- 41.Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, Monack DM, Tsolis RM, Vance RE. 2008. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diaz MR, King JM, Yahr TL. 2011. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front Microbiol 2:89. doi: 10.3389/fmicb.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brutinel ED, Vakulskas CA, Brady KM, Yahr TL. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol 68:657–671. doi: 10.1111/j.1365-2958.2008.06179.x. [DOI] [PubMed] [Google Scholar]
- 44.Cohen TS, Prince AS. 2013. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest 123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faure E, Mear JB, Faure K, Normand S, Couturier-Maillard A, Grandjean T, Balloy V, Ryffel B, Dessein R, Chignard M, Uyttenhove C, Guery B, Gosset P, Chamaillard M, Kipnis E. 2014. Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. Am J Respir Crit Care Med 189:799–811. doi: 10.1164/rccm.201307-1358OC. [DOI] [PubMed] [Google Scholar]
- 46.Schultz MJ, Knapp S, Florquin S, Pater J, Takeda K, Akira S, van der Poll T. 2003. Interleukin-18 impairs the pulmonary host response to Pseudomonas aeruginosa. Infect Immun 71:1630–1634. doi: 10.1128/IAI.71.4.1630-1634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arlehamn CS, Evans TJ. 2011. Pseudomonas aeruginosa pilin activates the inflammasome. Cell Microbiol 13:388–401. doi: 10.1111/j.1462-5822.2010.01541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kung VL, Khare S, Stehlik C, Bacon EM, Hughes AJ, Hauser AR. 2012. An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc Natl Acad Sci U S A 109:1275–1280. doi: 10.1073/pnas.1109285109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A 107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L, Ledizet M, Kar K, Koski RA, Kazmierczak BI. 2005. An indirect enzyme-linked immunosorbent assay for rapid and quantitative assessment of Type III virulence phenotypes of Pseudomonas aeruginosa isolates. Ann Clin Microbiol Antimicrob 4:22. doi: 10.1186/1476-0711-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soscia C, Hachani A, Bernadac A, Filloux A, Bleves S. 2007. Cross talk between type III secretion and flagellar assembly systems in Pseudomonas aeruginosa. J Bacteriol 189:3124–3132. doi: 10.1128/JB.01677-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koizumi Y, Toma C, Higa N, Nohara T, Nakasone N, Suzuki T. 2012. Inflammasome activation via intracellular NLRs triggered by bacterial infection. Cell Microbiol 14:149–154. doi: 10.1111/j.1462-5822.2011.01707.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.