

Abstract
mRNA decapping is a central step in eukaryotic mRNA decay that simultaneously shuts down translation initiation and activates mRNA degradation. A major complex responsible for decapping consists of the decapping enzyme Dcp2 in association with decapping enhancers. An important question is how the activity and accumulation of Dcp2 are regulated at the cellular level to ensure the specificity and fidelity of the Dcp2 decapping complex. Here, we show that human Dcp2 levels and activity are controlled by a competition between decapping complex assembly and Dcp2 degradation. This is mediated by a regulatory domain in the Dcp2 C terminus, which, on the one hand, promotes Dcp2 activation via decapping complex formation mediated by the decapping enhancer Hedls and, on the other hand, targets Dcp2 for ubiquitin-mediated proteasomal degradation in the absence of Hedls association. This competition between Dcp2 activation and degradation restricts the accumulation and activity of uncomplexed Dcp2, which may be important for preventing uncontrolled decapping or for regulating Dcp2 levels and activity according to cellular needs.
INTRODUCTION
Proper control of gene expression requires multiple levels of regulation. In eukaryotic cells, several steps in gene expression are affected by the 5′ N7-methylguanosine (m7G) cap of mRNAs. The m7G cap is added to RNA polymerase II transcripts cotranscriptionally and associates with proteins in the nucleus that stimulate pre-mRNA splicing and RNA nuclear export (1). Once an mRNA enters the cytoplasm, the m7G cap is required for initiation of translation of the majority of mRNAs and at the same time protects mRNAs from degradation from the 5′ end (2). Therefore, the m7G cap plays an essential role in the processing and function of eukaryotic mRNAs.
Removal of the m7G cap by the process of decapping is a central step in mRNA turnover that simultaneously shuts down translation initiation and activates degradation of the mRNA from the 5′ end (3–6). The Nudix hydrolase family member Dcp2 is an important cytoplasmic decapping enzyme that is conserved among eukaryotes (7–14). Dcp2 forms a decapping complex with its cofactor, Dcp1 (13), which activates Dcp2 by promoting a Dcp2 conformational change, as evidenced by structural studies of the proteins from Saccharomyces cerevisiae and Schizosaccharomyces pombe (15–17). In metazoans, an additional decapping complex component, Hedls (also called Edc4 or Ge-1), interacts with Dcp2 and promotes Dcp2-Dcp1 complex formation (11, 18–20), but the exact role of Hedls in decapping remains poorly understood.
Several decapping enhancers that interact with the Dcp2 decapping complex and stimulate Dcp2 activity by various mechanisms have been identified. These include Edc3, Pat1, and Scd6 (called Lsm14A/RAP55 in humans), all of which are conserved in eukaryotes, as well as yeast-specific Edc1 and Edc2. These decapping enhancers can directly interact with and enhance the catalytic activity of the Dcp2-Dcp1 complex, as evidenced by in vitro studies (21–26). In addition, Pat1 and Scd6, as well as an additional decapping enhancer, the RNA helicase Dhh1 (called Rck/p54 in humans), may promote decapping by interfering with the m7G cap-associated eukaryotic initiation factor (eIF) 4F complex, as evidenced by the ability of these factors to repress translation initiation (24, 27–29). Despite the current knowledge of these decapping modulators, little is known about how the network of decapping factors controls the specificity and fidelity of the Dcp2 decapping enzyme.
A common cellular strategy to prevent the uncontrolled activity of enzymes utilizes regulatory domains that function to prevent enzymes from acting outside their regulatory complexes. Here, we present evidence that the C terminus of human Dcp2 acts as such a regulatory domain. This domain promotes decapping complex assembly and Dcp2 activation by interacting with the decapping enhancer Hedls. The same domain restricts cellular Dcp2 levels by targeting uncomplexed Dcp2 for ubiquitin-mediated proteasomal degradation. Therefore, the cellular activity of Dcp2 is controlled by a competition between decapping complex formation and ubiquitination. This two-pronged mechanism to control Dcp2 function might serve to restrict the activity of Dcp2 outside the decapping complex and to modulate Dcp2 levels according to cellular needs.
MATERIALS AND METHODS
Plasmid constructs.
Expression plasmids, created using derivatives of pcDNA3 (Invitrogen), for tetracycline-regulated expression of a β-globin reporter for AU-rich element (ARE)-mediated mRNA decay (β-globin mRNA with the ARE from granulocyte-macrophage colony-stimulating factor [β-GMCSF mRNA]) and constitutively expressed internal control mRNA (a chimeric β-globin-glyceraldehyde 3-phosphate dehydrogenase mRNA [β-GAP]), as well as expression plasmids for N-terminally Myc- and FLAG-tagged Dcp2, Dcp2 E148Q, Hedls, Dcp1a, Edc3, Rck/p54, DsRed, and hnRNP A1, have been previously described (7, 19, 30–32). Plasmids expressing Myc-Dcp2 containing deletion or point mutations were created using the QuikChange site-directed mutagenesis method (Stratagene). DsRed fusions were created by subcloning DsRed into the BamHI site of Dcp2 expression plasmids. Tetracycline-inducible stable cell lines containing Myc- or 5× Myc-tagged Dcp2 were created using the Flp-In T-REx system (Invitrogen), according to the manufacturer's instructions: the Myc-tagged Dcp2 plasmids used for integration were generated by inserting annealed Myc oligonucleotides into the HindIII site of pcDNA5-frt-TO (Invitrogen). Then, Dcp2 was subcloned between the BamHI and NotI sites. To generate the 5× Myc-tagged Dcp2 plasmid, a PCR product containing the sequence for 4 repeating Myc tags was inserted between the HindIII and BamHI sites. Sequences are available upon request. Stable human embryonic kidney (HEK) 293T T-REx cell lines expressing FLAG-tagged Dcp2 were described earlier (19).
Antibodies.
The following antibodies were used for Western blotting at the indicated dilutions in TBST (100 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) containing 5% milk: anti-hDcp1a (1:2,000) (31), anti-Hedls (1:1,000) (19), anti-Rck/p54 (1:1,000; catalog number A300-461A; Bethyl Laboratories), anti-Edc3 (1:1,000) (19), anti-HuR (1:25,000) (33), anti-HuR (1:1,000; Santa Cruz Biotechnology), anti-Dcp2 (1:400) (9), anti-Nudt16 (1:200) (34), anti-Myc 9B11 (1:1,000; Cell Signaling), anti-FLAG M2 (1:1,000; Sigma), anti-TRIM21 (1:500; Santa Cruz Biotechnology), anti-Cdc34A (1:500; Santa Cruz Biotechnology), and anti-Cdc34B (1:500; Cell Signaling). Rabbit anti-Dcp2 and rat anti-Nudt16 were generous gifts from Megerditch Kiledjian (9, 34).
siRNAs.
All small interfering RNAs (siRNAs) were purchased from Dharmacon. The siRNA sequences were as follows: for luciferase (Luc) control siRNA, 5′-CGUACGCGGAAUACUUCGAUU-3′ and 5′-UCGAAGUAUUCCGCGUACGUU-3′; for Hedls open reading frame (ORF) siRNA (see Fig. 1), 5′-GAGUUAAAGAUGUGGUGUAUU-3′ and 5′-UACACCACAUCUUUAACUCUU-3′; for the Hedls 3′ untranslated region (UTR) siRNA pool (see Fig. 2B), On-Target modified, 5′-CACUGAAGGCCAGCAGACAUU-3′, 5′-UGUCUGCUGGCCUUCAGUGUU-3′, 5′-GUGUGGUAGUCAGAAGGUUUU-3′, and 5′-AACCUUCUGACUACCACACUU-3′; for Edc3 siRNA, 5′-GCACUGAAAUAAAGCUGAAUU-3′ and 5′-UUCAGCUUUAUUUCAGUGCUU-3′; for the Dcp2 ORF siRNA pool, siGenome SMARTpool M008425; and for the Dcp2 3′ UTR siRNA pool, On-Targetplus SMARTpool L008425. The Xrn1 siRNA was described by Eberle et al. (35). The following siRNAs were designed on the basis of the short hairpin RNAs described previously (36): for Cdc34A, 5′-GGAAGUGGAAAGAGAGCAAUU-3′ and 5′-UUGCUCUCUUUCCACUUCCUU-3′; for Cdc34B, 5′-GGAAAUGGAGAGACAGUAAUU-3′ and 5′-UUACUGUCUCUCCAUUUCCUU-3′; and for TRIM21, 5′-GGAAAUUGCAAUAAAGAGAUU-3′ and 5′-UCUCUUUAUUGCAAUUUCCUU-3′.
FIG 1.

Hedls depletion causes accumulation of a deadenylated ARE-containing mRNA intermediate. (A) Northern blots monitoring the degradation of β-globin reporter mRNA (β-GMCSF mRNA) containing an ARE from granulocyte-macrophage colony-stimulating factor mRNA in tetracycline-controlled pulse-chase mRNA decay assays in HeLa Tet-off cells incubated with the siRNAs indicated at the tops of the panels. The Hedls siRNA is targeted against the Hedls ORF. Numbers above the lanes refer to the time (in minutes) after transcription was stopped by addition of tetracycline. β-GAP is a constitutively expressed internal control mRNA. β-GMCSF-A0, position of a faster-migrating deadenylated species. (B) Northern blots of samples as for panel A taken at 0 min of chase (i.e., time zero [t = 0]) and treated or not treated with RNase H and oligo(dT) (RH/dT).
FIG 2.
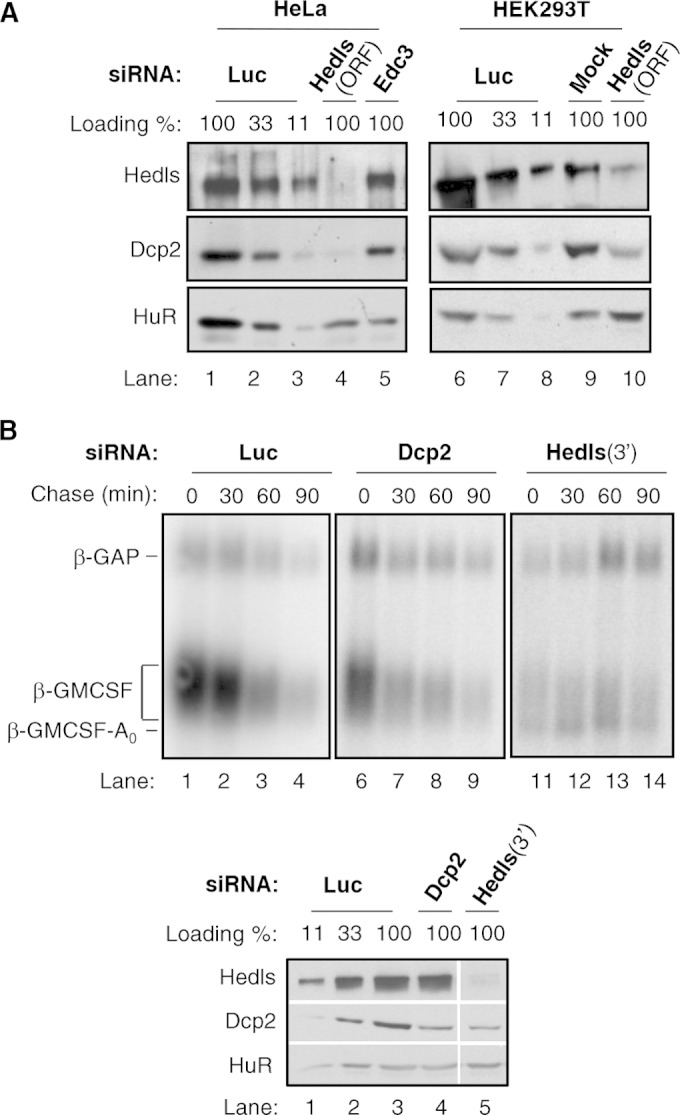
Hedls promotes the accumulation and activity of Dcp2. (A) Western blots monitoring the levels of the indicated proteins in HeLa (left) and HEK 293T (right) cells either transfected in the absence of siRNA (Mock) or transfected with siRNAs targeting the Hedls ORF, Edc3, or Luc. Antibodies against endogenous proteins were used to detect Hedls and Dcp2. HuR was used as a loading control. Control samples from luciferase siRNA transfections were titrated at 100%, 33%, and 11% in lanes 1 to 3, respectively, and 6 to 8, respectively. (B) (Top) Northern blots monitoring the degradation of β-GMCSF mRNA in HeLa Tet-off cells treated with siRNAs targeting Luc, Dcp2, or the Hedls 3′ UTR (3′). (Bottom) Western blots showing the efficiency of depletion. Lane 5 is from the same gel and exposure as lanes 1 to 4.
mRNA decay and Northern blot assays.
Pulse-chase mRNA decay assays were performed as described earlier (31). HeLa Tet-off cells (Clontech) cultured in 3.5-cm wells in 2 ml of Dulbecco modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin-streptomycin (PS) solution (Invitrogen) were transfected 72 h before reporter mRNA induction with siRNAs at a final concentration of 20 nM using the siLentFect lipid reagent (Bio-Rad) according to the manufacturer's protocol. In Dcp2 siRNA titration experiments for which the results are presented in Fig. S2B in the supplemental material, the total amount of siRNA was adjusted to 20 nM with addition of Luc siRNA. On the following day, cells were transfected with plasmids using the TransIT-HeLaMonster transfection reagent (Mirus) per the manufacturer's protocol and maintained in 50 ng/ml of tetracycline to repress transcription of reporter mRNAs. Cells for which the results are presented in Fig. S1B in the supplemental material were transfected with plasmids expressing β-GMCSF mRNA (1 µg), β-GAP (50 ng), and Myc-Dcp2 E148Q (2.5 µg) (for samples for which the results are presented in lanes 6 to 10 in Fig. S1B in the supplemental material), in addition to the empty pcDNA3 vector to a total of 4 μg. Cells for which the results are presented in Fig. 1A and B and 2B were transfected with plasmids expressing 1 μg β-GMCSF mRNA and 30 ng (or 50 ng for Fig. 1A) β-GAP, in addition to the empty pcDNA3 vector to a total of 2 μg. Two days later (3 days for assays in which the results are presented in Fig. S1B in the supplemental material), to induce β-GMCSF mRNA transcription, cells were washed once with phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, pH 7.4) and incubated in DMEM–10% FBS–1% PS lacking tetracycline for 6 h, before tetracycline was added back at 1 μg/ml to stop transcription and start the chase experiment. The first time point (time zero) at which RNA samples were collected was 30 min after tetracycline addition to ensure complete transcriptional arrest. Total cellular RNA was prepared by addition of the TRIzol reagent (Invitrogen) directly to cells, followed by RNA isolation per the manufacturer's protocol and analysis of the RNA by Northern blotting as previously described (31). Deadenylated control RNA samples were produced using oligo(dT)24 and RNase H (NEB) as described previously (37). Total cellular protein was collected after the cells were washed once with PBS by addition of sodium dodecyl sulfate (SDS) load buffer (100 mM Tris-HCl, pH 6.8, 200 mM dithiothreitol, 4% SDS, 0.2% bromophenol blue, 20% glycerol) directly to cells, followed by rigorous pipetting to shear genomic DNA. Protein samples were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) followed by Western blotting, to assess exogenous protein expression and siRNA knockdown efficiencies.
Translation shutoff and Western blotting assays.
HEK 293T cells seeded in 2.2-cm wells in 1 ml of DMEM–10% FBS–1% PS were transfected using the TransIT-293 transfection reagent (Mirus) per the manufacturer's protocol. Cells for which the results are presented in Fig. 3A to C were transfected with a total of 1.5 μg of DNA (adjusted with the empty pcDNA3 vector) with 50 ng FLAG- or Myc-hnRNP A1 expression plasmid and 1.45 μg FLAG- or Myc-Dcp2 (see Fig. 3A and B and C, lanes 1 to 4), 1 μg Myc-Dcp2 plus 0.45 μg Myc-Hedls expression plasmids (see Fig. 3C lanes 5 to 8), 0.5 μg FLAG-Dcp1a, -Hedls, -Edc3, or -Rck/p54 (see Fig. 3A), or 0.5 μg pSuperPuro and 0.5 μg pcDNA Myc or pcDNA Myc-Hedls (see Fig. 3D). Cells for which the results are presented in Fig. 5A were transfected with 0.8 μg wild-type or mutant Myc-Dcp2 expression plasmids and 0.2 μg the empty pcDNA3 vector. Cells for which the results are presented in Fig. 5B and 6A were transfected with 1.25 μg wild-type or mutant Myc-Dcp2 and 50 ng Myc-hnRNP A1 expression plasmids and the empty pcDNA3 vector to a total of 1.5 μg. Two days after transfection, translation was arrested with puromycin (Sigma) at 5 μg/ml or cycloheximide (Sigma) at 50 μg/ml, and total cellular protein was isolated at the times indicated in the figures by addition of SDS load buffer as described above. For the assays in which the results are presented in Fig. 3D, at 24 h after transfection the cells were treated with 5 μg/ml puromycin for 16 h to select transfected cells before addition of SDS load buffer. For the assays in which the results are presented in Fig. 6A, the proteasome inhibitor MG132 (Sigma) or lactacystin (Sigma) was added at 10 μM for 2 h prior to translational arrest. For the assays in which the results are presented in Fig. 2A, steady-state protein levels were analyzed after siRNA transfection as described above, a second transfection was performed 48 h after the first one, and samples were collected 48 h later. Samples were analyzed by SDS-PAGE followed by Western blotting. Quantification of signal intensities for half-life calculations was performed using the ECL Plus detection reagent (Amersham), and the signals were scanned on a Typhoon Trio scanner (Amersham).
FIG 3.

Hedls stabilizes Dcp2. (A) Translation shutoff assays in HEK 293T cells transiently expressing the indicated FLAG-tagged decapping factors and hnRNP A1 as a control. The numbers above the lanes refer to the times (in hours) after translation shutoff with cycloheximide (Chx). (B) The same as panel A, but monitoring Myc-tagged proteins and using puromycin (Puro) in place of cycloheximide for translation shutoff. (C) (Top) The same as panel B, but monitoring the effect of transiently expressing Dcp2 in the absence (lanes 1 to 4) or presence (lanes 5 to 8) of coexpressed Hedls. (Bottom) The levels of Dcp2 with or without Hedls relative to the amount of the internal hnRNP A1 control were calculated for each time point and graphed. Error bars represent standard errors of the means (SEMs) from three independent experiments, and the P value (determined by a paired two-tailed Student's t test) refers to the difference between the calculated half-lives from the individual experiments. (D) Western blots showing the levels of endogenous Dcp2 in HEK 293T cells in the absence (−) or presence (+) of exogenous Hedls. Transfected cells were enriched by puromycin selection. The average ± SEM Dcp2 level in Hedls-overexpressing over control cells from three independent experiments is shown below the blots; the P value was calculated using a paired two-tailed Student's t test. (E) Western blots of lysates from HEK 293T T-REx cells with stably integrated tetracycline (Tet)-inducible 5× Myc-tagged Dcp2 grown in the absence (−) or presence (+) of tetracycline. The 5× Myc-tagged Dcp2 and endogenous Dcp2 were detected using an antibody against endogenous Dcp2. HuR served as a loading control.
FIG 5.
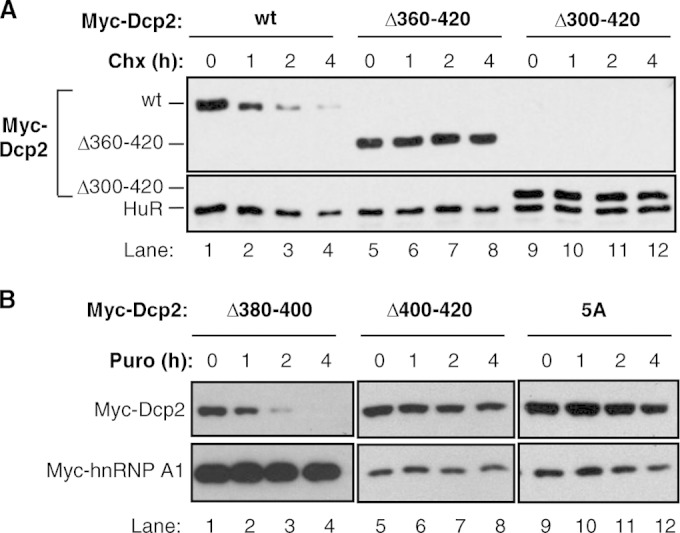
The Dcp2 C terminus is required for Dcp2 instability. (A) Translation shutoff assays in HEK 293T cells transiently expressing the indicated Myc-tagged Dcp2 proteins. wt, wild-type Dcp2. The times above the lanes refer to the times (in hours) after translation shutoff using cycloheximide (Chx). Endogenous HuR was monitored as a control. (B) The same as panel A, but monitoring different Dcp2 mutant proteins, as indicated. Translation shutoff was performed with puromycin (Puro).
FIG 6.
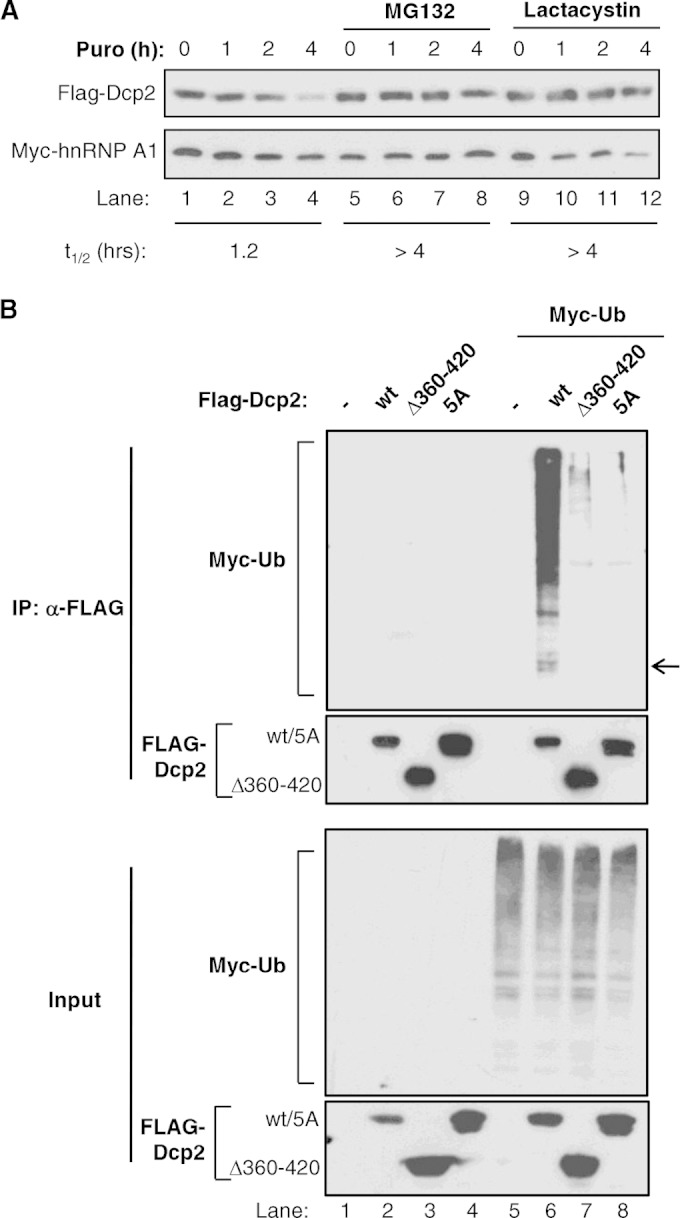
Dcp2 is ubiquitinated and degraded by the proteasome. (A) Translation shutoff assays in HEK 293T cells transiently expressing FLAG-tagged Dcp2 in the absence or presence of MG132 (lanes 5 to 8) or lactacystin (lanes 9 to 12), added 2 h prior to translation shutoff with puromycin (Puro). Cotransfected Myc-hnRNP A1 was used as a stable protein control, and numbers at the top refer to the times (in hours) after translation shutoff. The half-lives (t1/2s) of the Myc-Dcp2 protein were calculated after normalization of signal intensities to Myc-hnRNP A1 and are indicated at the bottom. (B) Western blots monitoring the ubiquitination of the FLAG-tagged Dcp2 wild type (wt) or mutants (Dcp2 Δ360–420 and 5A) transiently expressed in the absence (lanes 1 to 4) or presence (lanes 5 to 8) of Myc-tagged ubiquitin (Ub) and subjected to anti-FLAG IP. (Top) Immunoprecipitated samples; (bottom) input samples. The arrow indicates the position of FLAG-Dcp2 migration.
Co-IP assays.
Immunoprecipitation (IP) assays for which the results are presented in Fig. 4A and C were performed as described previously (7). HEK 293T cells seeded in 10-cm plates in 10 ml DMEM–10% FBS–1% PS were cotransfected using the TransIT-293 transfection reagent (Mirus) with 2 μg FLAG-Hedls expression vector and either 7 μg Myc-tagged Dcp2 from which amino acids 360 to 420 were deleted (Myc-Dcp2 Δ360–420) plus 6 μg the empty pcDNA3 vector or 13 μg of the other Myc-Dcp2 expression vectors, as indicated in the figures. At 48 h after transfection, the cells were washed with 10 ml PBS and then lysed by incubating the cells on ice for 10 min in 800 μl of ice-cold hypotonic lysis buffer (0.1% Triton X-100, 10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 2 mM EDTA, 1 μM aprotinin [Sigma], 1 μM leupeptin [Sigma], 1 mM phenylmethylsulfonyl fluoride [Sigma]). The lysates were incubated on ice for another 5 min with RNase A (Sigma), added to 125 μg/ml, and NaCl, added to 150 mM. Cell debris was removed by centrifugation at 14,000 rpm for 15 min at 4°C, and an input sample was collected before the remaining supernatant was nutated for 2 to 3 h at 4°C with 40 μl of anti-FLAG M2–agarose (Sigma). Beads were washed 8 times with NET-2 buffer (50 mM Tris-HCl, pH 6.5, 150 mM NaCl, 0.05% Triton X-100). Protein complexes were eluted with 30 μl SDS load buffer and analyzed by SDS-PAGE followed by Western blotting. Coimmunoprecipitation (co-IP) assays for which the results are presented in Fig. 4D were performed in the same way, except that we used HEK 293T T-REx cell lines in 15-cm plates stably expressing tetracycline-inducible FLAG-tagged Dcp2 proteins at nearly endogenous levels, using 2 μg/ml and 16 ng/ml tetracycline for Dcp2 and Dcp2 Δ360–420, respectively, and probed for copurifying endogenous proteins. Co-IP assays for which the results are presented in Fig. 6B were performed from HEK 293T cells in 15-cm plates transiently transfected using the TransIT-293 transfection reagent (Mirus) with 12.5 μg a Myc-tagged ubiquitin expression vector and FLAG-tagged Dcp2 (25 μg), Dcp2 Δ360–420 (12.5 μg), or Dcp2 5A (18.75 μg) and the empty pcDNA3 vector to a total of 50 μg. Forty hours later, cells were treated 10 μM MG132 for 2 h, prior to lysis in 2 ml of 0.5% Na deoxycholate, 150 mM NaCl, 0.1% SDS, 50 mM Tris-HCl, pH 7.5. IPs were performed as described above using anti-FLAG M2, and samples were subjected to Western blotting.
FIG 4.

The Dcp2 C terminus is required for Hedls interaction and decapping complex assembly. (A) Co-IP assays with RNase-treated lysates from HEK 293T cells transiently coexpressing Myc-tagged wild-type (wt) or mutant Dcp2 proteins, as indicated, with FLAG-tagged Hedls and immunoprecipitated (IP) using anti-FLAG antibody. Total input samples (IN) were collected prior to immunoprecipitation. HuR served as a negative control. Mouse light-chain IgG was detected in the immunoprecipitated samples, as indicated. (B) Schematic of Dcp2 showing the catalytic region in light gray and the C-terminal (Ct) region in dark gray. Segments that were deleted are delineated with boxes. The Hedls interaction domain is shown, and the conserved hydrophobic residues that were mutated are shown in red and shaded. H.s, Homo sapiens; M.m, Mus musculus; G.g, Gallus gallus; X.l, Xenopus laevis; D.r, Danio rerio. (C) The same as panel A, but monitoring the indicated Dcp2 mutant proteins. (D) The same as panel A, but using HEK 293T T-REx cells stably expressing the indicated FLAG-tagged Dcp2 proteins and monitoring the copurification of endogenous proteins, as indicated. Total, 5% of total extract.
RESULTS
Hedls promotes the accumulation and catalytic activity of the Dcp2 decapping enzyme.
Previous studies have implicated Hedls as an important component of the human Dcp2 decapping complex, which stimulates Dcp2 activity in vitro (19, 20) and is important for the activation of decapping by tethered mRNA decay activators (20). Consistent with this, the mRNA decay assays whose results are presented in Fig. 1A, which monitor the degradation of an mRNA (β-GMCSF) targeted for AU-rich element (ARE)-mediated mRNA decay, show the accumulation of a fast-migrating mRNA decay intermediate 0 to 60 min after transcription shutoff in the presence of an siRNA that efficiently targets Hedls (Fig. 1A, lanes 6 to 10, band indicated by β-GMCSF-A0; see also Fig. S1A in the supplemental material). This intermediate accumulates as a consequence of Hedls depletion, as it is also observed in the presence of an independent siRNA targeting Hedls (see Fig. 2B) but is not observed when cells are treated with control luciferase siRNA (Fig. 1A, lanes 1 to 5). The intermediate corresponds to a deadenylated mRNA decay intermediate, as evidenced by the accumulation of a similarly sized intermediate when 5′-to-3′ exonucleolytic decay is impaired by siRNA-mediated depletion of Xrn1 (lanes 11 to 15), when decapping is impaired by transient expression of a dominant-negative mutant Dcp2 protein (Dcp2 E148Q) (see Fig. S1B in the supplemental material), and when RNA samples collected from the first time points of the Hedls and luciferase knockdown assays are treated with oligo(dT) and RNase H to remove the poly(A) tail (Fig. 1B). The overall decay rate of the β-GMCSF mRNA was not noticeably affected upon Hedls or Xrn1 depletion or upon expression of Dcp2 E148Q, consistent with degradation from the mRNA 3′ end compensating for the defect in 5′-to-3′ decay (38–40). We conclude that depletion of Hedls causes a stall in ARE-mediated mRNA decay after deadenylation, consistent with an important role for Hedls in cellular decapping.
A defect in decapping upon Hedls depletion could be due to impaired Dcp2 activity or to effects on cellular Dcp2 levels. To discriminate between these possibilities, we monitored the effect of Hedls depletion on Dcp2 accumulation. As seen in Fig. 2A (lanes 4 and 10), depletion of Hedls caused a strong reduction in the level of endogenous Dcp2 protein, as observed in two independent human cell lines. In contrast, Nudt16, another known cytoplasmic decapping enzyme (41, 42), was unaffected by Hedls knockdown (see Fig. S2A in the supplemental material). Therefore, cellular levels of Dcp2 are limited by Hedls, suggesting that the accumulation of the deadenylated ARE-containing mRNA intermediate observed upon Hedls depletion in Fig. 1 is, at least in part, a result of reduced Dcp2 levels.
We and others previously observed that Hedls stimulates decapping by Dcp2 in vitro (11, 19, 20). To determine whether Hedls, in addition to its effect on Dcp2 levels, also enhances the catalytic activity of Dcp2 in cells, we assayed the effect on β-GMCSF mRNA of using an siRNA to deplete Dcp2 to levels similar to those observed upon Hedls knockdown (Fig. 2B, bottom; compare lanes 4 and 5; see also Fig. S2B in the supplemental material). As seen in the mRNA decay assay in Fig. 2B (top), depletion of Dcp2 alone failed to cause significant accumulation of the deadenylated β-GMCSF mRNA intermediate, despite the strong accumulation of this intermediate when Dcp2 is present at similarly low levels due to Hedls knockdown (compare lanes 11 to 14 with lanes 6 to 9 in Fig. 2B, top; see also Fig. S2B in the supplemental material). These observations suggest that Hedls enhances cellular Dcp2 activity by at least two mechanisms: by promoting the cellular accumulation of Dcp2 as well as the activity of Dcp2.
Hedls stabilizes the Dcp2 protein.
We next investigated the mechanism by which Hedls promotes the cellular accumulation of Dcp2. We previously observed that transient expression of Dcp2 is strongly enhanced in human cells when Hedls is coexpressed (19), suggesting a possible role for Hedls in stabilizing the Dcp2 protein. To test whether Dcp2 is regulated by proteolysis, we monitored the degradation of exogenously expressed Dcp2 in translation shutoff assays. As can be seen in Fig. 3A and B, exogenously expressed Dcp2 is rapidly degraded when translation is shut off by the addition of the translation inhibitor cycloheximide or puromycin. This is in contrast to the findings obtained with other decapping complex factors, Dcp1a, Hedls, Edc3, and Rck/p54, all of which are degraded with half-lives well beyond the 4-h time course (Fig. 3A).
Next, we tested whether Hedls affects Dcp2 protein stability. As can be seen in the translation shutoff assay whose results are presented in Fig. 3C, coexpression of Hedls had a strong stabilizing effect on exogenously expressed Dcp2. Moreover, exogenous expression of Hedls caused increased accumulation of endogenous Dcp2 (Fig. 3D). Taken together, the effects of Hedls on exogenous and endogenous Dcp2 observed in Fig. 2 and 3 show that Hedls stabilizes exogenous as well as endogenous Dcp2 and thereby functions as a limiting factor for Dcp2 protein accumulation in human cells. To further investigate this idea, we tested the prediction that exogenous expression of Dcp2 should titrate Hedls and result in reduced levels of endogenous Dcp2. Indeed, as seen in Fig. 3E, tetracycline-induced expression of 5× Myc-tagged Dcp2 in a stable HEK 293T T-REx cell line caused reduced levels of endogenous Dcp2 compared to the levels obtained in the same cell line in which exogenous 5× Myc-Dcp2 was not induced (compare Dcp2 levels with and without tetracycline in Fig. 3E).
The Dcp2 C terminus is critical for Hedls association and decapping complex assembly.
To further investigate the impact of Hedls on Dcp2, we wished to identify mutant forms of Dcp2 that fail to interact with Hedls. A previous study using a cellular fluorescence colocalization assay suggested that Hedls interacts with the Dcp2 C terminus (43). This was recently confirmed by biochemical studies (20). Consistent with this, the coimmunoprecipitation assays whose results are presented in Fig. 4A showed complex formation in RNase-treated HEK 293T cell extracts of Hedls with wild-type Dcp2 but not with Dcp2 with deletion of the C-terminal 60 amino acids (compare lanes 1 and 2 with lanes 3 and 4 in Fig. 4A). Moreover, indirect immunofluorescence assays revealed that the C-terminal region of Dcp2 is both necessary and sufficient for Dcp2 accumulation in P bodies in response to Hedls coexpression (see Fig. S3 in the supplemental material). To further map the region of Dcp2 responsible for Hedls association, 20-amino-acid deletion mutations and point mutations in conserved residues within the Dcp2 C terminus were generated (Fig. 4B). As seen in Fig. 4C, deletion of amino acids 380 to 400 or 400 to 420 of Dcp2 prevented coprecipitation with Hedls, whereas deletion of amino acids 360 to 380 had no effect (compare lanes 2, 4, and 6 in Fig. 4C). Moreover, mutation of five conserved hydrophobic residues within the C-terminal 20 amino acids of Dcp2 to either arginines (5R) or alanines (5A) prevented the Hedls association (compare lanes 8 and 10 with lane 2 in Fig. 4C). Therefore, consistent with recent reports (20, 43), the Dcp2 C-terminal 40 amino acids and conserved residues within this region are critical for complex formation with Hedls.
Evidence that Hedls serves as a scaffold for the association of Dcp2 with its cofactor, Dcp1, has been presented (19, 20). Consistent with this, deletion of the Dcp2 C-terminal region responsible for the Hedls association caused a loss of the association of Dcp2 with Dcp1a, as monitored by co-IP of RNase-treated cell lysates from HEK 293T cells stably expressing Dcp2 at nearly endogenous levels (Fig. 4D, lanes 5 and 6). Notably, decapping enhancers Edc3 and Rck/p54 were lost from the complex as well (Fig. 4D). Therefore, the Dcp2 C terminus plays a central role not only in the interaction with Hedls and Dcp1a but also in the assembly of human Dcp2 with the remainder of the decapping complex.
The C terminus of Dcp2 promotes Dcp2 instability.
We predicted that if Hedls stabilizes Dcp2 through its interaction with the Dcp2 C terminus, then mutations that prevent the Hedls association should result in a constitutively unstable Dcp2 protein. Surprisingly, however, despite the inability of C-terminally truncated Dcp2 to assemble with Hedls or the remainder of the decapping complex (Fig. 4), Dcp2 proteins lacking the C-terminal 60 or 120 amino acids (Dcp2 Δ360–420 and Dcp2 Δ300–420) were highly stabilized compared to the stability of wild-type Dcp2 (Fig. 5A; compare lanes 5 to 8 and 9 to 12 with lanes 1 to 4). Similarly, deletion of the Dcp2 C-terminal 20 amino acids (Dcp2 Δ400–420; Fig. 5B, lanes 5 to 8) and mutation of the five C-terminal conserved hydrophobic residues to alanines (Fig. 5B, lanes 9 to 12) resulted in strong stabilization of the Dcp2 protein. These observations suggest that Dcp2 contains a C-terminal region that actively targets wild-type Dcp2 for degradation and that this instability region overlaps with the binding site for Hedls.
The instability and Hedls-binding regions of Dcp2 do not fully overlap, as Dcp2 Δ380–400, which fails to interact with Hedls (Fig. 4C), was unstable when exogenously expressed, similar to full-length Dcp2 (Fig. 5B, lanes 1 to 4). Consistent with the observation that Hedls stabilizes Dcp2 (Fig. 3C), the Dcp2 Δ380–400 mutant protein that could not bind Hedls remained unstable even when Hedls was coexpressed (see Fig. S4 in the supplemental material). Collectively, these observations suggest that Dcp2 contains a region within its C-terminal 20 amino acids that actively targets Dcp2 for rapid degradation and that the assembly of Dcp2 with Hedls, which requires a larger region of the C-terminal domain, protects the Dcp2 protein from decay.
Dcp2 is degraded by the ubiquitin-proteasome pathway.
Recent studies have presented evidence that proteasome-mediated degradation pathways exist to control the stoichiometry of protein complexes by specifically targeting those components of protein complexes that fail to assemble with their complex partners (44) (45). Our observation that the Dcp2 C terminus promotes decapping complex formation in competition with protein degradation (Fig. 4 and 5) motivated us to test whether a ubiquitin-mediated degradation mechanism exists to limit cellular levels of uncomplexed Dcp2. We therefore tested whether the Dcp2 degradation promoted by the Dcp2 C terminus involves the ubiquitin-mediated proteasomal degradation pathway. Indeed, exogenously expressed Dcp2 is stabilized by two proteasome inhibitors, MG132 and lactacystin (Fig. 6A). Moreover, immunoprecipitation of wild-type Dcp2 coexpressed with Myc-tagged ubiquitin reveals the polyubiquitination of Dcp2, as observed by high-molecular-weight bands in the anti-Myc Western blot (Fig. 6B, top, lane 6). Importantly, this signal is strongly diminished with the stabilized Dcp2 Δ360–420 and 5A mutant proteins, despite equal expression levels (compare lane 6 with lanes 7 and 8 in Fig. 6B, top). Collectively, our observations suggest that the levels and activity of the Dcp2 decapping enzyme are controlled by a competition between Hedls and the ubiquitin-proteasome pathway for the Dcp2 C terminus. The outcome of this competition dictates whether Dcp2 forms a decapping complex and is activated or is subjected to ubiquitin-mediated proteasomal degradation.
DISCUSSION
Decapping is an important step in mRNA turnover that simultaneously shuts down translation initiation and initiates 5′-to-3′ exonucleolytic degradation of the mRNA body. Therefore, decapping needs to be tightly regulated to promote the decapping of mRNAs specifically targeted for decay. Here, we present evidence that the activity and levels of the human Dcp2 decapping enzyme are controlled by a C-terminal regulatory region, which plays a dual role in Dcp2 regulation by stimulating the assembly of Dcp2 with the decapping complex via Hedls and promoting ubiquitin-mediated proteasomal degradation of uncomplexed Dcp2 (Fig. 7). This competition between Dcp2 degradation and complex formation controls the cellular levels and activity of the Dcp2 decapping enzyme.
FIG 7.
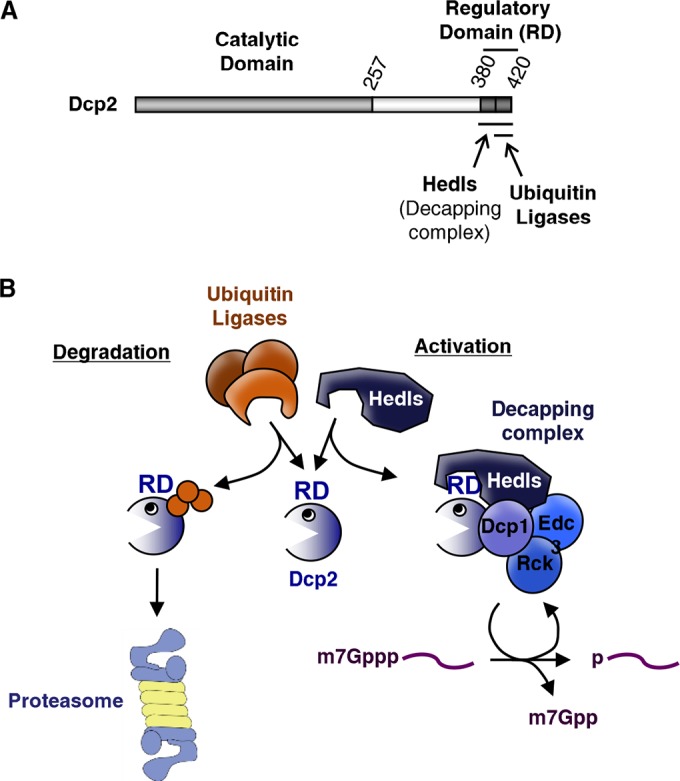
Competition between Hedls and ubiquitin ligases for a Dcp2 C-terminal regulatory domain controls Dcp2 activity and stability. (A) Schematic of Dcp2 showing the regulatory domain and binding sites for Hedls and ubiquitin ligases. (B) Competition between Hedls and ubiquitin ligases for the Dcp2 regulatory domain (RD) controls Dcp2 stability and activity. Orange spheres, ubiquitin moieties.
Multiple lines of evidence support a central role for Hedls in the activity of the human Dcp2 decapping complex. First, depletion of Hedls causes the accumulation of a deadenylated intermediate in the ARE-mediated mRNA decay pathway beyond that observed with reduced levels of Dcp2 alone (Fig. 1 and 2). Second, deletion and substitution mutation of conserved hydrophobic residues within the C terminus of Dcp2, which disrupt the Dcp2 interaction with Hedls, result in the loss of an association of Dcp2 with Dcp1 (Fig. 4D) (20) and with other decapping complex components (Fig. 4D). These observations, along with previous observations that Hedls promotes Dcp2 decapping activity in vitro (11, 19, 20) and of mRNAs tethered to mRNA decay activators in cells (20), place Hedls as a central component of the decapping complex. An important topic for future study is whether Hedls stimulates Dcp2 activity solely by serving as a scaffold for the decapping complex or whether Hedls also more directly stimulates the Dcp2 enzyme, for example, by promoting Dcp2 catalytic activity. A scaffolding function for Hedls is consistent with our previous observation that overexpression of Hedls causes inhibition of decapping (19), since overexpression of scaffolds can lead to the physical separation of cofactors.
A surprising finding from our studies is that the same Dcp2 C-terminal region responsible for the Hedls interaction also actively targets Dcp2 for ubiquitin-mediated proteolysis. This conclusion is supported by the observations that full-length Dcp2 is unstable, ubiquitinated, and stabilized by proteasome inhibitors (Fig. 3 and 6). In contrast, mutant Dcp2 proteins containing deletions or point mutations within the C terminus show reduced ubiquitination (Fig. 6) and are strongly stabilized (Fig. 5). Therefore, cellular Dcp2 levels are regulated by the Dcp2 C terminus. The specific mechanism by which the Dcp2 C terminus promotes Dcp2 instability remains to be determined. Either it could promote a conformation of the Dcp2 protein that renders it vulnerable to ubiquitination or it could serve as a platform for recruitment of cellular ubiquitin ligases. Consistent with the latter, IP followed by liquid chromatography and tandem mass spectrometry (LC-MS/MS) assays revealed ubiquitin ligases in complex with Dcp2 dependent on the Dcp2 C terminus (see Fig. S5A and B in the supplemental material); however, depletion of these ligases showed only modest effects on Dcp2 stability, suggesting redundancy in the ligases that act on Dcp2 (see Fig. S5C in the supplemental material). Degradation of Dcp2 likely requires regions in addition to the C terminus, as mutation of all lysines within this region failed to stabilize Dcp2 (see Fig. S6A in the supplemental material) and the Dcp2 60 C-terminal amino acids were not sufficient to cause destabilization of a DsRed fusion protein (see Fig. S6B in the supplemental material). An important goal of future studies is to reveal the detailed mechanism of Dcp2 ubiquitination and degradation and the responsible ubiquitin ligases.
The targeting of Dcp2 for degradation is in competition with decapping complex formation, as evidenced by the overlapping Hedls-binding and ubiquitin-targeting regions within the Dcp2 C terminus (Fig. 4 and 5). Moreover, exogenous Dcp2 and endogenous Dcp2 are stabilized when coexpressed with Hedls, whereas depletion of endogenous Hedls or expression of exogenous Dcp2 causes a concomitant reduction in endogenous Dcp2 levels (Fig. 2 and 3). This suggests that Hedls and ubiquitin ligases are competing for the Dcp2 C-terminal regulatory region (Fig. 7B). This competition could be either physical or kinetic and would dictate whether Dcp2 is assembled into a decapping complex and activated or targeted for destruction by the proteasome.
Why do human cells have a mechanism to actively degrade uncomplexed Dcp2? The observation that point mutations and small deletions in the Dcp2 C terminus are sufficient to stabilize Dcp2 (Fig. 5), despite these mutant Dcp2 proteins being unable to assemble into a decapping complex (Fig. 4), suggests that there is evolutionary pressure to maintain proteasomal targeting of uncomplexed Dcp2. Recent reports have presented evidence for proteolytic pathways that ensure correct complex stoichiometry by targeting uncomplexed components of protein complexes (45). In a similar manner, the pathway uncovered here could serve to ensure an appropriate abundance of Dcp2 relative to the amounts of the remaining decapping complex components. Interestingly, Dcp2 appears to be the only decapping complex component subject to rapid proteolysis (Fig. 3A), suggesting that it is critical to specifically control the level of the catalytic component of this complex and maintain it at low levels compared to the levels of the other subunits. Consistent with this, global proteomics analyses suggest that in human HeLa cells, Dcp2 is ≈10-fold less abundant than other decapping complex components (46), and we have previously observed that overexpression of Dcp2 accelerates ARE-mediated mRNA decay (31). This dual mechanism to restrict Dcp2 activity and levels according to cofactor levels could serve to prevent accumulation of unregulated decapping enzyme and promiscuous targeting of G-capped RNAs. Additionally, the ubiquitin-mediated proteolysis of Dcp2 could serve as a mechanism to regulate Dcp2 levels according to cellular cues. An exciting possibility is that Dcp2 proteolysis could serve to adjust cellular Dcp2 levels according to the levels of substrate mRNAs, for example, if decapping complex assembly—and, therefore, Dcp2 stabilization—is mRNA substrate stimulated. Our findings also raise the question of whether other mRNA decay enzymes are under similar proteolytic control. Interestingly, the Escherichia coli RNA decay enzyme RNase R has previously been observed to be controlled by proteolysis (47–51). Future studies should reveal whether active repression of uncomplexed RNA decay enzymes is a general principle that serves to restrict cellular levels of uncomplexed and potentially unregulated nucleases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant R01 GM077243 from the National Institutes of Health to J.L.-A. S.L.E. was supported by National Institutes of Health Institutional Training Grant in Signaling and Cellular Regulation no. T32 GM08759.
We thank Min Han for use of the fluorescence microscope and Megerditch Kiledjian for the anti-Dcp2 and anti-Nudt16 antibodies. We thank the J. Lykke-Andersen lab for helpful discussions; in particular, we thank Sebastien Durand and Rea Lardelli for critical comments on the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01517-14.
REFERENCES
- 1.Lewis JD, Izaurralde E. 1997. The role of the cap structure in RNA processing and nuclear export. Eur J Biochem 247:461–469. doi: 10.1111/j.1432-1033.1997.00461.x. [DOI] [PubMed] [Google Scholar]
- 2.Cougot N, van Dijk E, Babajko S, Seraphin B. 2004. ‘Cap-tabolism.’ Trends Biochem Sci 29:436–444. doi: 10.1016/j.tibs.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Franks TM, Lykke-Andersen J. 2008. The control of mRNA decapping and P-body formation. Mol Cell 32:605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coller J, Parker R. 2004. Eukaryotic mRNA decapping. Annu Rev Biochem 73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- 5.Eulalio A, Behm-Ansmant I, Izaurralde E. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- 6.Simon E, Camier S, Seraphin B. 2006. New insights into the control of mRNA decapping. Trends Biochem Sci 31:241–243. doi: 10.1016/j.tibs.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Lykke-Andersen J. 2002. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol 22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. 2002. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J 21:6915–6924. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. 2002. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A 99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen LS, Mikhli C, Jiao X, Kiledjian M, Kunkel G, Davis RE. 2005. Dcp2 decaps m2,2,7GpppN-capped RNAs, and its activity is sequence and context dependent. Mol Cell Biol 25:8779–8791. doi: 10.1128/MCB.25.20.8779-8791.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Yang JY, Niu QW, Chua NH. 2006. Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell 18:3386–3398. doi: 10.1105/tpc.106.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwasaki S, Takeda A, Motose H, Watanabe Y. 2007. Characterization of Arabidopsis decapping proteins AtDCP1 and AtDCP2, which are essential for post-embryonic development. FEBS Lett 581:2455–2459. doi: 10.1016/j.febslet.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 13.Dunckley T, Parker R. 1999. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J 18:5411–5422. doi: 10.1093/emboj/18.19.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beelman CA, Stevens A, Caponigro G, LaGrandeur TE, Hatfield L, Fortner DM, Parker R. 1996. An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature 382:642–646. doi: 10.1038/382642a0. [DOI] [PubMed] [Google Scholar]
- 15.Deshmukh MV, Jones BN, Quang-Dang DU, Flinders J, Floor SN, Kim C, Jemielity J, Kalek M, Darzynkiewicz E, Gross JD. 2008. mRNA decapping is promoted by an RNA-binding channel in Dcp2. Mol Cell 29:324–336. doi: 10.1016/j.molcel.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 16.She M, Decker CJ, Svergun DI, Round A, Chen N, Muhlrad D, Parker R, Song H. 2008. Structural basis of Dcp2 recognition and activation by Dcp1. Mol Cell 29:337–349. doi: 10.1016/j.molcel.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Floor SN, Jones BN, Hernandez GA, Gross JD. 2010. A split active site couples cap recognition by Dcp2 to activation. Nat Struct Mol Biol 17:1096–1101. doi: 10.1038/nsmb.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu JH, Yang WH, Gulick T, Bloch KD, Bloch DB. 2005. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA 11:1795–1802. doi: 10.1261/rna.2142405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fenger-Grøn M, Fillman C, Norrild B, Lykke-Andersen J. 2005. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell 20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Chang CT, Bercovich N, Loh B, Jonas S, Izaurralde E. 2014. The activation of the decapping enzyme DCP2 by DCP1 occurs on the EDC4 scaffold and involves a conserved loop in DCP1. Nucleic Acids Res 42:5217–5233. doi: 10.1093/nar/gku129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz D, Decker CJ, Parker R. 2003. The enhancer of decapping proteins, Edc1p and Edc2p, bind RNA and stimulate the activity of the decapping enzyme. RNA 9:239–251. doi: 10.1261/rna.2171203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R. 2003. Analysis of recombinant yeast decapping enzyme. RNA 9:231–238. doi: 10.1261/rna.2151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Decker CJ, Teixeira D, Parker R. 2007. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol 179:437–449. doi: 10.1083/jcb.200704147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nissan T, Rajyaguru P, She M, Song H, Parker R. 2010. Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol Cell 39:773–783. doi: 10.1016/j.molcel.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borja MS, Piotukh K, Freund C, Gross JD. 2011. Dcp1 links coactivators of mRNA decapping to Dcp2 by proline recognition. RNA 17:278–290. doi: 10.1261/rna.2382011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fromm SA, Truffault V, Kamenz J, Braun JE, Hoffmann NA, Izaurralde E, Sprangers R. 2012. The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. EMBO J 31:279–290. doi: 10.1038/emboj.2011.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holmes LEA, Campbell SG, De Long SK, Sachs AB, Ashe MP. 2004. Loss of translational control in yeast compromised for the major mRNA decay pathway. Mol Cell Biol 24:2998–3010. doi: 10.1128/MCB.24.7.2998-3010.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coller J, Parker R. 2005. General translational repression by activators of mRNA decapping. Cell 122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilkington GR, Parker R. 2008. Pat1 contains distinct functional domains that promote P-body assembly and activation of decapping. Mol Cell Biol 28:1298–1312. doi: 10.1128/MCB.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lykke-Andersen J, Shu MD, Steitz JA. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121–1131. doi: 10.1016/S0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 31.Lykke-Andersen J, Wagner E. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev 19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh G, Jakob S, Kleedehn MG, Lykke-Andersen J. 2007. Communication with the exon-junction complex and activation of nonsense-mediated decay by human Upf proteins occur in the cytoplasm. Mol Cell 27:780–792. doi: 10.1016/j.molcel.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 33.Gallouzi IE, Brennan CM, Stenberg MG, Swanson MS, Eversole A, Maizels N, Steitz JA. 2000. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci U S A 97:3073–3078. doi: 10.1073/pnas.97.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song MG, Li Y, Kiledjian M. 2010. Multiple mRNA decapping enzymes in mammalian cells. Mol Cell 40:423–432. doi: 10.1016/j.molcel.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH. 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16:49–55. doi: 10.1038/nsmb.1530. [DOI] [PubMed] [Google Scholar]
- 36.Popov N, Schulein C, Jaenicke LA, Eilers M. 2010. Ubiquitylation of the amino terminus of Myc by SCF(beta-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat Cell Biol 12:973–981. doi: 10.1038/ncb2104. [DOI] [PubMed] [Google Scholar]
- 37.Clement SL, Scheckel C, Stoecklin G, Lykke-Andersen J. 2011. Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol Cell Biol 31:256–266. doi: 10.1128/MCB.00717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107:451–464. doi: 10.1016/S0092-8674(01)00578-5. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z, Kiledjian M. 2001. Functional link between the mammalian exosome and mRNA decapping. Cell 107:751–762. doi: 10.1016/S0092-8674(01)00592-X. [DOI] [PubMed] [Google Scholar]
- 40.Mukherjee D, Gao M, O'Connor JP, Raijmakers R, Pruijn G, Lutz CS, Wilusz J. 2002. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J 21:165–174. doi: 10.1093/emboj/21.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghosh T, Peterson B, Tomasevic N, Peculis BA. 2004. Xenopus U8 snoRNA binding protein is a conserved nuclear decapping enzyme. Mol Cell 13:817–828. doi: 10.1016/S1097-2765(04)00127-3. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Song M, Kiledjian M. 2011. Differential utilization of decapping enzymes in mammalian mRNA decay pathways. RNA 17:419–428. doi: 10.1261/rna.2439811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bloch DB, Nobre RA, Bernstein GA, Yang WH. 2011. Identification and characterization of protein interactions in the mammalian mRNA processing body using a novel two-hybrid assay. Exp Cell Res 317:2183–2199. doi: 10.1016/j.yexcr.2011.05.027. [DOI] [PubMed] [Google Scholar]
- 44.Hwang CS, Shemorry A, Varshavsky A. 2010. N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shemorry A, Hwang CS, Varshavsky A. 2013. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol Cell 50:540–551. doi: 10.1016/j.molcel.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. 2011. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C, Deutscher MP. 2010. RNase R is a highly unstable protein regulated by growth phase and stress. RNA 16:667–672. doi: 10.1261/rna.1981010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang W, Deutscher MP. 2010. A novel mechanism for ribonuclease regulation: transfer-messenger RNA (tmRNA) and its associated protein SmpB regulate the stability of RNase R. J Biol Chem 285:29054–29058. doi: 10.1074/jbc.C110.168641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang W, Deutscher MP. 2012. Post-translational modification of RNase R is regulated by stress-dependent reduction in the acetylating enzyme Pka (YfiQ). RNA 18:37–41. doi: 10.1261/rna.030213.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang W, Deutscher MP. 2013. Ribosomes regulate the stability and action of the exoribonuclease RNase R. J Biol Chem 288:34791–34798. doi: 10.1074/jbc.M113.519553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang W, Malhotra A, Deutscher MP. 2011. Acetylation regulates the stability of a bacterial protein: growth stage-dependent modification of RNase R. Mol Cell 44:160–166. doi: 10.1016/j.molcel.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.