

Abstract
The aryl hydrocarbon receptor (AhR) is well known as a ligand binding transcription factor regulating various biological effects. Previously we have shown that long-term exposure to estrogen in breast cancer cells caused not only down regulation of estrogen receptor (ER) but also overexpression of AhR. The AhR interacts with several cell signaling pathways associated with induction of tyrosine kinases, cytokines and growth factors which may support the survival roles of AhR escaping from apoptosis elicited by a variety of apoptosis inducing agents in breast cancer. In this study, we studied the anti-apoptotic role of AhR in different breast cancer cells when apoptosis was induced by exposure to UV light and chemotherapeutic agents. Activation of AhR by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in AhR overexpressing breast cancer cells effectively suppressed the apoptotic response induced by UV-irradiation, doxorubicin, lapatinib and paclitaxel. The anti-apoptotic response of TCDD was uniformly antagonized by the treatment with 3′methoxy-4′nitroflavone (MNF), a specific antagonist of AhR. TCDD’s survival action of apoptosis was accompanied with the induction of well-known inflammatory genes, such as cyclooxygenase-2 (COX-2) and NF-κB subunit RelB. Moreover, TCDD increased the activity of the immunosuppressive enzyme indoleamine 2, 3-dioxygenase (IDO), which metabolizes tryptophan to kynurenine (Kyn) and mediates tumor immunity. Kyn also acts as an AhR ligand like TCDD, and kyn induced an anti-apoptotic response in breast cancer cells. Accordingly, our present study suggests that AhR plays a pivotal role in the development of breast cancer via the suppression of apoptosis, and provides an idea that the use of AhR antagonists with chemotherapeutic agents may effectively synergize the elimination of breast cancer cells.
Keywords: Breast cancer, AhR, Apoptosis, COX-2, IDO, Kynurenine, NF-κB, TCDD
1. Introduction
The recently found phenomenon of aryl hydrocarbon receptor (AhR) overexpression in breast cancer [1] has raised several important questions. The immediate question being raised by us is “does the AhR contribute to cancer progression?” There is a plethora of knowledge regarding this question. Brooks and Eltom [2] addressed this question by artificially overexpressing AhR in a human mammary epithelial cell, and thereby showed that they indeed convert this type of cells into that exhibiting the phenotypic characteristics of highly transformed breast cancer cells such as increased proliferation, matrigel invasion, epithelial to mesenchymal transition and apoptosis resistance. Among them, several mechanisms of anti-apoptotic action of AhR have already been reported [3]. In some mammary epithelial cells, for instance, AhR activation promotes cells to rod c transforming growth factor α (TGFα) and/or activate epidermal growth factor receptor (EGFR) signaling, which helps cells to survive under apoptotic conditions [4]. In addition, modulation of epithelial and endothelial tyrosine kinase (ETK) [5], p53 [6], NF-κB, phosphatidyl-inositol-3-kinase (PI3K)/AKT and extracellular signal-regulated kinase (ERK), TGFβ [7] and E2F [8] have been reported as mechanisms underlying AhR action for anti-apoptosis. Interestingly, inhibition of AhR in keratinocytes has been found to result in down-regulation of E2F1 and checkpoint kinase-1 (CHK1) protein expression, which was accompanied by an increase in UV-induced apoptosis [9]. These data provide the information for the possibility of AhR’s function as a broad-spectrum pro-survival factor for cancer cells to enhance survival via multiple means.
As for the probable cause for such an effect of AhR, while the simplest assumption may be that AhR is making cancer cells more susceptible to dioxin-like environmental pollutants, there is now an increasing body of evidence indicating that the phenomenon of losing ER (estrogen receptor) α is intimately ssociated with overexpression of AhR. During the course of our previous studies, we found that the increase of AhR expression is closely linked to continuous exposure of breast cancer cells to 1 nM estradiol (E2) for a number of passages as shown in both MCF-7 [10] and MCF10AT1 [11] cells. Contrary to AhR expression, this long-term exposure to E2 resulted in the reduction of ERα. Although it has not been fully investigated, it may be based on the fundamental negative relationship between the functions and expression of these two receptors [12].
The current recommendation for first line chemotherapy includes an anthracycline-based (including doxorubicin) and/or taxane-based (including paclitaxel) regimen. Of these two therapies, the anthracycline-based approach appears to be more effective against ErbB2-overexpressing subtypes of breast cancer than the taxane-based therapy, particularly in older patients [13]. Unfortunately, the effective duration typically lasts less than 1 year for over 90% of patients. For those patients with ErbB2 positive breast cancer, trastuzumab (=Herceptin) is considered as one of the most effective treatments. However, this effect is usually not long-lasting because resistance typically develops within one year. In addition, it has been found that ErbB2 protein level is frequently elevated in ER-negative breast tumors associated with increased levels of AhR, RelB and Interleukin 8 (IL-8) [14]. These results also indicate that the AhR-overexpressing tumors are found more frequently than previously thought.
The primary objective of the present study is the investigation about the role of AhR in the development of breast cancer and the possibility that AhR becomes a therapeutic target of breast cancer treatment. In assessing the influence of AhR we chose 6 different cultured breast cancer cell lines. P35E and P20E were generated by exposure to E2 from MCF-7 and MCF10AT1 cells, respectively, and express high levels of AhR while P20C and P35C were mock selected control cells [10, 11]. MDA-MB-231 and SKBR3 cells also have relatively high level of AhR, and especially SKBR3 cells are known as ErbB2 over-expressing type of breast cancer cells [15]. We also used the AhR antagonist MNF (3′methoxy-4′nitroflavone) in order r to elucidate AhR-dependent function of anti-apoptosis in breast cancer cells.
2. Material and methods
2.1. Materials
Doxorubicin, dimethyl sulfoxide (DMSO), E2, L-kynurenine (kyn) and paclitaxel were purchased from Sigma-Aldrich (St. Louis, MO). 3′-Methoxy-4′nitroflavone (MNF) was kindly provided by Dr. Gabriele Vielhaber (Symrise GmbH & Co.KG, Holzminden, Germany). TCDD (>99 99 % purity) was obtained from Dow Chemicals Co. (Midland, MI). Polyclonal antibody against actin of goat origin and polyclonal antibodies against human IDO1 and IDO2 of rabbit origin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Lapatinib was purchased from LC laboratories (Woburn, MA).
2.2. Breast cancer cell lines and culture conditions
The details of E2 selection process for MCF10AT1 cells (generation of P20E and the mock selected control; P20C), and for MCF-7 cells (generation of P35E and the mock selected control; P35C) have been described previously [10, 11]. Briefly, MCF10AT1 and MCF-7 cells were cultured with 1 nM E2 for 20 and 35 passages in order to establish AhR overexpressing breast cancer cells P20E and P35E, respectively. P20C and P35C cells were generated as the corresponding control cells by culturing each cell line for the same number of passages with the vehicle only. MDA-MB-231 cells were obtained from Dr Josef Abel. SKBR3 (overexpressing ErbB2, derived from MCF-7 cells) was obtained from American Type Culture Collection (ATCC) (Manassas, VA). Both P20C and P20E cell lines were cultured in phenol red free DMEM/F12 medium (Invitrogen Co., Carlsbad, CA) containing 2.5% fetal bovine serum (FBS) (Invitrogen) treated with charcoal dextran (Gemini Bio-Products, West Sacramento, CA), 20 ng/ml human recombinant epidermal growth factor (EGF) (EMD Chemicals, Gibbstown, NJ), 100 μg/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Serum-free medium for P20C and P20E were MEM/F12 contains no serum but still included 20 ng/ml of EGF, 100 μg/ml penicillin and 100 μg/ml streptomycin. Both P35C and P35E cell lines were cultured in phenol red free DMEM medium (Mediatech, Inc., Manassas, VA) containing 5 % FBS 100 μg/ml penicillin and 100 μg/ml streptomycin. MDA-MB-231 and SKBR3 cell lines were cultured in DMEM medium supplemented with 10% FBS, 100 μg/ml penicillin and 100 ug/ml streptomycin. All cell lines were maintained at 37°C with 5% CO2 and medium was changed every 3 days.
2.3. Apoptosis assay on UV-irradiated or apoptosis-inducing agent-treated cells
For UV-irradiation study, cells (5×105 cells) were seeded in a 6 cm dish. After 24 hr, cells were exposed to TCDD 1 h prior to UV-irradiation with changing the medium. UV was irradiated for 3 min at 3mJ/cm2 by using UV Crosslinker (FB-UVXL-1000, Fisher Scientific; emission peak at 254 nm), followed by being incubated for additional 4 h, and the number of cells showing apoptosis was counted. For apoptosis studies each chemical (doxorubicin, lapatinib or paclitaxel), was added to the cell culture at approximately 70% confluence after receiving the last medium change and incubated for additional 24 h. Apoptosis was detected by Annexin V staining as described previously [10].
2.4. Knockdown of AhR by siRNA transfection
Knockdown of AhR by siRNA was performed as described previously with slight modifications [16]. Briefly, siRNA specific for human AhR was transfected into P20E and P20C cells in 6 cm dishes by using jetPEI (PolyTransfection; Qbiogene, Irvine, CA). After 24 h, medium was changed, and cells were further incubated for 24 h with fresh medium. The effect of AhR knockdown on apoptosis induction was estimated by Annexin V staining as described [10].
2.5. Quantitative RT-PCR (qPCR)
Total RNA was extracted from cells using Quick RNA MiniPrep Kit (Zymo Research Co., Orange, CA). Reverse transcription was conducted with the High Capacity cDNA Reverse Transcription Kit (Applied Bioasystems Inc., Foster City, CA) according to manufacturer’s protocol. Two μl of each cDNA was mixed with Fast SYBR Green Master Mix (Applied Biosystems) and 10 pmol of each primer pair in a 20 μl total reaction volume, and then real-time PCR was performed using LC480 (Roche, Indianapolis, IN). The primer pairs for each gene were the following: GAPDH FP: 5′-GAGTCAACGGATTTGGTCGT-3′, GAPDH RP: 5′-TTGATTTTGGAGGGATCTCG-3′, COX-2 FP: 5′-TTTGTTGAGTCATTCACCAGACAGAT-3′, COX-2 RP: 5′-CAGTATTGAGGAGAACAGATGGGATT-3′, RelB FP: 5′-TGATCCACATGGAATCGAGA-3′, RelB RP: 5′-CAGGAAGGGATATGGAAGCA-3′ IDO1 FP: 5′-CAGGCAGATGTTTAGCAATGA-3′, IDO1 RP: 5′-GATGAAGAAGTGGGCTTTGC-3′, IDO2 FP: 5′-GGCTCTTGGGAAACTCCTTC-3′, IDO2 RP: 5′-TCAGGACATCACCAAAACCTT-3′
2.6. Western blot
In order to prepare whole cell lysates, cells were lysed on ice with RIPA buffer containing protease inhibitor cocktail (Sigma-Aldrich) for 30 min. The lysates were centrifuged at 16,000 g at 4°C for 10 min, and the supernatants were collected as whole cell lysates. Protein concentration in the whole cell lysate was determined by BIO-RAD protein assay (Bio-Rad Laboratories, Inc., Hercules, CA). Whole cell lysates were separated on 8% SDS-polyacrylamide gel and blotted onto a PVDF membrane (Immuno-Blot, Bio-Rad). The antigen-antibody complexes were visualized using the chemiluminescence substrate SuperSignal®, West Pico (Pierce, Rockford, IL) as recommended by the manufacture. For quantitative analysis, respective bands were quantified using a ChemilmagerTM 4400 (Alpha Innotech Corporation, San Leandro, CA).
2.7. Generation of the ido1 and ido2 reporter gene constructs and transient transfection
A 1000 bp fragment of the human ido1 promoter containing one putative DRE site (−814) was cloned for a luciferase reporter construct (SwitchGear Genomics, Carlsbad, CA). A 432 bp fragment of the human ido 2 promoter (−2475 to −2906) containing two putative DRE sites was amplified by PCR using proofreading polymerase (Peqlab, Erlangen, Germany) and the following linker primer: 5′-GTGGTACCAGCGGGTGTATCATGAGGTCA-3′ and 5′-GTCTCGAGACTGGGATGTGCAGAAAATGCC-3′. The resulting DNA fragment and the pGL3-basic vector (Promega, Mannheim, Germany) were double-digested with KpnI and XhoI and subsequently ligated using T4 DNA ligase (Promega Corp., Madison, WI). The ligation mixture was used to transform XL-1 blue competent cells (Stratagene, La Jolla, CA) using the heat-shock method. Subsequently, plasmid DNA was isolated using a plasmid isolation kit (Qiagen, Hilden, Germany).
For transient transfection of P20C and P20E cells, cells were plated in 24-well plates (1 × 105 cells per well) and transfected using jetPEI, according to the manufacturer’s instructions. Briefly, 0.3 μg of the IDO1 and IDO2 construct was suspended in 25 μl of 150 mm sterile NaCl solution. Also 0.3 μl of jetPEI solution was suspended in 25 μl of 150 mm sterile NaCl solution. The jetPEI/NaCl solution was then added to the DNA/NaCl solution and incubated at room temperature for 30 min. The medium in the wells was changed to fresh medium, and 50 μl of the DNA/jetPEI was added to each well. The transfection was allowed to proceed for 24 h, and cells were treated with 10 nM TCDD or 0.1% Me2SO (control) for 24 h. To control the transfection efficiency, cells were cotransfected with 0.1 μg per well β-galactosidase reporter construct. Luciferase activities were measured with the Luciferase Reporter Assay System (Promega) using a luminometer (Berthold Lumat LB 9501/16; Pittsburgh, PA). Relative light units were normalized to β-galactosidase activity and to protein concentration, using Bradford dye assay (Bio-Rad Laboratories).
2.8. Statistical analysis
All quantitative experiments were repeated at least three times and results were expressed as means ± standard deviations. Data were evaluated statistically by one-way ANOVA followed by post-hoc test at the significant level of p < 0.05.
3. Results
3.1. Effects of TCDD on the apoptosis induced by UV-irradiation
UV-irradiation was selected as the first apoptosis-inducing treatment because of the uniformity of its effects on those different cell lines as well as the absence of complicating factors often associated with chemical apoptotic agents such as cellular uptake, elimination and metabolism. Fig. 1 shows that the addition of TCDD to the medium prior to UV treatment reduced UV-induced apoptosis, and the effect was more significant in P20E and P35E than in P20C and P35C. As expected, pretreatment with MNF clearly antagonized the resistant effects of TCDD on apoptosis by UV-irradiation in all cell lines tested (Fig. 1). In addition, knockdown of AhR expression by siRNA specific for human AhR resulted in an increased level of UV-induced apoptosis, especially in P20E (Fig. 2), indicating the critical role of AhR for the anti-apoptotic effect.
Figure 1.
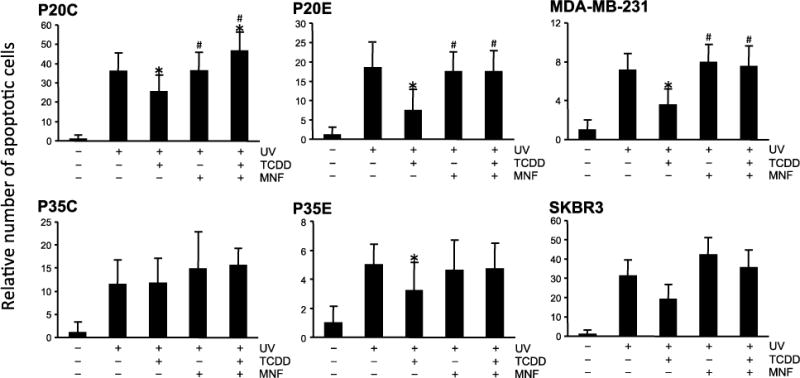
Comparison of TCDD’s effects on UV-induced apoptosis among various breast cancer cell lines. Cells were exposed to TCDD with or without MNF 1 h prior to UV-irradiation. Cells were irradiated by UV (3 mJ/cm2, 3 min), followed by incubation for additional 4 h. Apoptosis was detected by Annexin V staining. Statistical significance (*p < 0.05 vs. UV irradiation alone, #p < 0.05 vs. UV + TCDD).
Figure 2.
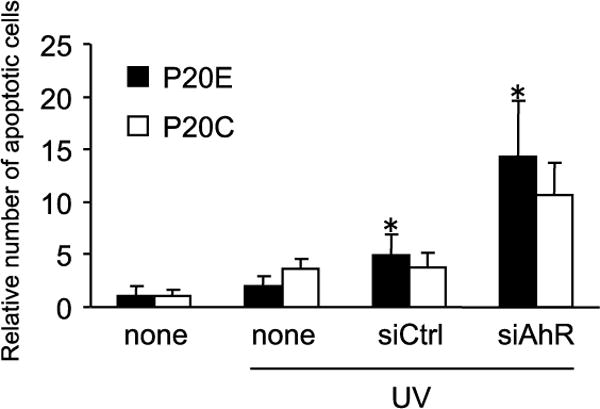
Increase of apoptosis induction by knockdown of Ahr expression. siRNA against AhR was transfected in P20E and P20C cells. Twenty four after transfection, cells were exposed to TCDD with or without MNF 1 h prior to UV-irradiation. Cells were irradiated by UV (3 mJ/cm2, 3 min), followed by incubation for additional 4 h. Apoptosis was detected by Annexin V staining. Statistical significance (*p < 0.05 vs. none (UV-irradiation alone)).
3.2. Effects of TCDD on the apoptosis induced by chemical agents
Next, we tested whether TCDD affects the effect of other apoptosis inducing chemotherapeutic agents, such as doxorubicin and lapatinib. As shown in Fig. 3, in most cell lines TCDD exposure reduced apoptosis induced by doxorubicin. Basically, susceptibility to TCDD in AhR overexpressing cells, such as P20E and P35E, was higher than in P20C and P35C with a relatively low expression level of AhR. MDA-MB-231 and SKBR3 also showed same results like the AhR overexpressing cells P20E and P35E. As expected, these actions of AhR were effectively reversed by MNF in all cell lines. Since the level of doxorubicin-induced apoptosis in P20E without TCDD was lower than that in P20C, it is suggested that AhR is constitutively activated in AhR overexpressing breast cancer cells. A similar trend was also observed when lapatinib was used as the apoptosis-inducing chemical (Fig. 4). On the other hand, the pattern of paclitaxel’s effectiveness among various cell lines was quite different from that of doxorubicin as well as lapatinib (Fig. 5). The action of TCDD in paclitaxel-induced apoptosis was minimally effective in P20E and P35E cells, even though TCDD showed anti-apoptotic effects in P20C and P35C. It appears that AhR activation is more effective against apoptotic agents that are based on their DNA damaging action by UV-irradiation, doxorubicin and lapatinib [17] than agents based on tubulin polymerization mechanism such as paclitaxel [18]. The most significant anti-apoptotic response by TCDD was found in doxorubicin-treated P20E and P20C cells.
Figure 3.
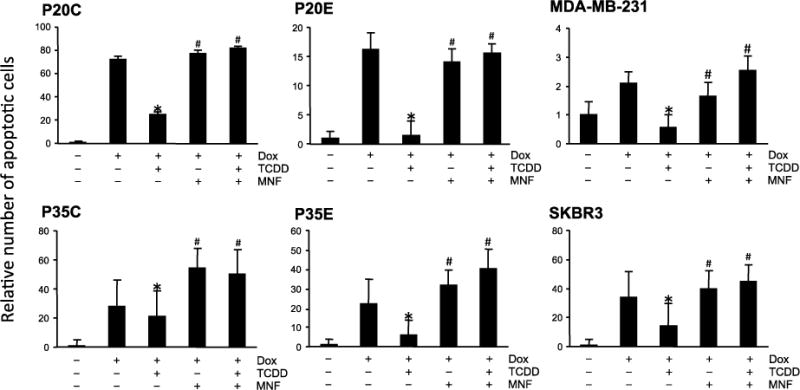
Comparison of TCDD’s effects on doxorubicin-induced apoptosis among various breast cancer cell lines. Cells were exposed to TCDD with or without MNF 1 h prior to treatment with doxorubicin (Dox). Cells were treated with Dox for 24 h, and then apoptosis was detected by Annexin V staining. Statistical significance (*p < 0.05 vs. Dox alone, #p < 0.05 vs. Dox + TCDD).
Figure 4.
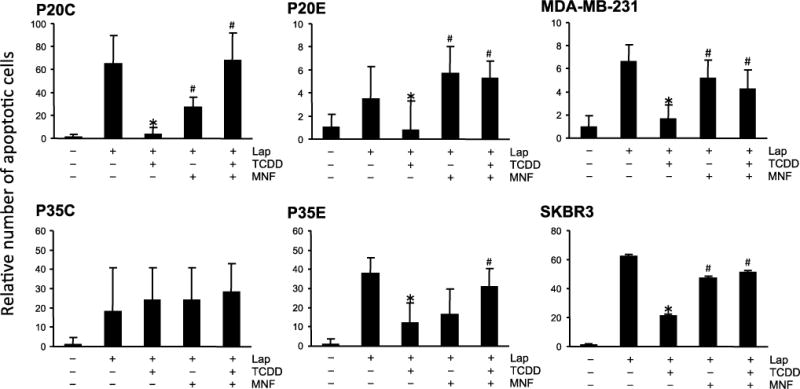
Comparison of TCDD’s effects on lapatinib-induced apoptosis among various breast cancer cell lines. Cells were exposed to TCDD with or without MNF 1 h prior to treatment with lapatinib (Lap). Cells were treated with Lap for 24 h, and then apoptosis was detected by Annexin V staining. Statistical significance (*p < 0.05 vs. Lap alone, #p < 0.05 vs. Lap + TCDD).
Figure 5.
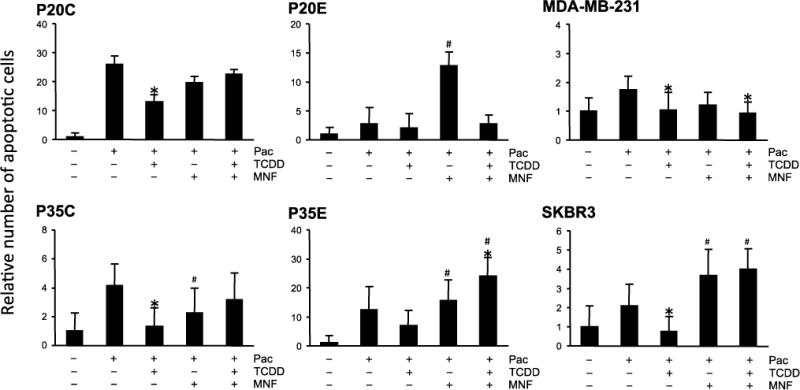
Comparison of TCDD’s effects on paclitaxel-induced apoptosis among various breast cancer cell lines. Cells were exposed to TCDD with or without MNF 1 h prior to treatment with paclitaxel (Pac). Cells were treated with Pac for 24 h, and then apoptosis was detected by Annexin V staining. Statistical significance (*p < 0.05 vs. Pac alone, #p < 0.05 vs. Pac + TCDD).
3.3. Expression of AhR-dependent inflammation markers
Our previous studies demonstrated that TCDD induces the inflammatory enzyme COX-2 in mammalian cells via alternative AhR pathways involving CCAAT/enhancer-binding protein β (C/EBPβ) [19, 20]. Other important information in the alternative AhR pathway is that RelB plays a pivotal role in the expression of some inflammatory cytokines, such as IL-8, and its overexpression was found in ER-negative breast cancer [21, 22]. Under the experimental condition in the current study, TCDD significantly increased COX-2 mRNA expression in the presence of doxorubicin in P20E cells, and MNF treatment completely inhibited its up-regulation (Fig. 6A). A similar effect of TCDD and MNF was found on mRNA expression of RelB (Fig. 6B). We further investigated whether NS-398, a COX-2 inhibitor, is able to abolish the TCDD-mediated inhibition of apoptosis. The chemoprotective effect of NS-398 eliminated the anti-apoptotic effect of TCDD by 70 % in P20E cells (Fig. 6C).
Figure 6.
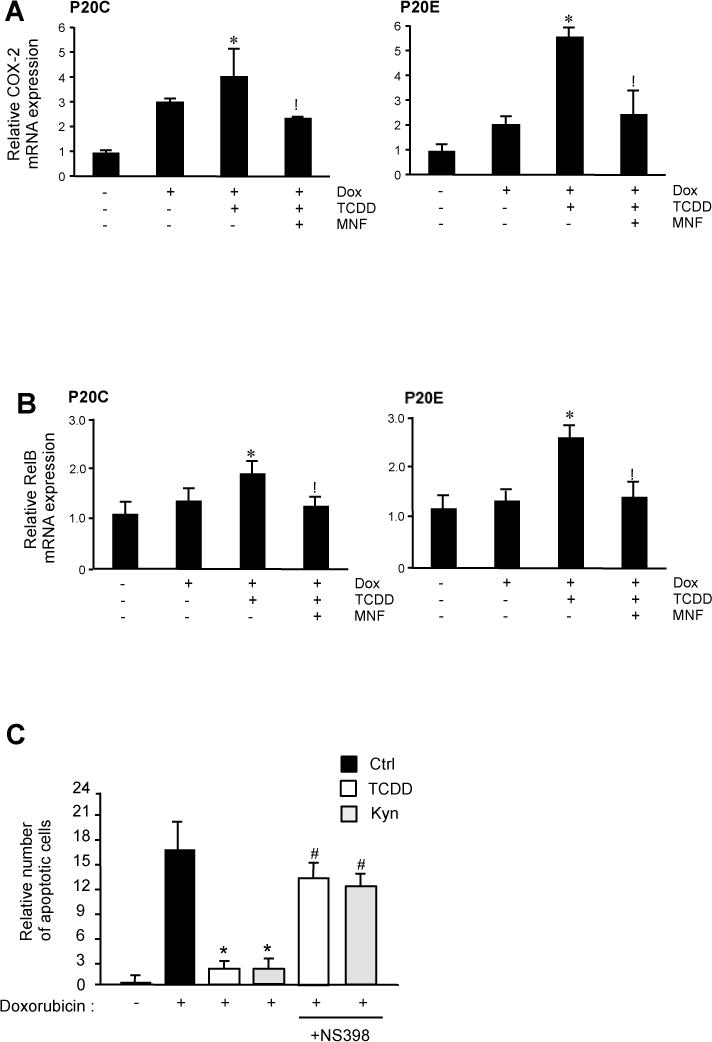
mRNA expression of COX-2 and RelB in P20E cells. Cells were treated with doxorubicin (Dox) for 24 h in absence or presence of TCDD and/or MNF. mRNA expression levels of COX-2 (A) and RelB (B) were expressed as the ratio to that of GAPDH. Statistical significance (*p < 0.05 vs. Dox alone, #p < 0.05 vs. Dox + TCDD).
3.4. IDO induction by TCDD and apoptosis inhibition by kyn
Basically, IDO expression in immune cells, such as dendritic cells and macrophages can regulate the development of regulatory T cells, leading to immune suppression [23]. Initially we have shown that the expression of both isoforms IDO1 and IDO2 is induced by TCDD in an AhR-dependent manner [24]. Other reports indicate that IDO is also expressed in breast cancer tissue [25, 26] although it’s physiological role has not been fully elucidated. The IDO enzyme catalyzes the rate-limiting step of tryptophan degradation along with the kyn pathway [23]. Kyn and related metabolites have been reported to act as an AhR agonist [27, 28].
In this study, we detected IDO1 and IDO2 protein in P20C and P20E cells, and TCDD exposure led to elevated protein levels of IDO1 and IDO2 in both cell lines (Fig. 7A). Transfection studies with reporter plasmids containing promoter sequences of IDO1 and IDO2 confirmed the transcriptional activation of both enzymes in P20C cells. The AhR overexpressing P20E cells showed an increased reporter activity of IDO1 and IDO2 compared to P20C after treatment with TCDD (Fig. 7B). Furthermore, treatment with kyn, a product of the IDO pathway, exerted apoptosis resistance, similar to the case of TCDD (Fig. 7C). These results suggest an interaction between AhR activation and the kyn pathway in the breast cancer cell lines under our experimental condition.
Figure 7.
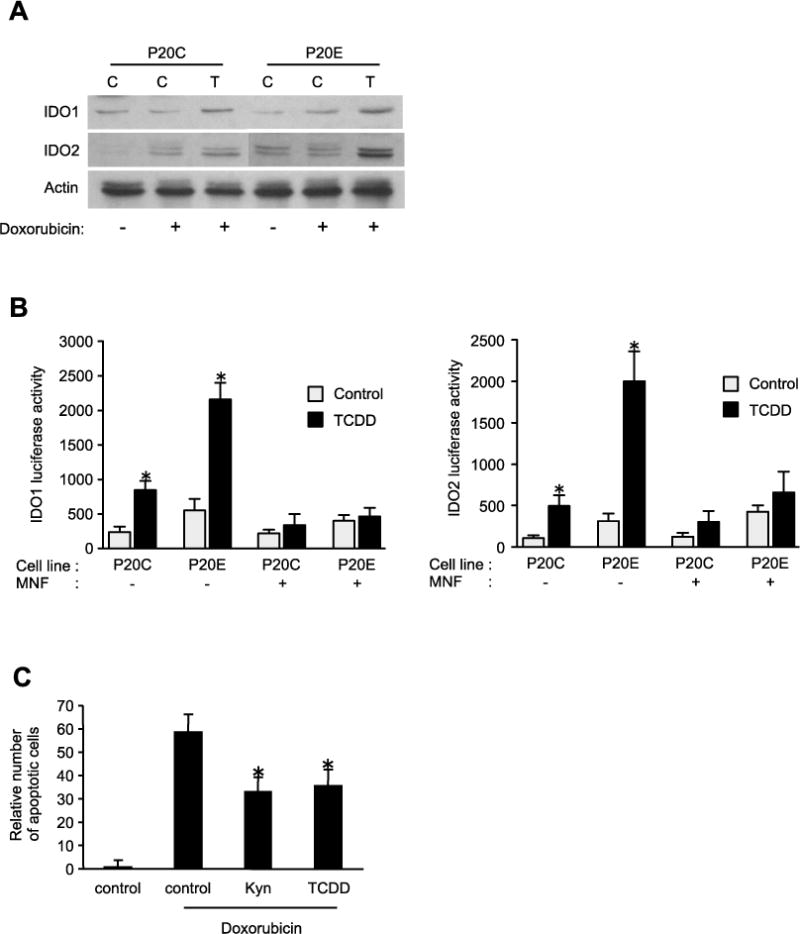
TCDD induction of IDO in breast cancer cells. (A) Expression of IDO1 and IDO2 protein in P20C and P20E after treatment with TCDD and doxorubicin for 24h. (B) IDO1 and IDO2 luciferase promoter activity in P20C and P20E cells. Cells were transfected for 24h with IDO1 and IDO2 reporter plasmids, and treated with TCDD for 24h. Statistical significance (*p < 0.05 vs. parent control). (C) The effects of kynurenine (Kyn) on apoptosis induced by doxorubicin in P20E cells. In parallel experiments, cells were co-cultured with the COX-2 inhibitor NS-398. Statistical significance (*p < 0.05 vs. control + Doxorubicin).
4. Discussion
We could demonstrate that AhR is an effective contributor in conferring the resistance to apoptosis against several apoptosis-inducing treatments on different breast cancer cell lines. Based on the results of the current study the question arose: why does the AhR act in such broad spectrum capacity? The reasons why AhR mediates a broad apoptosis resistance on breast cancer cells, particularly when it is activated by its prototypical ligand TCDD, may be obtained from a review by Chopra and Schrenk [3], who pointed out that the routes through which AhR saves cells from apoptosis are indeed multiple. In a variety of cells AhR is capable of interfering with both extrinsic (receptor mediated) and intrinsic (mitochondria-mediated) caspase activation (particularly 3 and 7), which can cause apoptosis in a variety of cell types. Mitochondrial apoptosis is also affected by AhR activation via induction of Bcl-2 as well as p53 [29]. In the case of mammary epithelial cells we have shown that TCDD activates a number of pro-survival factors such as EGFR, TGFα, Src kinase and ERK [4] as well as PI3K, Akt and ETK kinases [5], and at the same time inhibits the function of apoptosis-inducing factors such as PKCδ and EGF withdrawal-induced PARP cleavage [30]. One of the possible explanations may be that AhR has evolved as an overall pro-survival factor against environmental stress, which is also effective against stress-induced apoptosis.
In this study, we analyzed the expression of COX-2 gene as an inflammatory gene that is regulated by TCDD in an AhR-dependent manner. COX-2 is well known as a key enzyme in the arachidonic acid cascade, resulting in the production of a variety of inflammatory metabolites, such as prostaglandins and the role of COX-2 in preventing apoptosis has been shown previously [31]. The induction of COX-2 in breast cancer cell lines of the current study confirms previous findings that TCDD induces prolonged up-regulation of COX-2 in MCF10A mammary epithelial cells [16]. Among AhR-inducible genes, our earlier reports show that COX-2 is induced by TCDD via AhR and CEBPβ [19, 20], and is involved in AhR-induced stress response [32] as well as apoptosis resistance during the development of lymphoma [33]. In this study, we found that a COX-2 inhibitor may suppress TCDD-mediated inhibition of apoptosis, suggesting a link of TCDD-elicited inflammatory response and an anti-apoptotic response in AhR-overexpressing breast cancer cells. Besides COX-2, the results showed an increased expression of NF-κB RelB in P20C and P20E cells after exposure to TCDD. The fact that the effect of TCDD-induced COX-2 and RelB is stronger in P20E than in P20C cells is indicating that the overexpression of AhR may increase the sensitivity of P20E cells. The overexpression of RelB in association with IL-8 in primary breast cancer tissue has been shown recently [23]. It is interesting to note that in addition to COX-2 and AhR, both IL-8 [34] and RelB [35] are regarded as pro-survival factors contributing to carcinogenesis.
It is likely that breast cancer cell lines reflect many biological and genomic properties of in vivo tumors, but cell lines may also have different characteristics, such as changes in expression levels of AhR and ER. In this study, our data suggest that the effectiveness of AhR against apoptosis is linked to the level of AhR expression. For instance, when UV-irradiation, doxorubicin or lapatinib was used as the apoptosis-inducing agent, TCDD induced a more powerful inhibitory effect on apoptosis in P20E and P35E, compared with P20C and P35C. MDA-MB-231 and SKBR3 cells expressing relatively high levels of AhR were also sensitive to TCDD. The effects of TCDD were dependent on AhR activation, because MNF completely antagonized TCDD-induced apoptosis resistance in almost all breast cancer cell lines used in this study. However, in some cases, the results show a decrease of apoptotic cells by MNF alone. It is possible that antagonists like MNF have also agonistic action depending on the specific cell line and cell culture conditions. Furthermore, our data suggest the important role of a constitutive active AhR in AhR-overexpressing breast cancer cells, since the relative number of apoptotic cells in P20E was clearly lower than those in P20C, when cells were treated with UV, doxorubicin or lapatinib in the absence of TCDD. We also used trastuzumab as an apoptosis inducer of breast cancer cells. Trastuzumab is a specific antibody against ErbB2, and is being used in therapy for ErbB2 over-expressing breast cancer. Similar to the results obtained by using UV, doxorubicin and lapatinib, TCDD was effective in the apoptosis induced by trastuzumab in ErbB2 overexpressing SKBR3 cells (unpublished data). These results strongly indicate that breast cancer cells expressing high level of AhR develop resistance to various apoptosis inducer, even without adding an external AhR ligand. This is supported by our previous findings in which ETK plays a pivotal role in the induction of resistance to apoptosis inducer in P20E with or without AhR ligand [5]. In addition, COX-2 seems to be a key enzyme mediating the inhibition of apoptosis induced by TCDD as shown earlier in lymphoma cells [33].
Furthermore, the current study shows that breast cancer cells express IDO activity, which is involved in immune tolerance by dendritic cells [23]. Interestingly, expression of IDO has been demonstrated to cause the escape from immunosurveillance of breast tumors in mice [25, 26] highlighting the critical role of IDO in tumorigenesis. Another important point is that IDO generates kyn known as an endogenous AhR ligand through tryptophan catabolism [27] and previously we have shown that the expression of both isoforms IDO1 and IDO2 is induced by TCDD in an AhR-dependent manner [24]. The results show that kyn clearly reduced apoptosis in breast cancer cells, similar to the effect of TCDD. Accordingly, the data suggest that the expression of IDO may be an important step in the development of breast cancer, especially of AhR overexpressing cancer type, since high levels of AhR and IDO in breast cancer may contribute to the resistance of apoptosis and escape from tumor rejection, respectively. The importance of AhR overexpression including the increased activity of AhR in carcinogenesis has been discussed recently [36]. Besides IDO, COX-2 might also contribute to suppression of tumor immunity since deletion of COX-2 has been shown to restore, at least partially, immunosurveillance of mammary tumors in mice [37].
On the other hand, it appears that AhR does not effectively function as an anti-apoptotic mediator against the action of paclitaxel. In case of paclitaxel, TCDD promoted apoptosis resistance in P20C and P35C, but not in P20E and P35E cells, in contrast to the exposure with UV light, doxorubicin and lapatinib. The reason why the inhibitory effects of TCDD on paclitaxel-induced apoptosis did not correlate with the level of AhR is unclear. Mechanism under which paclitaxel induces apoptosis in cancer cells is controversy, but it seems that this mechanism differs from those of UV exposure, doxorubicin or lapatinib at the point of cell cycle phase. Basically, paclitaxel is known as an antimicrotubule agent, and inhibits cell cycle progression at G2-M phase followed by the induction of apoptosis [38], whereas it has been reported that UV light, doxorubicin, lapatinib and trastuzumab mainly act at G1 phase [15, 39–42]. Therefore, cellular consequences of AhR activation among each cell cycle phase might be different. Actually, Santini et al. reported a reduced AhR transcriptional activity at G2-M phase when compared with that at G1 phase [43]. Therefore, AhR may not act as an efficient survival factor in the case of paclitaxel, since it induced the accumulation of cell population at G2-M phase.
In conclusion, we could clearly establish in the current work, that AhR is more effective against apoptotic agents that are based on their DNA damaging action (UV, doxorubicin and lapatinib) than those based on tubulin polymerization mechanism (paclitaxel) in the breast cancer chemotherapy. Therefore, chemotherapy only by using a DNA damaging type drug is not fully effective in AhR overexpressing type of breast cancer cells because of the strong survival action of the AhR. That is why our data give rise to an idea that the combined treatment of AhR antagonists and chemotherapeutic agent may be synergistically effective to combat breast cancer. In addition, there are numerous AhR ligands, such as dioxins, polycyclic aromatic hydrocarbons and their derivatives in the environment, and humans are exposed daily to AhR ligands via inhalation and food intake. Therefore, the AhR presents an important target promoting the development of breast cancer in urban areas.
Highlights.
TCDD suppressed apoptosis induced by various apoptotic agents in breast cancer cells
TCDD increased expression of anti-apoptotic markers such as RelB and COX-2 in breast cancer cells
TCDD-induced inhibition of apoptosis is associated with expression of IDO
The natural AhR ligand Kynurenine is a product of the IDO pathway and inhibits the apoptotic response like TCDD
Acknowledgments
This work was supported by research grant #FAS0703859 from Susan G. Komen for the Cure, Breast Cancer Foundation and by National Institute of Environmental Health Sciences grant R01 ES019898-02 (CV).
Abbreviations
- AhR
aryl hydrocarbon receptor
- ER
estrogen receptor
- TCDD
2,3,7,8-Tetrachlorodibenzo-p-dioxin
- MNF
3′methyl-4′nitroflavone
- COX-2
cyclooxygenase-2
- IDO
indoleamine 2, 3-dioxygenase
- Kyn
kynurenine
- MNF
3′methoxy-4′nitroflavone
- TGFα
transforming growth factor α
- EGFR
epidermal growth factor receptor
- ETK
endothelial tyrosine kinase
- PI3K
phosphatidyl-inositol-3-kinase
- ERK
extracellular signal-regulated kinase
- IL-8
Interleukin 8
- CEBPβ
CCAAT/enhancer-binding protein β
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trombino AF, Near RI, Matulka RA, Yang S, Hafer LJ, Toselli PA, Kim DW, Rogers AE, Sonenshein GE, Sherr DH. Expression of the aryl hydrocarbon receptor/transcription factor (AhR) and AhR-regulated CYP1 gene transcripts in a rat model of mammary tumorigenesis. Breast Cancer Res Treat. 2000;63:117–131. doi: 10.1023/a:1006443104670. [DOI] [PubMed] [Google Scholar]
- 2.Brooks J, Eltom SE. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor, Curr. Cancer Drug Targets. 2011;11:654–669. doi: 10.2174/156800911795655967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chopra M, Schrenk D. Dioxin toxicity, aryl hydrocarbon receptor signaling, and apoptosis-persistent pollutants affect programmed cell death. Crit Rev Toxicol. 2011;41:292–320. doi: 10.3109/10408444.2010.524635. [DOI] [PubMed] [Google Scholar]
- 4.Park S, Matsumura F. Characterization of anti-apoptotic action of TCDD as a defensive cellular stress response reaction against the cell damaging action of ultra-violet irradiation in an immortalized normal human mammary epithelial cell line, MCF10A. Toxicology. 2006;217:139–146. doi: 10.1016/j.tox.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Fujisawa Y, Li W, Wu D, Wong P, Vogel C, Dong B, Kung HJ, Matsumura F. Ligand-independent activation of the aryl hydrocarbon receptor by ETK (Bmx) tyrosine kinase helps MCF10AT1 breast cancer cells to survive in an apoptosis-inducing environment. Biol Chem. 2011;10:897–908. doi: 10.1515/BC.2011.087. [DOI] [PubMed] [Google Scholar]
- 6.Pääjärvi G, Viluksela M, Pohjanvirta R, Stenius U, Högberg J. TCDD activates Mdm2 and attenuates the p53 response to DNA damaging agents. Carcinogenesis. 2005;26:201–208. doi: 10.1093/carcin/bgh289. [DOI] [PubMed] [Google Scholar]
- 7.Chang X, Fan Y, Karyala S, Schwemberger S, Tomlinson CR, Sartor MA, Puga A. Ligand-independent regulation of transforming growth factor beta1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol Cell Biol. 2007;27:6127–6139. doi: 10.1128/MCB.00323-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marlowe JL, Fan Y, Chang X, Peng L, Knudsen ES, Xia Y, Puga A. The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol Biol Cell. 2008;19:3263–3271. doi: 10.1091/mbc.E08-04-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frauenstein K, Sydlik U, Tigges J, Majora M, Wiek C, Hanenberg H, Abel J, Esser C, Fritsche E, Krutmann J, Haarmann-Stemmann T. Evidence for a novel anti-apoptotic pathway in human keratinocytes involving the aryl hydrocarbon receptor, E2F1, and checkpoint kinase 1. Cell Death Differ. 2013;10:1425–1434. doi: 10.1038/cdd.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou E, Matsumura F. Long-term exposure to beta-hexachlorocyclohexane (beta-HCH) promotes transformation and invasiveness of MCF-7 human breast cancer cells. Long-term exposure to beta-hexachlorocyclohexane (beta-HCH) promotes transformation and invasiveness of MCF-7 human breast cancer cells. Biochem Pharmacol. 2003;66:831–840. doi: 10.1016/s0006-2952(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 11.Wong PS, Li W, Vogel CF, Matsumura F. Characterization of MCF mammary epithelial cells overexpressing the arylhydrocarbon receptor (AhR) BMC Cancer. 2009;9:1–15. doi: 10.1186/1471-2407-9-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R, Romkes M, Biegel L. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and related compounds as antioestrogens: characterization and mechanism of action. Pharmacol Toxicol. 1991;69:400–409. doi: 10.1111/j.1600-0773.1991.tb01321.x. [DOI] [PubMed] [Google Scholar]
- 13.Fornier MN, Morris PG, Abbruzzi A, D’Andrea G, Gilewski T, Bromberg J, Dang C, Dickler M, Modi S, Seidman AD, Sklarin N, Chang J, Norton L, Hudis CA. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann Oncol. 2011;22:2575–2581. doi: 10.1093/annonc/mdr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogel CF, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, Fujisawa Y, Matsumura F. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys. 2011;512:78–86. doi: 10.1016/j.abb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Annals of Oncology. 2007;18:977–984. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 16.Dong B, Matsumura F. The conversion of rapid TCCD nongenomic signals to persistent inflammatory effects via select protein kinases in MCF10A cell. Mol Endocrinol. 2009;23:549–558. doi: 10.1210/me.2008-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forrest RA, Swift LP, Rephaeli A, Nudelman A, Kimura K, Phillips DR, Cutts SM. Activation of DNA damage response pathways as a consequence of anthracycline-DNA adduct formation. Biochem Pharmacol. 2012;83:1602–1612. doi: 10.1016/j.bcp.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 18.Sun X, Li D, Yang Y, Ren Y, Li J, Wang Z, Dong B, Liu M, Zhou J. Microtubule-binding protein CLIP-170 is a mediator of paclitaxel sensitivity. J Pathol. 2012;226:666–673. doi: 10.1002/path.3026. [DOI] [PubMed] [Google Scholar]
- 19.Vogel CF, Boerboom AM, Baechle C, El-Bahay C, Kahl R, Degen GH, Abel J. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway. Carcinogenesis. 2000;21:2267–2274. doi: 10.1093/carcin/21.12.2267. [DOI] [PubMed] [Google Scholar]
- 20.Vogel CF, Sciullo E, Park S, Liedtke C, Trautwein C, Matsumura F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J Biol Chem. 2004;279:8886–8894. doi: 10.1074/jbc.M310190200. [DOI] [PubMed] [Google Scholar]
- 21.Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21:2941–2955. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel CF, Li W, Wu D, Miller JK, Sweeney C, Lazennec G, Fujisawa Y, Matsumura F. Interaction of aryl hydrocarbon receptor and NF-κB subunit RelB in breast cancer is associated with interleukin-8 overexpression. Arch Biochem Biophys. 2011;512:78–86. doi: 10.1016/j.abb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munn DH, Mellor AL. Indoleamine 2, 3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2, 3-dioxygenase. Biochem Biophys Res Commun. 2008;375:331–335. doi: 10.1016/j.bbrc.2008.07.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soliman H, Rawal B, Fulp J, Lee JH, Lopez A, Bui MM, Khalil F, Antonia S, Yfantis HG, Lee DH, Dorsey TH, Ambs S. Analysis of indoleamine 2–3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol Immunother. 2013;62:829–837. doi: 10.1007/s00262-013-1393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nature Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 27.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 28.Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- 29.Pru JK, Kaneko-Tarui T, Jurisicova A, Kashiwagi A, Selesniemi K, Tilly JL. Induction of proapoptotic gene expression and recruitment of p53 herald ovarian follicle loss caused by polycyclic aromatic hydrocarbons. Reprod Sci. 2009;16:347–356. doi: 10.1177/1933719108327596. [DOI] [PubMed] [Google Scholar]
- 30.Davis JW, Melendez K, Salas VM, Lauer FT, Burchiel SW. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) inhibits growth factor withdrawal-induced apoptosis in the human mammary epithelial cell line, MCF-10A. Carcinogenesis. 2000;21:881–886. doi: 10.1093/carcin/21.5.881. [DOI] [PubMed] [Google Scholar]
- 31.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 32.Matsumura F, Vogel CF. Evidence supporting the hypothesis that one of the main functions of the aryl hydrocarbon receptor is mediation of cell stress responses. Biol Chem. 2006;387:1189–1194. doi: 10.1515/BC.2006.146. [DOI] [PubMed] [Google Scholar]
- 33.Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, Tuscano J, Matsumura F. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol. 2007;171:1538–1548. doi: 10.2353/ajpath.2007.070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benoy IH, Salgado R, Van Dam P, Geboers K, Van Marck E, Scharpé S, Vermeulen PB, Dirix LY. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin Cancer Res. 2004;10:7157–7162. doi: 10.1158/1078-0432.CCR-04-0812. [DOI] [PubMed] [Google Scholar]
- 35.Baud V, Jacque E. The alternative NF-kB activation pathway and cancer: friend or foe? Med Sci. 2008;24:1083–1088. doi: 10.1051/medsci/200824121083. [DOI] [PubMed] [Google Scholar]
- 36.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–814. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markosyan N, Chen EP, Smyth EM. Targeting COX-2 abrogates mammary tumorigenesis: Breaking cancer-associated suppression of immunosurveillance. Oncoimmunology. 2014;3:e29287. doi: 10.4161/onci.29287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Y, Shen D, Chen Z, Clayton S, Vadgama JV. Taxol induced apoptosis regulates amino acid transport in breast cancer cells. Apoptosis. 2007;12:593–612. doi: 10.1007/s10495-006-0007-y. [DOI] [PubMed] [Google Scholar]
- 39.Lüpertz R, Wätjen W, Kahl R, Chovolou Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology. 2010;271:115–121. doi: 10.1016/j.tox.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 41.Shao Z, Jiang M, Yu L, Han Q, Shen Z. p53 independent G1 arrest and apoptosis induced by adriamycin. Chin Med Sci J. 1997;12:71–75. [PubMed] [Google Scholar]
- 42.Sumikawa T, Shigeoka Y, Igishi T, Suyama H, Yamasaki A, Hashimoto K, Matsumoto S, Takeda K, Ueda Y, Shimizu E. Dexamethasone interferes with trastuzumab-induced cell growth inhibition through restoration of AKT activity in BT-474 breast cancer cells. Int J Oncol. 2008;32:683–688. [PubMed] [Google Scholar]
- 43.Santini RP, Myrand S, Elferink C, Reiners JJ., Jr Regulation of Cyp1a1 induction by dioxin as a function of cell cycle phase. J Pharmacol Exp Ther. 2001;299:718–728. [PubMed] [Google Scholar]