

Abstract
Human genetic variation is represented by the genetic differences both within and among populations, and most genetic variants do not cause overt diseases but contribute to disease susceptibility and influence drug response. During the last century, various genetic variants, such as copy number variations (CNVs), have been associated with diverse human disorders. Here, we review studies on the associations between CNVs and autoimmune diseases to gain some insight. First, some CNV loci are commonly implicated in various autoimmune diseases, such as Fcγ receptors in patients with systemic lupus erythemoatosus or idiopathic thrombocytopenic purpura and β-defensin genes in patients with psoriasis or Crohn's disease. This means that when a CNV locus is associated with a particular autoimmune disease, we should examine its potential associations with other diseases. Second, interpopulation or interethnic differences in the effects of CNVs on phenotypes exist, including disease susceptibility, and evidence suggests that CNVs are important to understand susceptibility to and pathogenesis of autoimmune diseases. However, many findings need to be replicated in independent populations and different ethnic groups. The validity and reliability of detecting CNVs will improve quickly as genotyping technology advances, which will support the required replication.
Keywords: Autoimmunity, Genetic variation, Genomewide, Association, Copy number variation
INTRODUCTION
Autoimmune diseases are mostly chronic and complex and arise when the immune system turns its antiforeign antigen defenses on normal healthy body components, such as pancreatic β-cells in patients with type 1 diabetes or myelin basic proteins in patients with multiple sclerosis [1,2,3]. Researchers have identified many genes responsible for autoimmune disorders during the last century. For example, since the first evidence supporting involvement of human leukocyte antigens (HLAs) in the pathogenesis of rheumatoid arthritis (RA) [4], the implications of HLA-DR4 subtypes are central knowledge, and HLA-DR genotyping is used widely to diagnose and treat RA. The association between HLA-B27 with ankylosing spondylitis (AS) remains the strongest compared with those of other common complex diseases; people homozygous for HLA-B27 have an odds ratio (OR) of ~100 and those who are heterozygous have an OR of ~50 [5]. Many tolerance checkpoints exist in the immune system to prevent self-components from activating self-reactive lymphocytes. Therefore, multiple defects in genes and molecular pathways are required for an autoimmune disease to emerge through bypassing these tolerance checkpoints. However, the mechanisms that contribute to disease pathogenesis are largely unknown.
Genetic variations, such as single nucleotide polymorphisms (SNPs), make each individual unique and determine our unique disease susceptibilities [6]. In addition, copy number variations (CNVs), which are genetic variations resulting in a coding gene dosage variation, have been recently discovered. CNVs influence interindividual differences in the risk for a disease via several mechanisms that affect gene expression, such as gene disruption or rearrangement [7,8]. CNVs are measured by whole-genome microarray-based comparative genomic hybridization (array-CGH), whole-genome SNP arrays, or whole-genome sequencing. Genome-wide approaches to study genetic variations help discover new genes that influence susceptibilities to various diseases [9,10]. Indeed, genetic variants have been associated with autoimmune disorders [11,12,13,14], which is useful for understanding the pathogenesis and discovering new drug targets [15].
In this review, we focus on CNVs newly identified to be associated with autoimmune diseases through genome-wide association studies (GWASs) as well as target gene studies and discuss their clinical implications.
COPY NUMBER VARIATIONS
What is a copy number variation?
Human genetic variations are the genetic sequence and/or structural differences within and among populations. Many genetic variants do not cause overt diseases but influence disease susceptibility and drug response. Genomic structural variants have been considered background noise in conventional genetic studies because they are difficult to distinguish from experimental errors or biases. Therefore, unverified genomic structural variants detected in the past were removed from the analysis. However, these variants drew attention of some scientists and have been recognized as genuine, novel genetic variations called CNVs [16,17,18].
CNVs are inherited or de novo structural variations, including all kinds of genomic variations > 1 kb, such as insertions, deletions, inversions, and translocations (Fig. 1) [19,20]. In total, 353,126 CNVs have been reported in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) as of October 2014. A study has estimated that ~68% of the human genome is covered by at least one CNV [21]. As 75.6% of exons and 91.2% of transcripts are overlapped by at least one CNV, CNVs could have significant biological implications [21]. The functional consequences of CNVs include changes in protein expression levels and truncation or alterations in protein sequences.
Figure 1. Different types of copy number variations (CNVs) and an example of genome-wide detection of CNVs. Upper plot illustrates relative deletion, duplication, and multiple segmental duplication of the "D" locus compared with the reference genome. Inversion of "C" and "D" loci is also illustrated. The lower plot shows an example of genome-wide CNV detection using oligoarray-comparative genomic hybridization. Red and blue arrows represent copy number gain- and loss-CNVs, respectively.

Studies using next-generation sequencing (NGS) technology show that nonallelic homologous recombination and nonhomologous end joining are the major mechanisms forming CNVs, whereas a retrotransposition contributes only partially [22,23]. More than 99% of CNVs are inherited, whereas others are generated de novo during meiosis. Van Ommen [24] measured the frequency of de novo large-segment CNVs in human newborns and estimated that the frequency of segmental deletions is 1 in 8 and that of segmental duplications is 1 in 50.
Identifying copy number variations
CNVs are detected using blood- or tissue-extracted DNA. CNV detection methods are categorized into targeted and genome-wide detection approaches [19,25]. Genome-wide approaches, in which the entire genome is scanned for CNVs, include microarray-based and NGS-based analyses (Fig. 2). Microarray-based analyses are divided into two approaches of array-CGH and SNP array analyses. In array-CGH, DNA from two individuals (reference and test subjects) are labeled with different dyes and co-hybridized onto DNA array spots representing the entire genome (Fig. 2A) [26]. Relative copy number differences (gain or loss) are calculated using relative signal intensities (test/reference). Most array-CGH DNA arrays are oligonucleotide arrays composed of ~70 oligomers specific for certain genomic locations across the entire genome. Identifying CNVs using SNP arrays is based on comparing signal intensity from whole-genome SNP genotyping data of a test individual with those of a reference group [27]. Whole-genome sequencing has facilitated the discovery of CNVs (Fig. 2B) [28,29]. NGS technology is used primarily to detect single nucleotide variants (or small indels), whereas NGS data can be an important resource to identify CNVs. Moreover, NGS data can be used to detect much smaller CNVs and define CNV breakpoints more precisely. In particular, inversion and translocation, which cannot be identified precisely by either conventional cytogenetics or array-CGH approaches, are definable by NGS [30].
Figure 2. Methods for genome-wide identification of copy number variations (CNVs). (A) Test and reference DNA samples are labeled with different fluorescent dyes (cyanine-3 and cyanine-5, respectively) to identify CNVs using array-comparative genomic hybridization (array-CGH). The mixture of labeled DNAs (reference and test) is hybridized onto the whole-genome microarray. Signal intensity ratios (cyanine-5/cyanione-3) are calculated after hybridization and image scanning and reveal the copy number differences between the test and reference genomes. (B) Test and reference DNA samples are sequenced independently to identify CNVs using next-generation sequencing. The sequenced reads are mapped to the reference genome by sequence alignment. The number of mapped reads in each sliding window is counted. The mapped reads ratios (test mapped reads count/reference mapped reads count) reflect the copy number differences between the test and reference genomes.

However, whole-genome CNV analyses may not be efficient for the validation or clinical translation of a relatively small set of known CNVs. Targeted approaches are more efficient for that purpose. Approaches targeting CNVs include quantitative polymerase chain reaction (qPCR) or Southern hybridization for single target screening and multiplex ligation-dependent probe amplification (MLPA), multiplex amplifiable probe hybridization, multiplex qPCR of short fluorescent fragments, or microsatellite genotyping for concurrent multitarget analyses [19]. The multiplexing ability of MLPA makes it attractive for DNA copy number analyses [31]. However, MLPA has its own shortcomings. For example, as MLPA depends on the length-based discrimination of the ligation products provided by capillary electrophoresis, "stuffer" sequence elements of different sizes are required. Long stuffer elements have potential problems, such as nonspecific hybridization and nonuniform amplification, which hinder widespread use of this technology. Shin et al. [32] developed a stuffer-free multiplex CNV detection method that combines the advantages of MLPA and capillary electrophoresis-strand conformation polymorphism to overcome these limitations (Fig. 3). The emergence of user-friendly and reliable multiplex CNV identification technologies will facilitate CNV-based clinical research and clinical application of the CNVs.
Figure 3. Multiplex ligation-dependent probe amplification (MLPA) technology for detecting copy number variations. (A) The amplified products are separated by conventional MLPA based on length differences in the "stuffer sequences." The stuffer sequence, which is required to separate multiple amplicons using length-based capillary electrophoresis analysis, has potential problems such as nonspecific hybridization and nonuniform amplification. (B) Multiple targets can be specifically amplified with equal efficiency using stuffer-free MLPA by eliminating the stuffer sequence because the ligated probes are of similar size and the amplification uses common primers. RFU, relative fluorescence unit.

COPY NUMBER VARIATIONS IN AUTOIMMUNE DISORDERS
Genetic variation and human autoimmune disease
Autoimmune disorders can be caused by several different mechanisms, including continuous stimulation of autoreactive lymphocytes by diverse self-antigens or inhibiting negative regulatory mechanisms for autoimmune lymphocyte growth. More than half of all antigen receptors generated by random V(D)J recombination recognize self-antigens [33]. The proliferation of autoimmune lymphocytes induced by self-antigens is normally blocked by mechanisms that suppress antigen receptor signaling to activate transcription factors and signaling pathways. However, autoimmune disorders emerge if these mechanisms fail, and many studies have reported the genetic diversity in human autoimmune disorders. Selection and regulation of lymphocytes are controlled by cell-signaling events that may vary among individuals, which is likely due to genetic or epigenetic diversity [34].
Thanks to the advance of technologies, especially genome-wide approaches, more than 200 genetic loci or variants have been suggested to be associated with various autoimmune disorders [34]. Although the causal genes have not been identified, the existence of some genes associated with multiple autoimmune disorders suggests that common pathways may exist among them. Genes within the major histocompatibility complex (MHC) are most strongly associated with autoimmune diseases and other genes likely have smaller, regulatory effects [34].
Associations between CNVs and autoimmune diseases
Several studies have reported associations between CNVs and various autoimmune diseases (Table 1). We review the major CNVs that are consistently associated with autoimmune disorders, although some inconsistencies appear among reports due to technical limitations, and not all of the suggested CNVs have been validated clearly through replicated studies.
Table 1. Copy number variant loci related to autoimmune diseases.
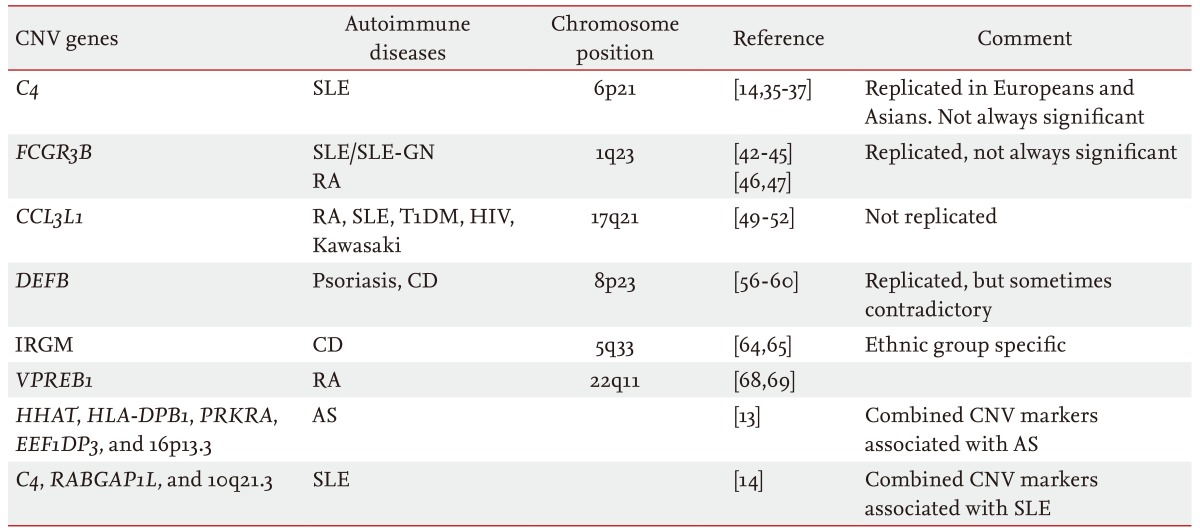
CNV, copy number variation; SLE-GN, systemic lupus erythematosus glomerulonephritis; RA, rheumatoid arthritis; T1DM, type 1 diabetes mellitus; HIV, human immunodeficiency virus; CD, Crohn's disease; AS, ankylosing spondylitis.
Complement component 4
Complement component 4 (C4) is a complement system protein consisting of > 30 proteins in plasma or on cell surfaces. The C4 gene resides in the MHC region as the C4A and C4B isotypes, both with copy numbers of 2 to 6 [35]. Yang et al. [36] studied C4 gene CNVs in 1,241 European and American patients with systemic lupus erythemoatosus (SLE), their first-degree relatives, and unrelated healthy controls and found that a low copy number of total C4 and C4A is associated with susceptibility to SLE. This association was successfully replicated in a study using 924 Han Chinese patients and 1,007 controls [37]. Kim et al. [14] studied SLE-associated CNVs in East Asians using genome-wide analysis and reported a significant correlation between low C4 copy number and SLE in Korean women.
However, the correlation between low C4 copy number and the risk of SLE is not always significant. Boteva et al. [38] investigated the C4 CNV in two large cohorts consisting of 2,207 subjects of northern and southern European ancestry (1,028 SLE cases and 1,179 controls) to determine whether a partial C4 deficiency is an independent risk factor for SLE. Multiple logistic regression was performed to control for the effects of well-known SLE-associated SNPs and risk alleles. They concluded that a genetically determined partial C4 deficiency is not an independent risk factor for SLE in either population and that the association seems to be caused by an unknown causal variant located somewhere else in the high linkage disequilibrium region with C4 [38]. A low C4B copy number, however, has been associated with RA [39].
Fcγ receptor locus
Human Fcγ receptors (FCGRs) are glycoproteins that bind to the immunoglobulin G (IgG) Fc region. The FCGR genes that encode the FcγRs are located on chromosome 1q23-24. Six classes of FCγ receptors (FcγRIA, FcγRIIB, FcγRIIA, FcγRIIC, FcγRIIIA, and FcγRIIIB) have been identified in humans [40]. Among them, Fcγ-RIIIB is a functional modulator of neutrophil activation that controls IgG binding [41]. Aitman et al. [42] detected a correlation between the FCGR3B CNV and SLE glomerulonephritis in United Kingdom (UK) families. A study of 161 SLE cases and 312 controls from the UK reported that individuals with less than two copies of the FCGR3B gene had a higher risk for SLE (OR, 2.43; p = 0.001) [43]. Subsequent studies have consistently reported a correlation between low FCGR3B copy number (< 2) and SLE [44,45].
However, this association is not consistent in patients with RA. One group reported a correlation between a FCGR3B CNV and RA in Caucasians [46]. Another study of 1,115 patients with RA and 654 controls also reported a correlation between low FCGR3B copy number and RA in Caucasians [47]. However, a third study on a population with Spanish ancestry showed that the FCGR3B CNV is correlated with SLE but not with RA [44]. A recent meta-analysis also showed that low FCGR3B copy number (< 2) is correlated with SLE, but not with RA [48]. These inconsistent findings could be due to different CNV analytical techniques or to the complex nature of RA.
Chemokine CC motif ligand 3 like-1
The chemokine CC motif ligand 3 like-1 (CCL3L1) protein binds to several proinflammatory cytokine receptors such as chemokine receptor 5. The CCL3L1 CNV has been well studied in many immune diseases, including RA [49], human immunodeficiency virus infection [50], SLE [51], and Kawasaki disease [52], but the results are conflicting. McKinney et al. [49] assessed the association of the CCL3L1 CNV with RA and type 1 diabetes mellitus (T1DM) susceptibility in Caucasians consisting of 1,136 RA cases and 1,470 controls from New Zealand (NZ) and the UK. They found that copy number more than two was a risk factor for RA in the NZ cohort (OR, 1.34) but not in the smaller UK cohort. Evidence for an association was found in the T1DM cohort (OR, 1.46) and in the combined RA/T1DM cohort (OR, 1.30). However, the Wellcome Trust Case Control Consortium study found no evidence of an association between the CCL3L1 CNV and RA or T1DM in 2,000 cases of each trait and 3,000 shared controls [10]. Thus, one cannot conclude anything about the role of the CCL3L1 CNV in autoimmunity because issues exist regarding the reliability of CNV detection assays, and the CCL3L1 locus is very complex.
β-Defensin
β-Defensins are small peptides encoded by DEFB genes in three main clusters, including 8p23.1, 20p13, and 20q11.1, which have antimicrobial activities against bacteria, fungi, and viruses [53]. They also have proinflammatory properties as chemotactic agents for dendritic cells, T cells, and neutrophils [54,55]. Hollox et al. [56] found that an increase in DEFB copy number was significantly correlated with psoriasis in 190 Dutch cases and 303 controls. Impaired induction of the epithelial β-defensins is related to Crohn's disease (CD), and defensins are underexpressed in patients with CD [57]. Fellermann et al. [58] hypothesized in 2006 that this result could be due to a low DEFB copy number and performed genome-wide DNA copy number profiling of the human β-defensin 2 (HBD-2) gene. They found that the copy number distribution in patients with CD was shifted to a lower number compared with the control group (median of four copies for healthy individuals and three copies for patients with colonic CD). They also found that a lower HBD-2 gene copy number was correlated with reduced expression of mucosal HBD-2 mRNA (p = 0.033) [58]. In 2010, Bentley et al. [59] tried to validate the findings of Fellermann et al. [58], which was the only report on the association between the DEFB CNV and CD in a larger case-control cohort of Europeans. However, the results were opposite to the previous report. As both studies used the same quantitative PCR assay, the discrepancy is unlikely to have been caused by differences in methodology. That population stratification caused the discrepancy is also unlikely, as both groups studied similar Caucasian subjects. Bentley et al. [59] suggested that the false-positive findings in the study of Fellermann et al. [58] may have been due to the relatively small, phenotypically ill-defined study population. About 1 year later, Aldhous et al. [60] tried to verify the relationship between the DEFB CNV and CD using a more accurate and reliable paralog ratio test in a larger study population consisting of 1,000 UK CD cases and 500 controls. However, neither of the associations between high and low DEFB4 copy numbers and CD [58,59] were replicated [60]. Nevertheless, the possibility may still exist that the DEFB CNV is generally related to autoimmune disorders. A large study in two Chinese cohorts detected associations of increased DEFB4 copy number with SLE, and antineutrophil cytoplasmic antibody-associated small vasculitis [61]. Thus, DEFB genes should be studied for their involvement in diverse autoimmune diseases in various populations.
Immunity-related GTPase family M
The human immunity-related GTPase family M (IRGM) gene induces autophagy as a mechanism to remove intracellular mycobacteria [62], and an association between SNPs near IRGM and CD was detected by a GWAS [63]. McCarroll et al. [64] examined the experimental data of 270 HapMap samples to assess the existence of common CNVs near IRGM and identified a common 20-kbp deletion located 2.7 kbp upstream of IRGM. They genotyped this deletion-CNV in 685 North American case-control samples and found an increased frequency of the CNV (allele frequency, 15%; OR, 1.6; p < 0.01), which has been proposed to be a likely causal variant [65]. However, a study in a Japanese population found no association between IRGM variants and CD, suggesting an influence of population stratification on the pathogenic effects [66]. One alternative explanation is that the causal variant at this locus arose after the European-Asian split.
VPREB1
B-lymphocytes are key players in innate and adaptive immunity, and their impaired function can lead to autoimmune diseases [67]. Pre-B cell receptor (pre-BCR), which is composed of VpreB and λ5, is involved in positive and negative selection of autoreactivity and shaping the B-cell repertoire [68]. Perturbation of the pre-BCR-mediated checkpoints can contribute to the development of autoimmune disorders. Yim et al. [69] reported that the VPREB1 CNV is associated with susceptibility to RA, observing that the proportion of individuals with less than two copies of the VPREB1 gene was significantly higher in the patient group than that in the controls. Similarly, the proportion of individuals with more than two copies was significantly lower in the patient group than that in the controls [69]. Considering the biological importance of pre-BCR on immunity and the association between the VPREB1 CNV and RA, more studies based on larger samples may facilitate clinical translation of this CNV.
Combined effects of CNVs associated with autoimmune diseases
Although many CNVs have been associated with various autoimmune diseases, the impact of a single CNV seems to be relatively small. However, a combination of multiple CNVs is logically expected to exert stronger effects on phenotypes. Kim et al. [14] reported that all C4, RABGAP1L, and 10q21.3 deletion variants are associated with the risk for SLE, with ORs of 1.0 to 1.3. Notably, individuals with deletion CNVs at all three loci had a 5.5 times higher risk for SLE than those who were diploid at all three loci, and individuals with deletion CNVs at two loci had a 1.78 times higher OR than those diploid at both loci [14]. This synergistic effect has also been observed in patients with AS. Jung et al. [13] reported that five deletion CNVs of HHAT (1q32.2), HLA-DPB1 (6p21.3), PRKRA (2q31.2), EEF1DP3 (13q13.1), and 16p13.3 are associated with the risk for AS, and that the ORs were 1.5 to 2.1. However, individuals with deletion CNVs in four or more loci had an 18 times higher risk for AS than those being diploid in all five loci, and individuals with deletion CNVs at three and two loci had 12.2 and 7.3 times higher ORs, respectively, than those of diploid subjects [13]. Combining CNV markers with multiplex CNV detection technology will facilitate CNV-based clinical research and application of the CNVs.
CONCLUSIONS
Results from genomics studies can be used to categorize diseases of previously unknown origin into auto-immune diseases and help identify new pathogenic mechanisms for known diseases. We provide some insight into the associations between CNVs and auto-immune diseases. First, we found that some CNV loci are commonly implicated in various autoimmune diseases, such as Fcγ receptors in SLE and ITP and β-defensin genes in psoriasis and CD. This observation suggests that some autoimmune diseases may share CNVs as common risk factors. Thus, when a CNV locus is associated with a particular autoimmune disease, its potential associations with other autoimmune diseases should be examined. Second, interpopulation or interethnic differences exist in the effects of CNVs on phenotypes including disease susceptibility. Thus, associations found in one population should be replicated in another population of interest to set up CNV databases containing information on significant CNVs in each population. Meaningful functional studies will follow only after reliable data on population-specific CNVs are produced, which can be and translated into clinical applications.
Although identifying the environmental components that interact with host genetic factors is very important to properly understand autoimmunity and develop preventive and therapeutic measures, evidence suggests that CNVs are important clues to understand the susceptibility and pathogenesis of autoimmune diseases. However, many of the findings need to be replicated in independent populations or in different ethnic groups. The validity and reliability of detecting CNVs will improve quickly as genotyping technologies advance, which will support the required replication. In addition, combined interpretation of CNVs and other types of genetic variants may help us understand disease susceptibility and pathogenesis.
Acknowledgments
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry for Health and Welfare, Republic of Korea (HI14C3417).
Footnotes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
References
- 1.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 2.Ohno S, Wolf U, Atkin NB. Evolution from fish to mammals by gene duplication. Hereditas. 1968;59:169–187. doi: 10.1111/j.1601-5223.1968.tb02169.x. [DOI] [PubMed] [Google Scholar]
- 3.Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584–589. doi: 10.1038/nature03723. [DOI] [PubMed] [Google Scholar]
- 4.Astorga GP, Williams RC Jr. Altered reactivity in mixed lymphocyte culture of lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1969;12:547–554. doi: 10.1002/art.1780120602. [DOI] [PubMed] [Google Scholar]
- 5.Robinson PC, Brown MA. Genetics of ankylosing spondylitis. Mol Immunol. 2014;57:2–11. doi: 10.1016/j.molimm.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 6.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39(7 Suppl):S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 8.Yim SH, Kim TM, Hu HJ, et al. Copy number variations in East-Asian population and their evolutionary and functional implications. Hum Mol Genet. 2010;19:1001–1008. doi: 10.1093/hmg/ddp564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 10.Craddock N, Hurles ME, et al. Wellcome Trust Case Control Consortium. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stahl EA, Raychaudhuri S, Remmers EF, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung SH, Yim SH, Hu HJ, et al. Genome-wide copy number variation analysis identifies deletion variants associated with ankylosing spondylitis. Arthritis Rheumatol. 2014;66:2103–2112. doi: 10.1002/art.38650. [DOI] [PubMed] [Google Scholar]
- 14.Kim JH, Jung SH, Bae JS, et al. Deletion variants of RABGAP1L, 10q21.3, and C4 are associated with the risk of systemic lupus erythematosus in Korean women. Arthritis Rheum. 2013;65:1055–1063. doi: 10.1002/art.37854. [DOI] [PubMed] [Google Scholar]
- 15.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCarroll SA, Hadnott TN, Perry GH, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 17.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 18.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 19.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 20.Eichler EE, Nickerson DA, et al. Human Genome Structural Variation Working Group. Completing the map of human genetic variation. Nature. 2007;447:161–165. doi: 10.1038/447161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Centre for Applied Genomics. Database of genomic variants [Internet] Toronto (ON): The Centre for Applied Genomics; 2014. [cited 2015 Feb 2]. Available from http://dgv.tcag.ca/dgv/app/statistics?ref= [Google Scholar]
- 22.Kidd JM, Cooper GM, Donahue WF, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Zhou W, Zhang L, Zhang F. Genome architecture and its roles in human copy number variation. Genomics Inform. 2014;12:136–144. doi: 10.5808/GI.2014.12.4.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Ommen GJ. Frequency of new copy number variation in humans. Nat Genet. 2005;37:333–334. doi: 10.1038/ng0405-333. [DOI] [PubMed] [Google Scholar]
- 25.Aten E, White SJ, Kalf ME, et al. Methods to detect CNVs in the human genome. Cytogenet Genome Res. 2008;123:313–321. doi: 10.1159/000184723. [DOI] [PubMed] [Google Scholar]
- 26.Kim TM, Yim SH, Chung YJ. Copy number variations in the human genome: potential source for individual diversity and disease association studies. Genomics Inform. 2008;6:1–7. [Google Scholar]
- 27.Lin CF, Naj AC, Wang LS. Analyzing copy number variation using SNP array data: protocols for calling CNV and association tests. Curr Protoc Hum Genet. 2013;79:Unit 1.27. doi: 10.1002/0471142905.hg0127s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JI, Ju YS, Park H, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abel HJ, Duncavage EJ. Detection of structural DNA variation from next generation sequencing data: a review of informatic approaches. Cancer Genet. 2013;206:432–440. doi: 10.1016/j.cancergen.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin GW, Jung SH, Yim SH, Chung B, Yeol Jung G, Chung YJ. Stuffer-free multiplex ligation-dependent probe amplification based on conformation-sensitive capillary electrophoresis: a novel technology for robust multiplex determination of copy number variation. Electrophoresis. 2012;33:3052–3061. doi: 10.1002/elps.201200334. [DOI] [PubMed] [Google Scholar]
- 33.Zerrahn J, Held W, Raulet DH. The MHC reactivity of the T cell repertoire prior to positive and negative selection. Cell. 1997;88:627–636. doi: 10.1016/s0092-8674(00)81905-4. [DOI] [PubMed] [Google Scholar]
- 34.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365:1612–1623. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 35.Chung EK, Yang Y, Rupert KL, et al. Determining the one, two, three, or four long and short loci of human complement C4 in a major histocompatibility complex haplotype encoding C4A or C4B proteins. Am J Hum Genet. 2002;71:810–822. doi: 10.1086/342778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Chung EK, Wu YL, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lv Y, He S, Zhang Z, et al. Confirmation of C4 gene copy number variation and the association with systemic lupus erythematosus in Chinese Han population. Rheumatol Int. 2012;32:3047–3053. doi: 10.1007/s00296-011-2023-7. [DOI] [PubMed] [Google Scholar]
- 38.Boteva L, Morris DL, Cortes-Hernandez J, Martin J, Vyse TJ, Fernando MM. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. Am J Hum Genet. 2012;90:445–456. doi: 10.1016/j.ajhg.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rigby WF, Wu YL, Zan M, et al. Increased frequency of complement C4B deficiency in rheumatoid arthritis. Arthritis Rheum. 2012;64:1338–1344. doi: 10.1002/art.33472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol. 2007;96:179–204. doi: 10.1016/S0065-2776(07)96005-8. [DOI] [PubMed] [Google Scholar]
- 41.Kocher M, Siegel ME, Edberg JC, Kimberly RP. Cross-linking of Fc gamma receptor IIa and Fc gamma receptor IIIb induces different proadhesive phenotypes on human neutrophils. J Immunol. 1997;159:3940–3948. [PubMed] [Google Scholar]
- 42.Aitman TJ, Dong R, Vyse TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 43.Fanciulli M, Norsworthy PJ, Petretto E, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mamtani M, Anaya JM, He W, Ahuja SK. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun. 2010;11:155–160. doi: 10.1038/gene.2009.71. [DOI] [PubMed] [Google Scholar]
- 45.Morris DL, Roberts AL, Witherden AS, et al. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur J Hum Genet. 2010;18:1027–1031. doi: 10.1038/ejhg.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinney C, Fanciulli M, Merriman ME, et al. Association of variation in Fcgamma receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann Rheum Dis. 2010;69:1711–1716. doi: 10.1136/ard.2009.123588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson JI, Carr IM, Cooper DL, et al. Confirmation of association of FCGR3B but not FCGR3A copy number with susceptibility to autoantibody positive rheumatoid arthritis. Hum Mutat. 2012;33:741–749. doi: 10.1002/humu.22031. [DOI] [PubMed] [Google Scholar]
- 48.McKinney C, Merriman TR. Meta-analysis confirms a role for deletion in FCGR3B in autoimmune phenotypes. Hum Mol Genet. 2012;21:2370–2376. doi: 10.1093/hmg/dds039. [DOI] [PubMed] [Google Scholar]
- 49.McKinney C, Merriman ME, Chapman PT, et al. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2008;67:409–413. doi: 10.1136/ard.2007.075028. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 51.Mamtani M, Rovin B, Brey R, et al. CCL3L1 gene-containing segmental duplications and polymorphisms in CCR5 affect risk of systemic lupus erythaematosus. Ann Rheum Dis. 2008;67:1076–1083. doi: 10.1136/ard.2007.078048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burns JC, Shimizu C, Gonzalez E, et al. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J Infect Dis. 2005;192:344–349. doi: 10.1086/430953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 54.Yang D, Chertov O, Bykovskaia SN, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 55.Niyonsaba F, Ogawa H, Nagaoka I. Human beta-defensin-2 functions as a chemotactic agent for tumour necrosis factor-alpha-treated human neutrophils. Immunology. 2004;111:273–281. doi: 10.1111/j.0019-2805.2004.01816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hollox EJ, Huffmeier U, Zeeuwen PL, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wehkamp J, Harder J, Weichenthal M, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2003;9:215–223. doi: 10.1097/00054725-200307000-00001. [DOI] [PubMed] [Google Scholar]
- 58.Fellermann K, Stange DE, Schaeffeler E, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bentley RW, Pearson J, Gearry RB, et al. Association of higher DEFB4 genomic copy number with Crohn's disease. Am J Gastroenterol. 2010;105:354–359. doi: 10.1038/ajg.2009.582. [DOI] [PubMed] [Google Scholar]
- 60.Aldhous MC, Abu Bakar S, Prescott NJ, et al. Measurement methods and accuracy in copy number variation: failure to replicate associations of beta-defensin copy number with Crohn's disease. Hum Mol Genet. 2010;19:4930–4938. doi: 10.1093/hmg/ddq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou XJ, Cheng FJ, Lv JC, et al. Higher DEFB4 genomic copy number in SLE and ANCA-associated small vasculitis. Rheumatology (Oxford) 2012;51:992–995. doi: 10.1093/rheumatology/ker419. [DOI] [PubMed] [Google Scholar]
- 62.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 63.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brest P, Lapaquette P, Souidi M, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 66.Prescott NJ, Dominy KM, Kubo M, et al. Independent and population-specific association of risk variants at the IRGM locus with Crohn's disease. Hum Mol Genet. 2010;19:1828–1839. doi: 10.1093/hmg/ddq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Viau M, Zouali M. B-lymphocytes, innate immunity, and autoimmunity. Clin Immunol. 2005;114:17–26. doi: 10.1016/j.clim.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 68.Zouali M. Transcriptional and metabolic pre-B cell receptor-mediated checkpoints: implications for autoimmune diseases. Mol Immunol. 2014;62:315–320. doi: 10.1016/j.molimm.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 69.Yim SH, Chung YJ, Jin EH, et al. The potential role of VPREB1 gene copy number variation in susceptibility to rheumatoid arthritis. Mol Immunol. 2011;48:1338–1343. doi: 10.1016/j.molimm.2010.11.009. [DOI] [PubMed] [Google Scholar]