

Abstract
Although transformation to Hodgkin lymphoma (HL) is a recognized complication in patients with chronic lymphocytic leukemia (CLL), its incidence, clinical characteristics and outcomes are not well defined. We used the Mayo Clinic CLL and Lymphoma Databases to identify CLL patients who developed biopsy-proven HL (CLL/HL) on follow-up, as well as cases of de novo HL (i.e., without prior CLL). Among 3887 CLL patients seen at Mayo Clinic from January 1995 through August 2011, 26 (0.7%) developed HL. In a nested cohort of 2,465 newly diagnosed CLL patients followed prospectively, the incidence of HL was 0.05%/year (10 year risk = 0.5%). The median overall survival (OS) from date of HL diagnosis in patients with CLL/HL was 3.9 years compared to not reached for de novo HL patients (n = 709) seen during the same time interval (P < 0.001). The shorter OS of CLL/HL patients persisted after adjusting for differences in age and Ann Arbor stage of disease. The International Prognostic score (IPS) developed for de novo HL stratified prognosis among CLL/HL patients with median survival of not reached, 6.2, 2.4, and 0.3 years (P = 0.006) for those with IPS scores of ≤2, 3, 4, and ≥5, respectively. In summary, approximately 1 of every 200 CLL patients will develop HL within 10 years. Survival after HL diagnosis in patients with CLL is shorter than de novo HL patients. The IPS for de novo HL may be useful for stratifying survival in CLL/HL patients.
Introduction
Although chronic lymphocytic leukemia (CLL) is considered a low-grade lymphoproliferative disorder, ∼5–10% of patients transform into a more aggressive lymphoma. The most common histologic subtype in patients who transform is diffuse large B-cell lymphoma (DLBCL) [1]. Less frequently, CLL can transform to Hodgkin lymphoma (HL). Although the incidence and risk factors of CLL patients who develop DLBCL are well described in the literature [2–4], the limited knowledge regarding HL in CLL patients (CLL/HL) is derived from case reports [5–9] or case series involving few patients [10-15]. While CLL patients that develop clonally related transformation to DLBCL are known to have a worse outcome than de novo DLBCL [3,16], there is no information regarding the outcome of HL arising as a complication of CLL compared to de novo HL (i.e., with no prior CLL). In the largest single-center experience published to date, investigators at the MD Anderson Cancer Center (MDACC) described 18 CLL patients who developed HL and reported a median survival after HL diagnosis of 0.8 years [17]. More recently, Bockorny and colleagues conducted a comprehensive review of the literature from 1975 to 2011 and identified 86 CLL patients (including the 18 from MDACC) who developed HL. Although the authors did not have access to the individual patient data for their analysis, they estimated the median survival for CLL/HL was 1.7 years, and reported the outcomes of HL patients who had received prior CLL therapy (especially therapy containing purine analogs) was worse compared to those who had not received prior CLL therapy [18]. We used the Mayo Clinic CLL and Mayo Clinic Lymphoma Databases to evaluate the incidence, clinical characteristics, and outcomes of CLL patients who developed HL. We also compared the outcomes of these patients to individuals with de novo HL seen at Mayo Clinic during the same time interval.
Methods
The Mayo Clinic CLL Database includes all patients with a pathologically confirmed diagnosis of CLL seen in the Division of Hematology at Mayo Clinic, Rochester, MN and who permit their records to be used for research purposes [2,19–21]. Baseline clinical and laboratory characteristics are recorded on all patients, including novel prognostic markers such as immunoglobulin heavy chain variable (IGHV) gene mutation status, expression of ZAP-70 and CD38, and genetic abnormalities detected by fluorescence in situ hybridization (FISH). In selected patients, in situ hybridization studies were performed on paraffin sections of the bone marrow biopsy or lymph node specimen using probes specific for EBV-encoded ribonucleic acid. Patients are prospectively followed, and data regarding therapy of CLL, secondary cancers, and overall survival (OS) are recorded for all patients on an ongoing basis.
After approval from the Mayo Clinic Institutional Review Board, CLL patients seen at Mayo Clinic from January 1995 through August 2011 were considered eligible for this study. CLL patients with biopsy-proven HL pathologically confirmed at Mayo Clinic were identified within this cohort. The Mayo Clinic Lymphoma Database was also used to identify a contemporaneous cohort of patients with de novo HL. The clinical and laboratory characteristics of patients at the time of HL diagnosis, therapy administered for HL and survival were abstracted from clinical records. The International Prognostic Score (IPS) for HL was applied to all patients at the time of HL diagnosis [22,23]. The original study by Hasenclever et al reported the following seven factors were independently associated with shorter OS: serum albumin <4 g dL−1, hemoglobin <10.5 g dL−1, male sex, age >45 years, stage IV disease, WBC ≥15 × 109/L, and lymphocytopenia (absolute lymphocyte count <0.6 × 109/L or <8% of differential) [23]. We also evaluated the recently proposed simplified prognosis score based on age >45 years, hemoglobin <10.5 g dL−1, and stage IV disease (3-Factor Prognostic Score, PS-3) [24] among CLL/HL patients. Patients were followed until death, loss to follow-up, or the study end date of December 31, 2013.
The time to development of HL was defined as date of CLL diagnosis to the date of diagnosis of HL. To reduce referral bias and calculate prevalence of HL over long-term follow-up of CLL patients accurately, only newly diagnosed (<12 months of diagnosis) CLL patients were included in the time-to-HL analyses. Fisher's exact and Kruskal–Wallis tests were used to define differences between the categorical and continuous variables, respectively. The OS was estimated by the Kaplan–Meier method and defined as the time from the date of HL diagnosis until death or last follow-up. Patients alive at the last recorded follow-up were censored. Multivariable Cox proportional hazards models were used to determine which factors affect OS from HL diagnosis; included in the model were age at HL diagnosis, Ann Arbor stage, and prior CLL. All statistical analyses were conducted using SAS 9.3 software package (SAS Institute, Cary, NC).
Results
CLL patients who develop HL
Among 3,887 CLL patients seen at Mayo Clinic from January 1995 through August 2011, 26 (0.7%) patients developed biopsy-proven HL. The baseline characteristics of these patients at the time of HL diagnosis are shown in Table I. Their median age was 67 years (range 45–88 years). All patients had classical HL (except in three patients in whom HL could not be further classified). Of nine CLL/HL patients in whom EBV in situ hybridization studies were performed, EBV was detected in 6 (67%) biopsy samples. No association was observed between prior purine analog therapy and EBV status (P = 1.0).
Table I. Characteristics of CLL Patients at the Time of Hodgkin Lymphoma Diagnosis.
| Characteristic | N (%) | |
|---|---|---|
| Number of patients | 26 | |
| Median age, years (range) | 67 (45–88) | |
| Male | 21 (81) | |
| Median WBC, ×109/L (range) | 7.8 (1.3–87.3) | |
| Median HB, g dL−1(range) | 11 (7.2–13) | |
| Median PLT, ×109/L (range) | 220.5 (53–627) | |
| Median LDH, U L−1(range) | 249 (133–658) | |
| Median creatinine, mg dL−1 (range) | 1 (0.6–2.1) | |
| Median albumin, g dL−1(range) | 3.5 (2.6–4.2) | |
| Ann Arbor stage | I | 0 |
| II | 10 (38) | |
| III | 3 (11) | |
| IV | 11 (42) | |
| Undetermined | 2 (8) | |
| Epstein–Barr virus statusa | Positive | 6 (67) |
| Negative | 3 (33) | |
| Not done | 17 | |
| Prior CLL therapy before HL diagnosis | No | 9 (35) |
| Yes | 17 (65) | |
| Purine analog containing regimen | 13 (50) | |
| Therapy administered for of HL | ABVD | 16 (61) |
| MOPP/ABV hybrid | 3 (11) | |
| BCVPP | 2 (8) | |
| CVPP | 1 (4) | |
| VP | 1 (4) | |
| Unknown | 3 (11) |
Performed clinically on select patients.
Abbreviations used: WBC; white cell count; HB: hemoglobin; PLT; platelet; LDH; lactate dehydrogenase; ABVD: doxorubicin, bleomycin, vinblastine and dacarbazine; MOPP/ABV hybrid: mechlorethamine, vincristine, procarbazine, and prednisone (MOPP) followed by doxorubicin, bleomycin and vinblastine (ABV); BCVPP: carmustine, cyclophosphamide, vinblastine, pro-carbazine, and prednisone; CVPP: chlorambucil, vincristine, procarbazine and prednisone; VP: vinblastine and prednisone.
The median time from CLL diagnosis to development of HL was 6.2 years (range, 0–24.5 years). In a nested cohort of 2,465 CLL patients who were seen at the time of CLL diagnosis and followed prospectively (median follow-up= 6.3 years), the prevalence of HL was 0.05%/year (10 year risk = 0.5%, 95% C.I. = 0.1–0.8%, Fig. 1).
Figure 1.
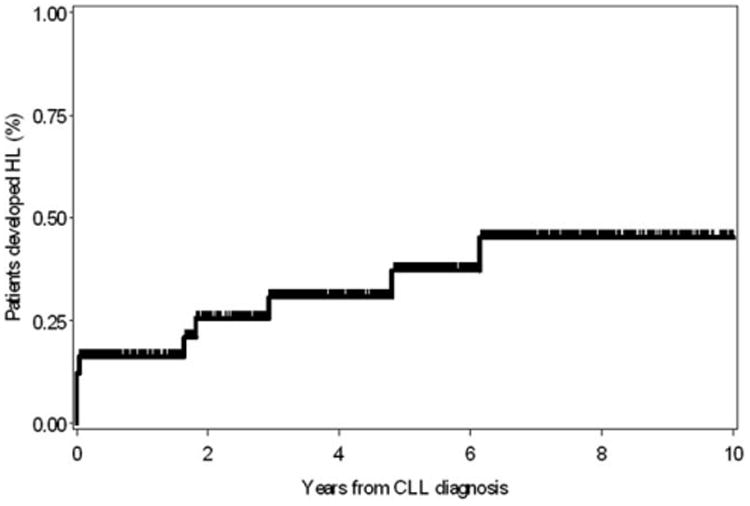
Risk of Hodgkin lymphoma in 2,465 newly diagnosed CLL patients seen at the time of diagnosis and followed prospectively.
Risk factors for development of HL in CLL patients
The baseline characteristics of CLL patients (at the time of CLL diagnosis) who developed subsequent HL (n = 26) versus those who did not (n = 3,861) are shown in Table II (Supporting Information). No significant differences in the baseline clinical and biological characteristics were noted between the two groups, though comparisons were limited by the small sample size of CLL/HL patients.
Survival of CLL/HL patients
After a median follow-up of 9.4 years from CLL diagnosis date, 17/26 (65.4%) CLL/HL patients have died. The median OS from date of HL diagnosis was 3.9 years. The median survival of CLL/HL patients who had previously received CLL therapy was significantly shorter compared to those who were untreated for CLL (2.8 years vs. 7.6 years, P = 0.02, Fig. 2A). The survival of CLL/HL patients who received prior purine analog therapy for CLL was also significantly shorter compared to those who did not receive purine analogs (1.8 years vs. 5 years, P = 0.03, Fig. 2B). There was no difference in survival according to the EBV in situ hybridization status of HL in the nine cases analyzed (data not shown).
Figure 2.
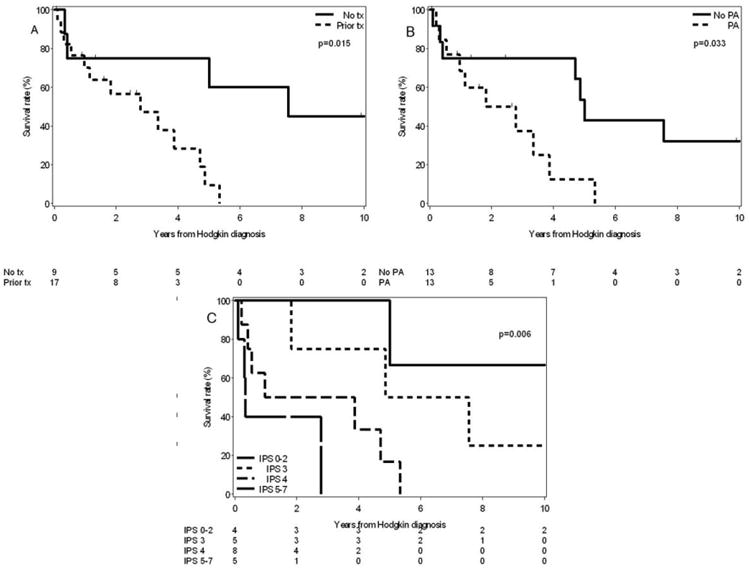
(A) Overall Survival of CLL/HL patients based on prior receipt of CLL therapy. (B) Overall survival of CLL/HL patients who had received previous purine analog therapy (PA) for CLL compared to those who did not receive PA therapy. (C) Overall survival of CLL/HL patients according to the International Prognostic Score (IPS).
The IPS for HL predicted median survival among CLL/HL patients as follows: score of ≤2 (18% of the patients), median survival not reached; score of 3 (23% of the patients), 6.2 years; score of 4 (36% of the patients), 2.4 years; and score ≥5 (23% of the patients), 0.3 years (P = 0.006, Fig. 2C). A recently proposed simplified prognostic score (PS-3 score) was also able to stratify median survival among CLL/HL patients as follows: score of 1 (36% of the patients), 0.3 years; score of 2 (36% of the patients), 1.4 years; and score of 3 (27% of the patients), 6.2 years (P = 0.02, data not shown).
Comparison of CLL/HL to de novo HL
Seven hundred and nine patients with de novo HL were seen at Mayo Clinic during the same time interval and entered into the Mayo Clinic Lymphoma Database. The median survival of CLL/HL patients was significantly shorter than those with de novo HL (3.9 years vs. not reached, P < 0.001; Fig. 3A). Older age (hazard ratio [HR] for each year older = 1.05, 95% CI = 1.04–1.06, P < 0.001), Ann Arbor stage (HR = 1.7, 95% CI = 1.2–2.4, P = 0.002) and prior CLL (HR = 1.7, 95% CI = 1.02–2.9, P = 0.04) were independently associated with worse survival on multivariate analysis. Among patients with an IPS of 0-3, there was a trend toward shorter survival in CLL/HL patients compared to de novo HL (median OS: 7.6 years vs. not reached, respectively, P = 0.06, Fig. 3B). Among patients with IPS of 4-7, OS was significantly shorter in CLL/HL patients compared to de novo HL patients (median OS: 1 year vs. not reached, respectively, P = 0.003, Fig. 3C).
Figure 3.
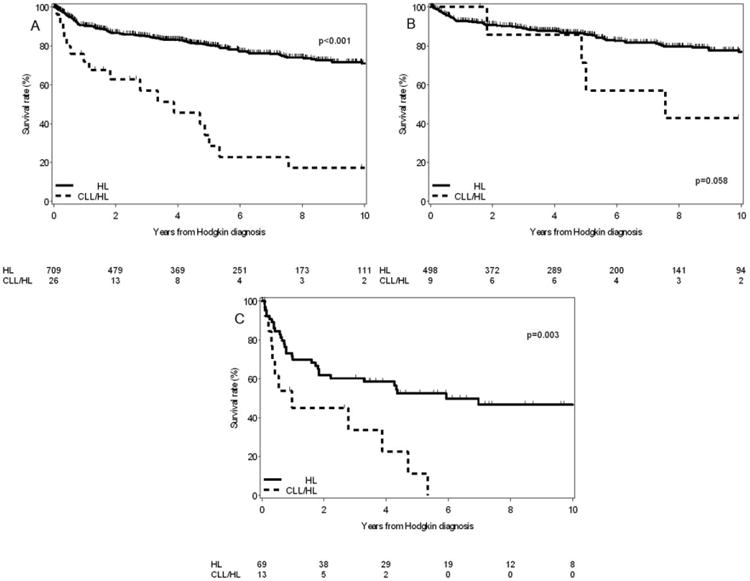
(A) Overall Survival of CLL/HL patients (from the time of diagnosis of Hodgkin lymphoma) compared to patients with de novo Hodgkin lymphoma. (B) Overall survival of CLL/HL patients (from the time of diagnosis of Hodgkin lymphoma) compared to de novo HL according to the International Prognostic Score of 0–3. (C) Overall survival of CLL/HL patients (from the time of diagnosis of Hodgkin lymphoma) compared to de novo HL according to the International Prognostic Score of 4–7.
Discussion
This is the largest single-institution study of HL in CLL patients and is the first, to our knowledge, to employ a prospective cohort design that allows calculation of incidence and compares the outcome of CLL/HL patients to those with de novo HL. The primary findings include the following: (1) the 10-year incidence of HL among newly diagnosed CLL patients was ∼0.5% (1/200 CLL patients), and the median time from CLL diagnosis to development of HL was 6.2 years; (2) the median OS after a diagnosis of HL was ∼4 years and patients who previously received CLL therapy (especially purine analogs) experienced a worse outcome; (3) the OS of CLL/HL patients was worse than de novo HL patients; and (4) the IPS score for de novo HL was able to effectively stratify survival of CLL/HL patients.
Several studies have reported that HL develops in ∼0.1–0.4% CLL patients on long-term follow-up [13,17,25]. This is the first study that describes the incidence of HL in a cohort of newly diagnosed CLL patients who were followed prospectively with adequate long-term follow-up. Our results show the risk of developing HL after CLL diagnosis is ∼0.25% (1 in 400) at 5-years and 0.5% (1 in 200) at 10-years (∼0.05%/year risk). This is in contrast to DLBCL transformation in CLL which is ∼ 10–20 times more common (10-year risk of DLBCL in CLL: 5-10%) [2–4]. The median time to development of HL was ∼6 years after CLL diagnosis in our study, slightly longer than the 2.6–4.6 years reported in other similar series [13,17,18]. Our study did not identify clinical or biological risk factors at the time of CLL diagnosis that were associated with the future development of HL, likely, at least in part, due to small sample size.
Among CLL/HL patients, prior therapy of CLL with purine analogs adversely impacted survival after HL diagnosis. Results from our study are consistent with this observation [13,18]. It is postulated that therapy with purine analogs leads to prolonged immunosuppression, which may increase the risk of EBV reactivation and subsequent development of secondary lymphoproliferative disorders, including HL [26]. In our series, 6/9 (67%) CLL/HL patients tested were EBV positive; and 4/6 (67%) EBV-positive patients previously received nucleoside analog. It is unlikely that EBV reactivation alone explains the worse prognosis in patients with prior purine analog exposure, since there was no difference in EBV positivity between those who received purine analogs versus those who did not.
Therapy with doxorubicin, bleomycin, vinblastine and dacarbazine (ABVD) currently represents a standard of care for newly diagnosed de novo HL patients [27]. In our study, ∼60% of CLL/HL patients were treated with ABVD. In contrast, in the MDACC study and in the series reported by Bockorny and colleagues, only 30% CLL/HL patients received ABVD. No patients in our study underwent stem cell transplantation or receive novel therapies for HL such as brentuximab vedotin (anti-CD30 monoclonal antibody) [28]. The median survival of patients in our study (3.9 years) was longer than previously reported by Tsimberidou et al. (0.8 years) [17], Bockorny et al. (1.7 years) [18], and Jamroziak et al. (0.3 years) [13]; although it was comparable to those reported by Tamdor et al. (3.3 years) [15]. Although the improved survival in our study may be in part related to a higher proportion of patients receiving ABVD, it could also be due to difference among other factors including age, IPS risk profile, or the clonal relationship of the HL to the underlying CLL. Unfortunately, like most previous studies, we do not have the information about clonal relationship between the CLL and HL phase to be able to comment on this aspect. It is unlikely that patients in our study were low risk Hodgkin transformations since ∼60% patients received prior therapy for CLL (including 50% patients who received purine analogs), ∼50% had an IPS of ≥4 (considered high-risk for de novo HL), and ∼40% patients had stage IV disease at HL diagnosis.
There is limited information about prognostic factors related to survival after HL transformation in CLL. Tsimberidou et al. applied the Richter prognosis score in their cohort of 18 transformed HL patients but were not able to segregate patients with different outcomes [17]. Similarly, the Richter prognosis score failed to predict survival in our cohort of CLL/HL patients. Hasenclever and Diehl developed the IPS for de novo HL based on 5,141 patients treated with combination chemotherapy and/or radiotherapy. Although the IPS did not include de novo HL patients >65 years of age, this scoring system was applied to our group of CLL/HL patients whose median age was 67 years. We were able to effectively stratify our cohort of CLL/HL patients, with median survival of not reached, 6.2, 2.4, and 0.3 years for those with IPS scores of ≤2, 3, 4, and ≥5, respectively. Additionally, the PS-3 score (age ≥45 years, hemoglobin <10.5 g dL−1, and stage IV disease), proposed by Diefenbach et al. from the US Intergroup E2496 study for de novo HL patients, accurately predicted outcomes in our cohort, suggesting that this prognostic score may be useful in CLL/HL patients also. In de novo HL, age >70 is associated with a significantly shorter survival compared to patients 60-69 years of age [29–32]. To investigate the impact of older age on survival, we conducted a multivariable analysis for all patients with HL (CLL/HL and de novo HL) in our study. After adjusting for age and Ann Arbor disease stage, prior CLL remained significantly associated with shorter survival, thereby suggesting that age alone does not explain the observed differences in survival between de novo HL and CLL/HL. It is unclear if newer prognostic markers for de novo HL such as response to therapy based on positron emission tomography can be readily applied to CLL patients with HL transformation at this time.
Our study has several limitations. Novel CLL prognostic markers were not available for all patients in the study during the entire study period; hence, we had limited power to study their impact on the development of HL. The lack of archived tissue for most patients precluded determining the clonal relationship between HL and the preceding CLL in this clinical study. Additionally, although it is the largest single-institution study to date, the relatively small number of HL patients did not allow a more comprehensive modeling of risk factor analysis. It should also be noted that the IPS was originally developed from patients <65 years treated in the pre-ABVD era [23]; nonetheless this staging system was subsequently validated in community-based cohort of HL patients up to age 85 years receiving ABVD-based therapy [22].
In summary, this large single-institution analysis of CLL patients shows that HL transformation is 0.5% at 10 years. The median survival after HL diagnosis was ∼4 years for the entire cohort; and 1.8 years for the subset of patients who had previously received purine analog CLL therapy. The IPS score may help in predicting survival after HL transformation in CLL patients.
Supplementary Material
Acknowledgments
This study was presented at the Annual American Society of Hematology Meeting at New Orleans, LA in December 2013.
Contract grant sponsor: NCI; Contract grant numbers: K23CA160345; CA95241.
Contract grant sponsor: Predolin Foundation.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of interest: Sameer Parikh is the recipient of the Mayo Clinic Department of Medicine Career Development Grant. Tait Shanafelt is a scholar of the Leukemia and Lymphoma Society. The following authors have financial disclosures: Tait D. Shanafelt: Has received research funding from Genentech, Glaxo-Smith-Kline, Cephalon, Hospira, Celgene, Jannsen, and Polyphenon E International. All other authors: None.
Author Contributions: Sameer Parikh and Tait Shanafelt were the principal investigators, collected and analyzed data and wrote the first draft of the manuscript. Kari Rabe and Susan Slager collected and analyzed data and performed statistical analyses. Thomas Habermann, Timothy Call, Wei Ding, Jose Leis, Luis Porrata and Neil Kay helped conceive the study, and participated in manuscript preparation. Susan Schwager and Kay Ristow collected and analyzed data, and participated in manuscript preparation. William Macon performed pathological review on specimens. All authors participated in data interpretation and critical review of the final article.
The following authors have financial disclosures: Tait D. Shanafelt: Has received research funding from Genentech, Glaxo-Smith-Kline, Cephalon, Hospira, Celgene, Jannsen, and Polyphenon E International. All other authors: None.
References
- 1.Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123:1647–1657. doi: 10.1182/blood-2013-11-516229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parikh SA, Rabe KG, Call TG, et al. Diffuse large B-cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): A cohort study of newly diagnosed patients. Br J Haematol. 2013;162:774–782. doi: 10.1111/bjh.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117:3391–3401. doi: 10.1182/blood-2010-09-302174. [DOI] [PubMed] [Google Scholar]
- 4.Tsimberidou AM, O'Brien S, Khouri I, et al. Clinical outcomes and prognostic factors in patients with Richter's syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24:2343–2351. doi: 10.1200/JCO.2005.05.0187. [DOI] [PubMed] [Google Scholar]
- 5.Krause JR, Drinkard LC, Keglovits LC. Hodgkin lymphoma transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma. Proc Baylor Univ Med Center. 2013;26:16–18. doi: 10.1080/08998280.2013.11928901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tseng D, Jones CD, Anderson M, et al. Clonally identical Hodgkin's disease develops after allogeneic hematopoietic cell transplant for CLL. Bone Marrow Transplant. 2011;46:1576–1578. doi: 10.1038/bmt.2010.340. [DOI] [PubMed] [Google Scholar]
- 7.Vasilj A, Kojic-Katovic S, Maricevic I, et al. Hodgkin's lymphoma variant of Richter's syndrome. Collegium Antropol. 2010;34:295–299. [PubMed] [Google Scholar]
- 8.Tzankov A, Fong D. Hodgkin's disease variant of Richter's syndrome clonally related to chronic lymphocytic leukemia arises in ZAP-70 negative mutated CLL. Med Hypotheses. 2006;66:577–579. doi: 10.1016/j.mehy.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Fabbri A, Cencini E, Bocchia M. Hodgkin's lymphoma as Richter's transformation in chronic lymphocytic leukemia. Med Oncol. 2014;31:904. doi: 10.1007/s12032-014-0904-9. [DOI] [PubMed] [Google Scholar]
- 10.Kazmierczak M, Kroll-Balcerzak R, Balcerzak A, et al. Hodgkin lymphoma transformation of chronic lymphocytic leukemia: Cases report and discussion. Med Oncol. 2014;31:800. doi: 10.1007/s12032-013-0800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fayad L, Robertson LE, O'Brien S, et al. Hodgkin's disease variant of Richter's syndrome: Experience at a single institution. Leuk Lymph. 1996;23:333–337. doi: 10.3109/10428199609054836. [DOI] [PubMed] [Google Scholar]
- 12.Dujardin F, Lefrancq T, Blechet C, et al. Hodgkin's disease variant of Richter's syndrome. Two cases and literature review. Ann Pathol. 2008;28:311–316. doi: 10.1016/j.annpat.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Jamroziak K, Grzybowska-Izydorczyk O, Jesionek-Kupnicka D, et al. Poor prognosis of Hodgkin variant of Richter transformation in chronic lymphocytic leukemia treated with cladribine. Br J Haematol. 2012;158:286–288. doi: 10.1111/j.1365-2141.2012.09127.x. author reply 289. [DOI] [PubMed] [Google Scholar]
- 14.Momose H, Jaffe ES, Shin SS, et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma with Reed-Sternberg-like cells and possible transformation to Hodgkin's disease. Mediation by Epstein–Barr virus. Am J Surg Pathol. 1992;16:859–867. doi: 10.1097/00000478-199209000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Tadmor T, Shvidel L, Goldschmidt N, et al. Hodgkin's variant of Richter transformation in chronic lymphocytic leukemia: A retrospective study from the Israeli CLL study group. Anticancer Res. 2014;34:785–790. [PubMed] [Google Scholar]
- 16.Mao Z, Quintanilla-Martinez L, Raffeld M, et al. IgVH mutational status and clonality analysis of Richter's transformation: Diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol. 2007;31:1605–1614. doi: 10.1097/PAS.0b013e31804bdaf8. [DOI] [PubMed] [Google Scholar]
- 17.Tsimberidou AM, O'Brien S, Kantarjian HM, et al. Hodgkin transformation of chronic lymphocytic leukemia: The M. D. Anderson cancer center experience. Cancer. 2006;107:1294–1302. doi: 10.1002/cncr.22121. [DOI] [PubMed] [Google Scholar]
- 18.Bockorny B, Codreanu I, Dasanu CA. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: A retrospective analysis of world literature. Br J Haematol. 2012;156:50–66. doi: 10.1111/j.1365-2141.2011.08907.x. [DOI] [PubMed] [Google Scholar]
- 19.Maddocks-Christianson K, Slager SL, Zent CS, et al. Risk factors for development of a second lymphoid malignancy in patients with chronic lymphocytic leukaemia. Br J Haematol. 2007;139:398–404. doi: 10.1111/j.1365-2141.2007.06801.x. [DOI] [PubMed] [Google Scholar]
- 20.Shanafelt TD, Rabe KG, Kay NE, et al. Age at diagnosis and the utility of prognostic testing in patients with chronic lymphocytic leukemia. Cancer. 2010;116:4777–4787. doi: 10.1002/cncr.25292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parikh SA, Rabe KG, Kay NE, et al. Chronic lymphocytic leukemia in young (≤55 years) patients: a comprehensive analysis of prognostic factors and outcomes. Haematologica. 2014;99(1):140–147. doi: 10.3324/haematol.2013.086066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moccia AA, Donaldson J, Chhanabhai M, et al. International prognostic score in advanced-stage Hodgkin's lymphoma: Altered utility in the modern era. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:3383–3388. doi: 10.1200/JCO.2011.41.0910. [DOI] [PubMed] [Google Scholar]
- 23.Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin's disease. International prognostic factors project on advanced Hodgkin's disease. N Engl J Med. 1998;339:1506–1514. doi: 10.1056/NEJM199811193392104. [DOI] [PubMed] [Google Scholar]
- 24.Diefenbach C, Li H, Hong F, et al. Evaluation of a novel 3 factor prognostic score (PS-3) for patients with advanced Hodgkin lymphoma (HL) treated on US intergroup E2496. ASH Annual Abstracts 2013. Blood. 122(21) Abstract 4277. [Google Scholar]
- 25.Travis LB, Curtis RE, Hankey BF, et al. Second cancers in patients with chronic lymphocytic leukemia. J Natl Cancer Inst. 1992;84:1422–1427. doi: 10.1093/jnci/84.18.1422. [DOI] [PubMed] [Google Scholar]
- 26.Roschewski M, Wilson WH. EBV-associated lymphomas in adults. Best Practice Res Clin Haematol. 2012;25:75–89. doi: 10.1016/j.beha.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodgkin Lymphoma. NCCN Clinical Guidelines in Oncology: Hodgkin Lymphoma Available at: www.nccn.org.
- 28.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363:1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 29.Jagadeesh D, Diefenbach C, Evens AMXII. Hodgkin lymphoma in older patients: Challenges and opportunities to improve outcomes. Hematol Oncol. 2013;31(Suppl 1):69–75. doi: 10.1002/hon.2070. [DOI] [PubMed] [Google Scholar]
- 30.Landgren O, Algernon C, Axdorph U, et al. Hodgkin's lymphoma in the elderly with special reference to type and intensity of chemotherapy in relation to prognosis. Haematologica. 2003;88:438–444. [PubMed] [Google Scholar]
- 31.Evens AM, Helenowski I, Ramsdale E, et al. A retrospective multicenter analysis of elderly Hodgkin lymphoma: Outcomes and prognostic factors in the modern era. Blood. 2012;119:692–695. doi: 10.1182/blood-2011-09-378414. [DOI] [PubMed] [Google Scholar]
- 32.Enblad G, Gustavsson A, Sundstrom C, et al. Patients above sixty years of age with Hodgkin's lymphoma treated with a new strategy. Acta Oncol. 2002;41:659–667. doi: 10.1080/028418602321028283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.