

Abstract
Background
Fischoederius elongates is an important trematode of Paramphistomes in ruminants. Animals infected with F. elongates often don’t show obvious symptoms, so it is easy to be ignored. However it can cause severe economic losses to the breeding industry. Knowledge of the mitochondrial genome of F. elongates can be used for phylogenetic and epidemiological studies.
Findings
The complete mt genome sequence of F. elongates is 14,120 bp in length and contains 12 protein-coding genes, 22 tRNA genes, two rRNA genes and two non-coding regions (LNR and SNR). The gene arrangement of F. elongates is the same as other trematodes, such as Fasciola hepatica and Paramphistomum cervi. Phylogenetic analyses using concatenated amino acid sequences of the 12 protein-coding genes by Maximum-likelihood and Neighbor-joining analysis method showed that F. elongates was closely related to P. cervi.
Conclusion
The complete mt genome sequence of F. elongates should provide information for phylogenetic and epidemiological studies for F. elongates and the family Paramphistomidae.
Keywords: Fischoederius elongates, Mitochondrial genome
Findings
Background
Paramphistomes are distributed worldwide and have been reported in many countries, such as Bulgaria, France, Poland, Hungary, Italy, India, Russia, Sardinia and Yugoslavia [1]. The paramphistome can infect fishes, reptiles, birds and mammals, some of which can lead to huge economic losses related to seriously gastrointestinal diseases, low producitivity or death in ruminants [2]. In Arumeru District, the prevalence rate of paramphistomes is as high as 56.7 % in cattle [3].
Fischoederius elongates is an important member of paramphistomes, the parasite usually inhabits the rumen of cattle, buffaloes, sheep and goats. Ruminants are usually infected by ingesting snails, such as Lymnaea acuminata, Lymnaea succinea or Gyraulus euphraticus [4]. Ruminants infected with F. elongates show weakness, mental fatigue and eventually death. More seriously, F. elongates maybe a zoonotic trematode, a Chinese woman from Guangdong Province was reported to be the first human infection case [5], but it is still unknown how she was infected.
Untill now, the most common diagnostic method for F. elongates is the microscopical examination, but it’s time-consuming, and hard to distinguish with other paramphistomes. As a useful marker, mt genome has been widely used for species identification [6–10]. The complete mt genome of F. elongates can provide alternative molecular markers for the species identification, epidemiology and genetic diversity of paramphistomes.
In the present study, we got the full sequence and gene arrangement of mt genome of F. elongates and compared it with selected trematodes. We found that F. elongates had the closest relationship with P. cervi.
Methods
Ethical approval
The study was performed under the instructions and approval of Laboratory Animals Research Centre of Hubei province in P. R. China and the ethics committee of Huazhong Agricultural University (Permit number: 4200695757).
Parasite collection and DNA isolation
F. elongates adults were collected from the rumen and reticulum of naturally infected cattle in Zhanggang, Tianmen, Hubei province, PR China, according to the Animal Ethics Guidelines of Huazhong Agricultural University. Then, the adult worms were washed extensively in 0.9 % sodium chloride solution, and identified through morphological examinations [2]. Subsequently, one worm was stained for identification [11], and the rest were fixed in 75 % alcohol (V/V) and stored at −20 °C until use [12]. Total genomic DNA was isolated from one worm [13]. The ITS-2 region of F. elongates was amplified and sequenced as reported previously [14], it was 100 % similar to that of F. elongates (GenBank accession no. JQ688410.1).
Amplification and sequencing of F. elongates mt genome
Firstly, we designed 12 oligonucleotide primers according to the conserved regions from reported mt genome sequences of F. hepatica [15], Clonorchis sinensis [16] and P. cervi [17] to amplify partial fragments from cox3, cytb, nad4, cox1, rrnS and nad5 (Table 1). PCRs (25 μl) were performed in the following reaction: 10 mM Tris–HCl (pH 8.4), 50 mM KCl, 4 mM MgCl2, 200 mM each of dNTP, 50 pmol of each primer,2 U Taq polymerase (Takara) and 2.5 μl genomic DNA. Reactions were run under the following conditions: 94 °C for 5 min, followed by 35 cycles of 94 °C/30 s, 50 °C/30 s and 72 °C/1 min. Amplicons were sent to Sangon Company (Shanghai, China) for sequencing.
Table 1.
Primers used in the present study
| Primer codes | Sequences (5′-3′) | Target gene | References |
|---|---|---|---|
| XCCOX3F | AGYACDGTDGGDTTRCATTT | cox31 | Present study |
| XCCOX3R | CANAYATAATCMACARAATGNCA | cox31 | Present study |
| nxccobF | ATGTCWTWTTGRGCKGCBACNGT | cytb1 | Present study |
| nxccobR | GADVCTCNGGRTGRCAVGCHCC | cytb1 | Present study |
| nxcND4F | GAKTCBCCDTATTCDGARCG | nad41 | Present study |
| nxcND4R | ACHCCNGCHGANANMCCRTGMCC | nad41 | Present study |
| TXCCOX1F | GGHTGAACHRTWTAYCCHCC | cox11 | Present study |
| TXCCOX1R | TGRTGRGCYCAWACDAYAMAHCC | cox11 | Present study |
| XC12SF | AAWAAYGAGAGYGACGGGCG | rrnS1 | Present study |
| XC12SR | TARACTAGGATTAGATACCC | rrnS1 | Present study |
| NxcND5F | TGKTTGCBTCNCGNTTBGGNGATG | nad51 | Present study |
| NxcND5R | TAACACTTRCANAHMCCRTGHGT | nad51 | Present study |
| 3CF1 | TGCATGTAGTGATAGGTTTGG | cox3- cytb2 | Present study |
| 3CR1 | AACTAACGTAACATTTGTCAC | cox3- cytb2 | Present study |
| 3CF2 | TTTGTTTTGTGGTTGCCTTC | cytb-nad42 | Present study |
| 3CR2 | AACGTAAATTAAACCTCCCCC | cytb-nad42 | Present study |
| 3CF3 | TGGCGTTTTTGAGTTTGTCTC | nad4-cox12 | Present study |
| 3CR3 | TCAACGAACTCAATATACTTG | nad4-cox12 | Present study |
| 3CF4 | TGGTTTCGGGGCTGTGAGAC | cox1-rrnS2 | Present study |
| 3CR4 | ACCAAGCAAAGAAAATTCTACC | cox1-rrnS2 | Present study |
| 3CF5 | TGTTAAAAGGCTTTGGTGTG | rrnS-nad52 | Present study |
| 3CR5-1 | ACCAACCAAACCTACACATC | rrnS-nad52 | Present study |
| 3CF6-1 | TTACGTTAGTTGGGTTGTTG | nad5-cox32 | Present study |
| 3CR6 | TTACATCTTTATAAAACACTTTC | nad5-cox32 | Present study |
1 short regions amplified by PCR from cox3 (139 bp), cytb (613 bp), nad4 (554 bp), cox1 (497 bp), rrnS (500 bp) and nad5(458 bp). 2 large fragments that were amplified by long-range PCR from cox3-cytb (724 bp), cytb-nad4 (1008 bp), nad4-cox1 (4675 bp), cox1-rrnS (2198 bp), rrnS-nad5 (1981 bp) and nad5-cox3 (1718 bp)
Then, 12 additional primers (Table 1) were designed based on the obtained sequencing results to amplify six regions from genomic DNA (~40-80 ng) by long-PCR. PCRs (50 μl) were performed in reactions containing 0.4 mM each of dNTPs, 5 μl 10× LA Taq buffer II(Mg2+ Plus), 2.5 μM of each primer, 2.5 U LA Taq polymerase (Takara) and 2.5 μl genomic DNA. And the reactions were run under the following program: 94 °C for 5 min, followed by 35 cycles of 94 °C/30 s, 50 °C/30 s and 72 °C/1-5 min (depending on the size of F. hepatica). Amplicons were cloned into pGEM-T-Easy vector (Promega, USA) and then sequenced using a primer-walking strategy [18].
Sequence analyses
F. elongates mt genome sequences were assembled manually and then aligned with the mt genome sequences of F. hepatica, C. sinensis and P. cervi using the program Clustal X 1.83 [19]. Open reading frames were identified by ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) using the echinoderm and flatworm mitochondrial code. Initiation and termination codons of the 12 protein-coding genes were identified as reported [15]. The 22 tRNA genes were predicted using tRNAscan-SE or manual adjustments [20,21]. The two rRNA genes were predicted by comparison with those of F. hepatica [15], C. sinensis [16] and P. cervi [17]. Amino acid sequences of 12 protein-coding genes were inferred using ExPASy Translate tool (http://web.expasy.org/translate/) using the echinoderm and flatworm mitochondrial codes, and aligned using MEGA 5.0 with default settings [22].
Nucleotide variation analysis
The nucleotide variation between F. elongates and P. cervi was analysed by sliding window analysis as reported [17].
Phylogenetic analysis
Amino acid sequences translated from individual genes of the mt genome of F. elongates were aligned with those predicted from mt genomes of selected trematodes, including C. sinensis (NC_012147) [16], Dicrocoelium dendriticum (NC_025280.1) [23], F. hepatica (NC_002546) [15], Haplorchis taichui (NC_022433.1) [24], Metagonimus yokogawai (KC330755.1), Opisthorchis viverrini (JF739555.1) [25], P. cervi (NC_023095.1) [17], Schistosoma haematobium (NC_008074) [26], Schistosoma japonicum (AF215860) [15], Schistosoma mekongi (NC_002529) [27], Schistosoma spindale (NC_008067) [26], and the cestode Taenia solium (outgroup) (NC_004022.1) [28]. The amino acid sequences of selected trematodes were aligned using MEGA 5.0 [22], and phylogenetic analysis of the aligned amino acid sequences was conducted in MEGA 5.0 using the Maximum Likelihood (ML) method.
Results and discussion
Features of the mt genome of F. elongates
The complete mitochondrial genome of F. elongates (GenBank accession no. KM_397348) is 14,120 bp in length. The length of the F. elongates mt genome is larger than the mtDNA genomes of C. sinensis (13,875 bp) and S. japonicum (14,085 bp), but smaller than D. dendriticum (14,884 bp), F. hepatica (14,462 bp), H. taichui (15,130 bp), M. yokogawai (15,258 bp), S. haematobium (15,003 bp), S. mekongi (14,072 bp) and S. spindale (16,901 bp).
The circular mt genome of F. elongates includes 12 protein-coding genes (cox1-3, nad1-6, nad4L, cytb and atp6), 22 tRNA genes, two rRNA genes (rrnS and rrnL) and two non-coding regions (SNR and LNR). All the 12 protein-coding genes are transcribed in the same direction (Fig. 1), which is the same as in F. hepatica [15], C. sinensis [16] and P. cervi [17]. The gene arrangement order is as follow: cox3-cytb-nad4L-nad4-atp6-nad2-nad1-nad3-cox1-rrnL-rrnS-cox2-nad6-nad5, which is consistent with F. hepatica, O. viverrini, P. cervi, S. japonicum and S. mekongi, except for S. haematobium and S. spindale [26].
Fig. 1.
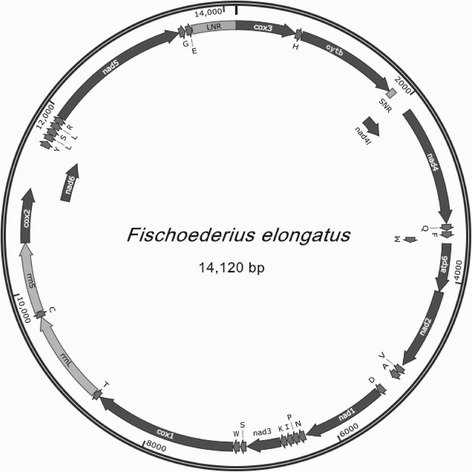
Arrangement of the mitochondrial genome of Fischoederius elongatus
Overlapping nucleotides between mt genes of F. elongates ranged from 1 to 53 bp (Table 2). The F. elongates mt genome has 26 intergenic spacers ranging from 1 bp to 148 bp in length (Table 2). The nucleotide contents of A, C, T and G in the mt genome are 19.78 %, 9.62 %, 44.10 % and 26.50 %, respectively (Table 3), with T being the most favored nucleotide, followed by G, A and C, which is also the same as the mt genomes of F. hepatica [15], C. sinensis [16] and P. cervi [17]. The A + T content of 12 protein coding genes and 22 rRNA genes of F. elongates ranged from 59.65 % (rrnS) to 66.97 % (cox3), and the overall A + T content of the mt genome is 63.88 %.
Table 2.
The organization of the mitochondrial genome of Fischoederius elongatus
| Gene/region | Positions | Size (bp) | Number of aa1 | Ini/Ter codons2 | Anticodons | In3 |
|---|---|---|---|---|---|---|
| cox3 | 1-645 | 645 | 215 | ATG/TAG | 0 | |
| trnH | 648-715 | 68 | GTG | +2 | ||
| cytb | 717-1829 | 1113 | 371 | ATG/TAA | +1 | |
| SNR | 1830-1892 | 63 | 0 | |||
| nad4L | 1893-2156 | 264 | 88 | ATG/TAG | 0 | |
| nad4 | 2117-3397 | 1281 | 427 | GTG/TAA | −38 | |
| trnQ | 3409-3471 | 63 | TTG | +11 | ||
| trnF | 3486-3549 | 65 | GAA | +14 | ||
| trnM | 3549-3612 | 64 | CAT | −1 | ||
| atp6 | 3613-4128 | 516 | 172 | ATG/TAG | 0 | |
| nad2 | 4133-5008 | 876 | 292 | GTG/TAG | +4 | |
| trnV | 5039-5102 | 64 | TAC | +30 | ||
| trnA | 5109-5179 | 71 | TGC | +6 | ||
| trnD | 5328-5397 | 70 | GTC | +148 | ||
| nad1 | 5400-6296 | 897 | 299 | ATG/TAG | +2 | |
| trnN | 6314-6379 | 66 | GTT | +17 | ||
| trnP | 6384-6447 | 64 | TGG | +4 | ||
| trnI | 6449-6511 | 63 | GAT | +1 | ||
| trnK | 6518-6582 | 65 | CTT | +6 | ||
| nad3 | 6587-6943 | 357 | 119 | ATG/TAG | +4 | |
| trnS1 | 6955-7014 | 60 | GCT | +11 | ||
| trnW | 7027-7091 | 65 | TCA | +12 | ||
| cox1 | 7095-8636 | 1542 | 514 | GTG/TAA | +3 | |
| trnT | 8646-8709 | 64 | TGT | +9 | ||
| rrnL4 | 8710-9704 | 995 | 0 | |||
| trnC | 9707-9767 | 61 | GCA | +2 | ||
| rrnS4 | 9768-10518 | 751 | 0 | |||
| cox2 | 10519-11100 | 582 | 194 | ATG/TAG | 0 | |
| nad6 | 11046-11546 | 501 | 167 | ATG/TAG | −53 | |
| trnY | 11568-11632 | 65 | GTA | +21 | ||
| trnL1 | 11652-11715 | 64 | TAG | +19 | ||
| trnS2 | 11717-11785 | 69 | TGA | +1 | ||
| trnL2 | 11792-11856 | 65 | TAA | +6 | ||
| trnR | 11860-11925 | 66 | TCG | +3 | ||
| nad5 | 11926-13506 | 1581 | 527 | GTG/TAG | 0 | |
| trnG | 13510-13574 | 65 | TCC | +3 | ||
| trnE | 13587-13651 | 65 | TTC | +12 | ||
| LNR | 13652-14120 | 469 | 0 |
The inferred length of amino acid sequence of 12 protein-coding genes: 1amino acid; 2initiation and termination codons; 3intergenic nucleotides; 4initiation or termination positions of ribosomal RNAs defined by adjacent gene boundaries
Table 3.
Nucleotide contents of genes and the non-coding region within the mitochondrial genome of Fischoederius elongatus
| Gene | A(%) | C(%) | G(%) | T(%) | A + T(%) |
|---|---|---|---|---|---|
| cox3 | 18.29 | 8.53 | 24.50 | 48.68 | 66.97 |
| cytb | 18.96 | 8.89 | 26.33 | 45.82 | 64.78 |
| SNR | 20.63 | 4.76 | 31.75 | 42.86 | 63.49 |
| nad4L | 21.97 | 8.33 | 25.38 | 44.32 | 66.29 |
| nad4 | 16.55 | 9.52 | 25.45 | 48.48 | 65.03 |
| atp6 | 17.64 | 10.08 | 24.42 | 47.87 | 65.50 |
| nad2 | 15.64 | 7.99 | 25.11 | 51.26 | 66.89 |
| nad1 | 16.39 | 7.47 | 28.21 | 47.94 | 64.33 |
| nad3 | 15.97 | 7.84 | 28.01 | 48.18 | 64.15 |
| cox1 | 18.87 | 11.02 | 24.51 | 45.59 | 64.46 |
| rrnL | 25.83 | 10.35 | 26.73 | 37.09 | 62.91 |
| rrnS | 24.37 | 12.25 | 28.10 | 35.29 | 59.65 |
| cox2 | 19.93 | 11.11 | 27.49 | 41.58 | 61.51 |
| nad6 | 17.44 | 8.61 | 26.71 | 47.24 | 64.68 |
| nad5 | 16.32 | 8.29 | 28.78 | 46.62 | 62.93 |
| LNR | 26.01 | 9.17 | 26.44 | 38.38 | 64.39 |
The present F. elongates mt genome can provide useful information for the studies of epidemiology, species identification and genetic diversity of Fischoederius spp. At the same, it will also make contribution to the taxonomy study of Fischoederius spp. With the full mt genome of F. elongates, we can undertake a study within F. elongates from different regions or among Fischoederius spp. by combining the morphological features with genetic analyses (with molecular markers from mitochondria or ribosome, such as cox1, nad4, 18S, ITS-1 and ITS-2). Meanwhile, the mt genome of F. elongates may also provide information for the prevention and diagnosis of Fischoederius spp. and perhaps, this mt genome information may assist in the new drug, since mitochondria is the target of some drugs, such as decoquinate.
Protein-coding genes
The F. elongates mt genome has 12 protein-coding genes, including cox3, cytb, nad4L, nad4, atp6, nad2, nad1, nad3, cox1, cox2, nad6 and nad5. For these protein coding genes, ATG (eight of 12 protein genes) is the most common initiation codon, followed by GTG (four of 12 protein genes) (Table 2), which is the same as other trematodes, such as F. hepatica [15], C. sinensis [16], P. cervi [17], S. mekongi [27]. TAG (seven of 12 protein genes) or TAA (five of 12 protein genes) are the termination codons, this is in agreement with other digeneans, except for P. cervi (Only TAG was used as termination codons). Excluding the termination codons, 10,107 nucleotides encode 3,369 amino acids of protein-coding genes in the F. elongates mt genome. The most frequently used amino acid is TTT (Phe), with the frequency of 9.65 %, followed by TTT (Phe), TTG (Leu: 8.61 %), GTT (Val: 5.25 %) and TAT (Tyr: 5.02 %) (Table 4). The least used codons are AAC (Asn: 0.06 %), GAC (Asp: 0.06 %) and CGC (Arg: 0).
Table 4.
Codon usage for 12 protein-coding genes in the mitochondrial genome of Fischoederius elongatus
| Amino acid | Codon | Number | Frequency(%) | Amino acid | Codon | Number | Frequency(%) |
|---|---|---|---|---|---|---|---|
| Phe | TTT | 325 | 9.65 | Ile | ATT | 127 | 3.77 |
| Phe | TTC | 28 | 0.83 | Ile | ATC | 6 | 0.18 |
| Leu | TTA | 167 | 4.96 | Ile | ATA | 71 | 2.11 |
| Leu | TTG | 290 | 8.61 | Met | ATG | 105 | 3.12 |
| Ser | TCT | 118 | 3.50 | Met | GTG | 165 | 4.90 |
| Ser | TCC | 6 | 0.18 | Thr | ACT | 54 | 1.60 |
| Ser | TCA | 22 | 0.65 | Thr | ACC | 3 | 0.09 |
| Ser | TCG | 25 | 0.74 | Thr | ACA | 19 | 0.56 |
| Tyr | TAT | 169 | 5.02 | Thr | ACG | 16 | 0.47 |
| Tyr | TAC | 11 | 0.33 | Asn | AAT | 54 | 1.60 |
| Stop | TAA | 3 | 0.09 | Asn | AAC | 2 | 0.06 |
| Stop | TAG | 9 | 0.27 | Asn | AAA | 23 | 0.68 |
| Cys | TGT | 112 | 3.32 | Lys | AAG | 50 | 1.48 |
| Cys | TGC | 9 | 0.27 | Ser | AGT | 92 | 2.73 |
| Trp | TGA | 41 | 1.22 | Ser | AGC | 9 | 0.27 |
| Trp | TGG | 72 | 2.14 | Ser | AGA | 31 | 0.92 |
| Leu | CTT | 43 | 1.28 | Ser | AGG | 35 | 1.04 |
| Leu | CTC | 3 | 0.09 | Val | GTT | 177 | 5.25 |
| Leu | CTA | 17 | 0.50 | Val | GTC | 12 | 0.36 |
| Leu | CTG | 23 | 0.68 | Val | GTA | 58 | 1.72 |
| Pro | CCT | 53 | 1.57 | Ala | GCT | 95 | 2.82 |
| Pro | CCC | 4 | 0.12 | Ala | GCC | 4 | 0.12 |
| Pro | CCA | 11 | 0.33 | Ala | GCA | 13 | 0.39 |
| Pro | CCG | 15 | 0.45 | Ala | GCG | 33 | 0.98 |
| His | CAT | 41 | 1.22 | Asp | GAT | 62 | 1.84 |
| His | CAC | 7 | 0.21 | Asp | GAC | 2 | 0.06 |
| Gln | CAA | 13 | 0.39 | Glu | GAA | 17 | 0.50 |
| Gln | CAG | 14 | 0.42 | Glu | GAG | 67 | 1.99 |
| Arg | CGT | 45 | 1.34 | Gly | GGT | 165 | 4.90 |
| Arg | CGC | 0 | 0 | Gly | GGC | 16 | 0.47 |
| Arg | CGA | 6 | 0.18 | Gly | GGA | 22 | 0.65 |
| Arg | CGG | 11 | 0.33 | Gly | GGG | 51 | 1.51 |
Transfer RNA and ribosomal RNA genes
The F. elongates mt genome encodes 22 tRNAs, and the length of 22 tRNA genes ranged from 60 bp to 71 bp (Table 2). There are two non-coding regions in F. elongates mt genome, rrnS (751 bp) and rrnL (995 bp) (Table 2). The location of rrnS is between tRNA-Cys and cox2 and the rrnL is between tRNA-Thr and tRNA-Cys, which is the same as other trematodes, such as F. hepatica [15], C. sinensis [16] and P. cervi [17].
Non-coding regions
Many flatworms have non-coding regions, it’s common to find two non-coding regions in trematodes: one long non-coding region (LNR) and one short non-coding region (SNR). In F. elongates, there is a short non-coding region (SNR: 62 nucleotides), which is located between cytb and nad4L. In addition, there is also a long non-coding region (LNR: 468 nucleotides) between tRNA-Phe and cox3 (Table 2), the LNR has two obvious features, one is microsatellite-like sequences, such as (TA)n (n <5); the other is homopolymer sequences, such as (T)n (n <7). People still don’t understand clearly why the non-coding regions exist, and the function of them, people just knew the non-coding regions may participate in the replication of mitochondria [26].
Nucleotide variability between F. elongates and P. cervi
A sliding window analysis of F. elongates and P. cervi using full mt genome sequences reflected the nucleotide diversity (π) for all the protein-coding genes (Fig. 2). The highest and lowest level of nucleotide variability was within nad6 and cox3, respectively. In our study, nad6 and cox2 are the most conserved genes, and cox3 and atp6 are the least conserved. With sliding window analysis, we could know the conserved regions of mt genome among species.
Fig. 2.
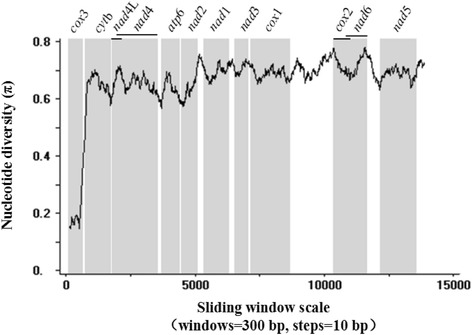
A sliding window analysis of complete mt genome sequences of Fischoederius elongatus and Paramphistomum cervi. The black line showed nucleotide diversity in a window of 300 bp (10 bp steps). Nad4L and nad4, cox2 and nad6 are overlapping genes. Gene regions are marked in grey boxes and boundaries are indicated
Genetic relationships
Concatenated amino acid sequence data representing 12 protein-coding genes of 11 digenean species (C. sinensis, D. dendriticum, F. hepatica, H. taichui, M. yokogawai, O. viverrini, P. cervi, S. haematobium, S. japonicum, S. mekongi and S. spindale) and one tapeworm (T. solium) were used for genetic relationship analysis (Fig. 3). In the tree, we can find two large clades with strong support (100 %): one clade consists of eight members representing five families (Heterophyidae, Opisthorchiidae, Fasciolidae, Paramphistomidae and Dicrocoeliidae); the other clade is Schistosomatidae. In the present analysis, F. elongates has the closest genetic relationship with P. cervi (100 %), followed by Fasciolidae, this is consistent with their relationship in the classification of biology. At the same time, we also used NJ method analysis (not shown), and there was no difference between these two methods.
Fig. 3.
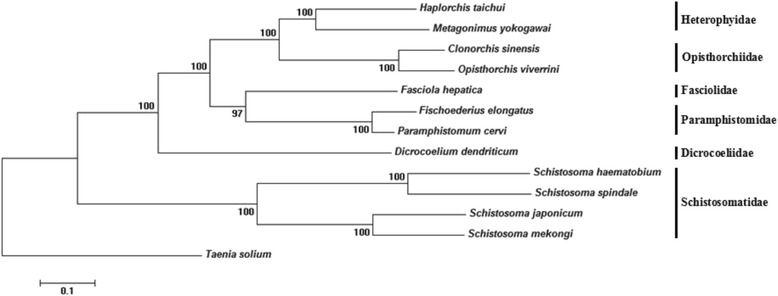
The phylogenetic relationships of Fischoederius elongatus and other trematodes based on concatenated amino acid sequence data representing 12 protein-coding genes by Maximum Likelihood analysis, using Taenia solium as an outgroup
Acknowledgements
Sincere thanks to Professor Bang Shen for comments on the manuscript. This work was supported in part by the “National Key Basic Research Program (973 Program) of China” (Grant No. 2015CB150300), the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201303037) and “Huazhong Agricultural University Students Research Fund” (Grant No. 2015054).
Footnotes
Xin Yang and Yunyang Zhao contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RF conceived and designed the study. XY and YYZ wrote the manuscript with input from other coauthors. XY, YYZ and LXW performed the experiments, HLF, LT, WQL and KXZ analyzed the data. MH assisted in study design and editing. All authors read and approved the final manuscript.
Contributor Information
Xin Yang, Email: 695237569@qq.com.
Yunyang Zhao, Email: 373715956@qq.com.
Lixia Wang, Email: 448472171@qq.com.
Hanli Feng, Email: 610579870@qq.com.
Li Tan, Email: 396322874@qq.com.
Weiqiang Lei, Email: 1017475188@qq.com.
Pengfei Zhao, Email: 2314776455@qq.com.
Min Hu, Email: mhu@mail.hzau.edu.cn.
Rui Fang, Email: fangrui19810705@163.com.
References
- 1.Horak IG. Paramphistomiasis of domestic ruminants. Adv Parasitol. 1971;9:33–72. doi: 10.1016/S0065-308X(08)60159-1. [DOI] [PubMed] [Google Scholar]
- 2.Li XR. Color atlas of animal parasitosis (Second Edition) Beijing: China Agriculture Press; 2011. [Google Scholar]
- 3.Nzalawahe J, Kassuku AA, Stothard JR, Coles GC, Eisler MC. Trematode infections in cattle in Arumeru District, Tanzania are associated with irrigation. Parasit Vectors. 2014;7:107. doi: 10.1186/1756-3305-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaguti S. A synoptical review of life histories of digenetic trematodes of vertebrates. Tokyo: Keigaku Publishing Co.; 1975. [Google Scholar]
- 5.Li D. A case of Fischoederius elongatus infection in China. Annual Bull Soc Parasitol Guangdong Province. 1991;12(11–13):155–6. [Google Scholar]
- 6.Ramesh A, Small ST, Kloos ZA, Kazura JW, Nutman TB, Serre D, et al. The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol Biochem Parasitol. 2012;183(1):32–41. doi: 10.1016/j.molbiopara.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gasser RB, Jabbar A, Mohandas N, Hoglund J, Hall RS, Littlewood DT, et al. Assessment of the genetic relationship between Dictyocaulus species from Bos taurus and Cervus elaphus using complete mitochondrial genomic datasets. Parasit Vectors. 2012;5:241. doi: 10.1186/1756-3305-5-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi KS, Koekemoer LL, Coetzee M. Population genetic structure of the major malaria vector Anopheles funestus s.s. and allied species in southern Africa. Parasit Vectors. 2012;5:283. doi: 10.1186/1756-3305-5-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jane EH, Bernard F. Echinostoma and Echinostomiasis. Adv Parasitol. 1990;29:215–69. doi: 10.1016/S0065-308X(08)60107-4. [DOI] [PubMed] [Google Scholar]
- 10.Ghatani S, Shylla JA, Roy B, Tandon V. Multilocus sequence evaluation for differentiating species of the trematode Family Gastrothylacidae, with a note on the utility of mitochondrial COI motifs in species identification. Gene. 2014;548(2):277–84. doi: 10.1016/j.gene.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 11.Wang GL. Laboratory diagnostic techniques of parasites. Feeding Livestock. 2013;3:39–43. [Google Scholar]
- 12.Liu GH, Wang Y, Song HQ, Li MW, Ai L, Yu XL, et al. Characterization of the complete mitochondrial genome of Spirocerca lupi: sequence, gene organization and phylogenetic implications. Parasit Vectors. 2013;6:45. doi: 10.1186/1756-3305-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, et al. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc. 2006;1(6):3121–8. doi: 10.1038/nprot.2006.485. [DOI] [PubMed] [Google Scholar]
- 14.Itagaki T, Tsumagari N, Tsutsumi K, Chinone S. Discrimination of three amphistome species by PCR-RFLP based on rDNA ITS2 markers. J Vet Med Sci. 2003;65(8):931–3. doi: 10.1292/jvms.65.931. [DOI] [PubMed] [Google Scholar]
- 15.Le TH, Blair D, McManus DP. Complete DNA sequence and gene organization of the mitochondrial genome of the liverfluke, Fasciola hepatica L. (Platyhelminthes; Trematoda) Parasitology. 2001;123(Pt 6):609–21. doi: 10.1017/s0031182001008733. [DOI] [PubMed] [Google Scholar]
- 16.Shekhovtsov SV, Katokhin AV, Kolchanov NA, Mordvinov VA. The complete mitochondrial genomes of the liver flukes Opisthorchis felineus and Clonorchis sinensis (Trematoda) Parasitol Int. 2010;59(1):100–3. doi: 10.1016/j.parint.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Yan HB, Wang XY, Lou ZZ, Li L, Blair D, Yin H, et al. The mitochondrial genome of Paramphistomum cervi (Digenea), the first representative for the family Paramphistomidae. PLoS One. 2013;8(8):e71300. doi: 10.1371/journal.pone.0071300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007;2(10):2339–44. doi: 10.1038/nprot.2007.358. [DOI] [PubMed] [Google Scholar]
- 19.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea) Int J Parasitol. 2002;32(2):145–58. doi: 10.1016/S0020-7519(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 21.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu GH, Yan HB, Otranto D, Wang XY, Zhao GH, Jia WZ, et al. Dicrocoelium chinensis and Dicrocoelium dendriticum (Trematoda: Digenea) are distinct lancet fluke species based on mitochondrial and nuclear ribosomal DNA sequences. Mol Phylogenet Evol. 2014;79:325–31. doi: 10.1016/j.ympev.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Lee D, Choe S, Park H, Jeon HK, Chai JY, Sohn WM, et al. Complete mitochondrial genome of Haplorchis taichui and comparative analysis with other trematodes. Korean J Parasitol. 2013;51(6):719–26. doi: 10.3347/kjp.2013.51.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai XQ, Liu GH, Song HQ, Wu CY, Zou FC, Yan HK, et al. Sequences and gene organization of the mitochondrial genomes of the liver flukes Opisthorchis viverrini and Clonorchis sinensis (Trematoda) Parasitol Res. 2012;110(1):235–43. doi: 10.1007/s00436-011-2477-2. [DOI] [PubMed] [Google Scholar]
- 26.Littlewood DT, Lockyer AE, Webster BL, Johnston DA, Le TH. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol Phylogenet Evol. 2006;39(2):452–67. doi: 10.1016/j.ympev.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Le TH, Blair D, Agatsuma T, Humair PF, Campbell NJ, Iwagami M, et al. Phylogenies inferred from mitochondrial gene orders-a cautionary tale from the parasitic flatworms. Mol Biol Evol. 2000;17(7):1123–5. doi: 10.1093/oxfordjournals.molbev.a026393. [DOI] [PubMed] [Google Scholar]
- 28.Nakao M, Sako Y, Ito A. The mitochondrial genome of the tapeworm Taenia solium: a finding of the abbreviated stop codon U. J Parasitol. 2003;89(3):633–5. doi: 10.1645/0022-3395(2003)089[0633:TMGOTT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]