

Abstract
Leukocyte chemotactic factor-2 (LECT2) amyloidosis has been described as being associated with kidney disease; however, no clinical manifestations outside of the kidney have been previously reported. We describe a patient presenting with pulmonary-renal syndrome found to have deposition of amyloidogenic LECT2 (ALECT2) within both the lung and the kidney. This case is unique in regard to both the patient's clinical presentation of pulmonary-renal syndrome in the setting of amyloidosis and the biopsy finding of ALECT2 deposition within the lung. It also emphasizes the importance of tissue diagnosis in such cases, given that amyloidosis was not initially considered in the differential diagnosis.
Keywords: amyloid, lect2, pulmonary-renal syndrome
Background
Amyloidosis represents an uncommon group of disorders which results from abnormal deposition of protein fibrils arranged in cross-β sheet quaternary structures [1]. An expanding list of native proteins has been implicated, with amyloidogenic leukocyte chemotactic factor 2 (ALECT2) being a recent addition [2]. Deposition of ALECT2 within the kidney is a known cause of kidney dysfunction; however, extra-renal manifestations and pulmonary ALECT2 deposition have not been reported. Herein, we report a patient presenting with pulmonary-renal syndrome found to have ALECT2 deposition within both the lung and the kidney.
Clinical report
An 84-year-old Mexican-American man with diabetes mellitus, hypertension, hyperlipidemia and prostatic adenocarcinoma, status-post prostatectomy presented with shortness of breath and fatigue, which had progressed over 2 weeks. Three days prior to presentation, he experienced an acute worsening of his respiratory status, prompting him to seek emergency care. The patient did not report any neurologic symptoms, rash, recent fever or infection. At presentation, he was hypoxic with oxygen saturations of 80% on room air. An arterial blood gas showed a pH 7.43 (reference range 7.35–7.45), partial carbon dioxide of 28 mm Hg (reference range 35–45 mm Hg) and partial oxygen of 61 mm Hg (reference range 80–100 mm Hg) on inspired oxygen of 60%. Physical examination revealed moderate respiratory distress and edema of the lower extremities. A computerized tomography scan of the chest showed bilateral opacities in a pattern that was felt to possibly represent diffuse alveolar hemorrhage (Figure 1). Fig.
1.

CT scan of chest demonstrates patchy and confluent, groundglass and consolidated opacities in both lungs, in a pattern consistent with diffuse alveolar hemorrhage.
Initial chemistries were significant for a carbon dioxide of 17 mmol/L (17 meq/L), blood urea nitrogen of 26.4 mmol/L (74 mg/dL) and a serum creatinine of 406.6 μmol/L (4.6 mg/dL) with an estimated glomerular filtration rate (eGFR) of 12 mL/min/1.73 m2 calculated by the four-variable MDRD (Modification of Diet in Renal Disease) study equation. A complete blood count showed a white blood cell count of 10.4 × 109/L (10.4 × 103/μL), hemoglobin of 52 g/L (5.2 g/dL) and a normal platelet count of 335 × 109 /L (335 × 103 /μL). Urinalysis revealed a specific gravity of 1.011, pH 5.0, 3+ protein, 1+ blood and negative leukocyte esterase and nitrites. Spot urine protein-to-creatinine ratio showed sub-nephrotic proteinuria with a ratio of 1.5. Serologic studies revealed the presence of perinuclear antineutrophil cytoplasmic antibody (P-ANCA) of 1:160 (reference range <1:20). Immunoglobulin G (IgG) myeloperoxidase antibody (MPO) was 385 AU/mL (normal: <19 AU/mL). Serum and urine protein electrophoresis demonstrated a monoclonal IgM kappa band. Given the patient's markedly elevated anti-MPO titer, a thorough review of his medications was performed. No medications associated with ANCA production were identified.
Hemodialysis was initiated due to acute kidney failure and volume overload, and the patient was transferred to the intensive care unit where he required non-invasive positive pressure ventilation and developed a transient episode of hemoptysis. Shortly after the initiation of hemodialysis, the patient was weaned from positive airway pressure ventilation. Pulse intravenous methylprednisolone was initiated and plasmapheresis was started as empiric therapy for a presumed diagnosis of microscopic polyangiitis with diffuse alveolar hemorrhage. Kidney and lung biopsies were performed for tissue diagnosis. A bone marrow biopsy was also obtained which showed no plasma cell dyscrasia.
The kidney biopsy showed renal cortex containing up to eight glomeruli per level, of which two were globally sclerosed. The non-sclerotic glomeruli showed segmental mesangial expansion by extracellular amorphous material which did not stain with Jones methenamine silver stain, stained weakly on periodic acid Schiff stain, and demonstrated a gray hue on the trichrome stain. Glomerular capillaries were patent without hypercellularity and no fibrinoid necrosis or crescents were identified. The interstitium and walls of arteries and arterioles demonstrated diffuse infiltration by the same amorphous material. There were no lesions of vasculitis. A Congo red stain was performed and showed strong staining of the mesangial, interstitial and vascular deposits (Figure 2A). Apple-green birefringence was demonstrated upon polarization of the Congo red stain, confirming the presence of amyloid (Figure 2B). Fig.
2.
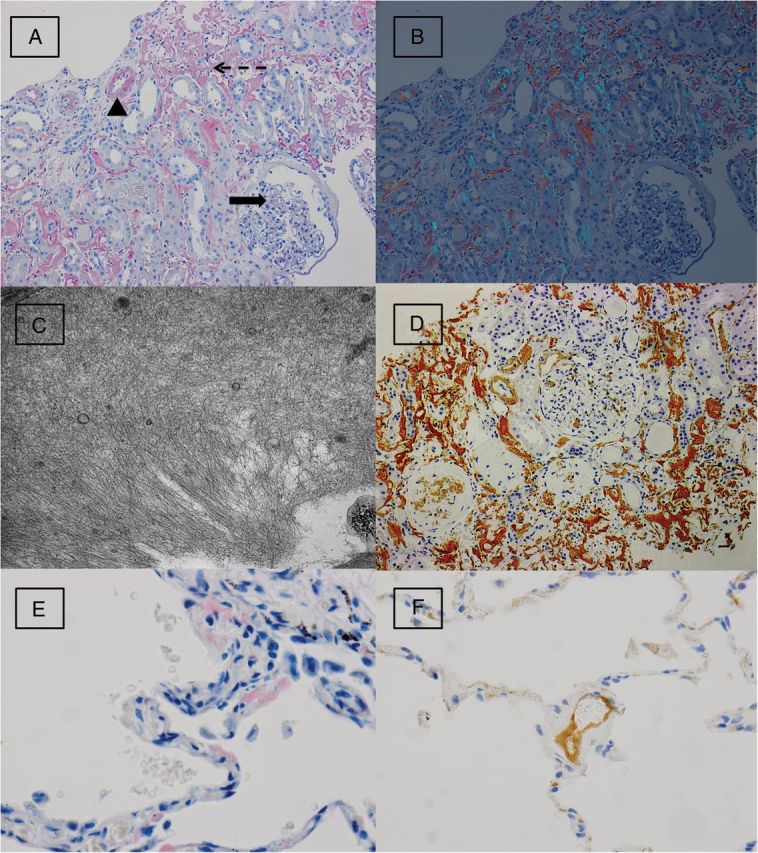
(A) A Congo red stain performed on kidney biopsy shows deposition of Congophilic material within the glomeruli (solid arrow), arterial wall (arrow head) and interstitium (dashed arrow) (Congo red stain, original magnification: ×200). (B) Polarization of the Congo red stain demonstrates apple-green birefringence, consistent with amyloid (original magnification: ×200). (C) Electron microscopy on the kidney biopsy shows haphazard deposition of fibrils measuring 6–10 nm in thickness, consistent with amyloid (original magnification: ×20 000). (D) Staining for LECT2 by immunohistochemistry shows strong and diffuse staining within glomerular, interstitial and vascular deposits within the kidney biopsy (original magnification: ×200). (E) Wedge biopsies of the lung show multifocal deposition of Congophilic material within alveolar septa (Congo red stain, original magnification: ×400). (F) Amyloid deposits within a small vessel of the lung show positive staining for LECT2 by immunohistochemistry (original magnification: ×400).
Immunofluorescence microscopy was performed using antisera to human immunoglobulin G (IgG), IgA, IgM, C1q, C3, albumin, fibrinogen and kappa/lambda light chains (DAKO Corporation - Carpinteria, CA) and graded on a scale of 0–4+. Weak and smudgy staining (1–2+) was seen in the interstitium and vessel walls for IgG, IgA, IgM and both kappa and lambda light chains. There was no significant staining for C3, C1q, fibrinogen or albumin or evidence of immunoglobulin restriction. Ultrastructural examination revealed extensive haphazard deposition of fibrils ranging in thickness from 6–10 nm within the interstitium and focally within mesangial areas (Figure 2C). The size and distribution of the fibrils were consistent with amyloid. An immunohistochemical workup was initiated to better classify the amyloid subtype, which included stains for kappa and lambda light chains, amyloid A and prealbumin (transthyretin), all of which were negative (DAKO Corporation). Further immunohistochemical evaluation with anti-LECT2 antibodies (performed at Nephropath Laboratories—Little Rock, AR) demonstrated diffuse positive staining within the amyloid deposits (Figure 2D).
Due to uncertainty in the lung pathology, wedge biopsies of the upper, middle and lower lobes of the right lung were performed. Biopsies showed multifocal, small deposits of the same extracellular amorphous material within the alveolar septa and walls of small and large vessels. These deposits were strongly positive on the Congo red stain (Figure 2E) and demonstrated apple-green birefringence upon polarization, consistent with amyloid. There were no lesions of vasculitis. Immunohistochemical staining with anti-LECT2 antibodies was positive within the amyloid deposits (Sigma Life Sciences—St. Louis, MO) (Figure 2F).
In summary, kidney and lung biopsies both demonstrated deposition of amyloid showing positive immunohistochemical staining for LECT2. No lesions of vasculitis were identified in any of the biopsy specimens and crescents were not seen in the kidney biopsy.
The patient was discharged 36 days after his initial presentation with a creatinine of 203.3 μmol/L (2.3 mg/dL, eGFR: 27 mL/min/1.73 m2). Steroids were gradually tapered off at 6 months, and the patient is currently maintained on azathioprine. Eight months after his initial presentation, his creatinine clearance improved to 20–25 mL/min and he no longer required hemodialysis.
Discussion
Leukocyte chemotactic factor 2 (LECT2) is a recently described amyloidogenic protein and potential cause of kidney disease. Originally characterized in 1998, human LECT2 is predominantly synthesized by hepatocytes. Along with neutrophil chemotactic properties, it is also believed that LECT2 plays a role in tissue growth and repair following damage. The latter function has been proposed given the similarities between human LECT2 and its bovine counterpart: chondromodulin-II [3–5]. LECT2 amyloidosis is now a well-described cause of kidney injury; however, extra-renal manifestations have not been reported [2]. Since 1971, when monoclonal light chain became the first chemically characterized amyloid protein, a growing list of amyloidogenic proteins has been amassed, including, but not limited to, monoclonal light chain (AL), amyloid A protein (AA), transthyretin (ATTR), apolipoproteins AI (AApoAI) and AII (AApoAII), lysozyme (ALys), gelsolin (AGel) and β-2 microglobulin [6, 7]. The mechanism of amyloidogenesis is quite variable, including hereditary mutation, protein overproduction and decreased protein excretion. In the case of ALECT2, the exact mechanism of disease has not been fully elucidated; however, a genetic component is suspected [1]. The LECT2 gene has been mapped to chromosome 5q31.1–32 and contains four exons and three introns. The four exons code for a 151 amino acid protein, which is 133 amino acids in its secreted form [3]. In one published series, four of four patients with LECT2 amyloidosis who underwent LECT2 gene analysis were identified as homozygous for the G allele, substituting valine at position 40 of the mature protein for isoleucine, which is found in the A allele [4]. This single amino acid switch may result in a propensity to form amyloid fibrils [4]. The vast majority of reported cases have occurred in patients of Mexican descent, also supporting a genetic role [8].
Published reports and case series of clinical disease due to LECT2 amyloidosis have thus far been limited to deposition within the kidney. One report has demonstrated incidentally discovered ALECT2 within hepatic, splenic, colonic and adrenal tissue [9]. Murphy et al. described a series of 10 patients with kidney-limited LECT2 amyloidosis, representing 2.5% of amyloid cases diagnosed by kidney biopsy over an 8.5-year period. Clinically, patients presented with varying degrees of kidney insufficiency, and the majority presented with only minimal proteinuria in contrast to AL and AA, which often present with nephrotic-range proteinuria. Histologically, ALECT2 deposition was predominantly localized within the interstitium and mesangial regions, although all compartments of the kidney parenchyma may be involved [4,10]. Other published case reports have shown similar clinical and histologic features of ALECT2 amyloidosis [10,11].
Like the kidney, the lung may be involved in systemic amyloidosis with the most frequent subtypes being AL and AA amyloid. ALECT2 has not been previously reported in the lung. Deposition of amyloid within the pulmonary parenchyma is the most common manifestation of amyloidosis within the respiratory system and may occur in a nodular or diffuse pattern [12]. In nodular pulmonary amyloidosis, deposition is typically localized and is often an incidental radiographic finding in the lung periphery [13]. Diffuse pulmonary involvement is most often seen in the setting of systemic AL amyloidosis, and although severe clinical manifestations are not common, hemoptysis and fatal pulmonary hemorrhage have been reported [14].
The case we present here is unique both in its clinical presentation and in the biopsy findings. The positive ANCA serology and the clinical picture of pulmonary-renal syndrome were highly suspicious for a systemic small vessel vasculitis. Reports of systemic amyloidosis presenting with pulmonary-renal syndrome are scant in the literature. A case of systemic AA amyloidosis in a patient with dystrophic epidermolysis bullosa resulting in recurrent pulmonary infections and nephrotic syndrome has been reported; however, hemoptysis was not present and creatinine clearance was normal [15]. Histopathologically, this case is unique both in the extent and distribution of the amyloid deposition and the amyloid protein itself. To our knowledge, this is the first description of LECT2 amyloidosis causing clinical disease outside of the kidney and the first description of LECT2 deposition in the lung.
Conflict of interest statement
None declared.
Acknowledgement
No support or funding was provided for the preparation of this manuscript.
References
- 1.Leung N, Nasr SH, Sethi S. How I treat amyloidosis: the importance of accurate diagnosis and amyloid typing. Blood. 2012;120:3206–3213. doi: 10.1182/blood-2012-03-413682. doi:10.1182/blood-2012-03-413682. [DOI] [PubMed] [Google Scholar]
- 2.Benson MD, James S, Scott K, et al. Leukocyte chemotactic factor 2: A novel renal amyloid protein. Kidney Int. 2008;74:218–222. doi: 10.1038/ki.2008.152. doi:10.1038/ki.2008.152. [DOI] [PubMed] [Google Scholar]
- 3.Yamagoe S, Kameoka Y, Hashimoto K, et al. Molecular cloning, structural characterization, and chromosomal mapping of the human LECT2 gene. Genomics. 1998;48:324–329. doi: 10.1006/geno.1997.5198. doi:10.1006/geno.1997.5198. [DOI] [PubMed] [Google Scholar]
- 4.Murphy CL, Wang S, Kestler D, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis. 2010;56:1100–1107. doi: 10.1053/j.ajkd.2010.08.013. doi:10.1053/j.ajkd.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagai H, Hamada T, Uchida T, et al. Systemic expression of a newly recognized protein, LECT2, in the human body. Pathol Int. 1998;48:882–886. doi: 10.1111/j.1440-1827.1998.tb03855.x. doi:10.1111/j.1440-1827.1998.tb03855.x. [DOI] [PubMed] [Google Scholar]
- 6.Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150–1151. doi: 10.1126/science.172.3988.1150. doi:10.1126/science.172.3988.1150. [DOI] [PubMed] [Google Scholar]
- 7.von Hutten H, Mihatsch M, Lobeck H, et al. Prevalence and origin of amyloid in kidney biopsies. Am J Surg Pathol. 2009;33:1198–1205. doi: 10.1097/PAS.0b013e3181abdfa7. doi:10.1097/PAS.0b013e3181abdfa7. [DOI] [PubMed] [Google Scholar]
- 8.Said SM, Sethi S, Cornell LD, et al. Renal ALECT2 amyloidosis: a clinicopathologic study of 17 patients. Mod Pathol. 2013;26:392A. [Google Scholar]
- 9.Dogan A, Thies J, Vrana J, et al. Clinical and pathological phenotype of leukocyte cell-derived chemotaxin-2 (LECT2) amyloidosis (ALECT2) [abstract OP-058] Amyloid. 2010;17:69–70. [Google Scholar]
- 10.Larsen CP, Walker PD, Weiss DT, et al. Prevalence and morphology of leukocyte chemotactic factor 2-associated amyloid in renal biopsies. Kidney Int. 2010;77:816–819. doi: 10.1038/ki.2010.9. doi:10.1038/ki.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holanda DG, Acharya VK, Dogan A, et al. Atypical presentation of atypical amyloid. Nephrol Dial Transplant. 2011;26:373–376. doi: 10.1093/ndt/gfq638. doi:10.1093/ndt/gfq638. [DOI] [PubMed] [Google Scholar]
- 12.Utz JP, Swensen SJ, Gertz MA. Pulmonary amyloidosis. The Mayo clinic experience from 1980 to 1993 . Ann Intern Med. 1996;124:407–413. doi: 10.7326/0003-4819-124-4-199602150-00004. [DOI] [PubMed] [Google Scholar]
- 13.Lachmann HJ, Hawkins PN. Amyloidosis and the lung chronic respiratory disease. Chronic Respir Dis. 2006;3:203–214. doi: 10.1177/1479972306070066. doi:10.1177/1479972306070066. [DOI] [PubMed] [Google Scholar]
- 14.Sterlacci W, Veits L, Moser P, et al. Idiopathic systemic amyloidosis primarily affecting the lungs with fatal pulmonary haemorrhage due to vascular involvement. Pathol Oncol Res. 2009;15:133–136. doi: 10.1007/s12253-008-9066-4. doi:10.1007/s12253-008-9066-4. [DOI] [PubMed] [Google Scholar]
- 15.Csikos M, Orosz Z, Bottlik G, et al. Dystrophic epidermolysis bullosa complicated by cutaneous squamous cell carcinoma and pulmonary and renal amyloidosis. Clin Exp Dermatol. 2003;28:163–166. doi: 10.1046/j.1365-2230.2003.01185.x. doi:10.1046/j.1365-2230.2003.01185.x. [DOI] [PubMed] [Google Scholar]