

Abstract
Dabigatran is a direct thrombin inhibitor used as an alternative to warfarin for long-term anticoagulation. We describe a patient who developed acute kidney injury (AKI) in the setting of warfarin conversion to dabigatran, and a renal biopsy demonstrating acute tubular injury. Although the patient had undiagnosed IgA nephropathy that may have predisposed him to bleeding, AKI was due to heme-associated tubular injury. We propose that severe hematuria in patients with underlying glomerular pathology treated with either dabigatran or warfarin may lead to toxic tubular injury through the accumulation of heme-proteins.
Keywords: acute kidney injury, acute tubular necrosis, dabigatran
Background
Dabigatran (Pradaxa, Boehringer-Ingelheim) is a direct thrombin inhibitor that is often substituted for warfarin because monitoring of anticoagulation is not required. In the RE-LY study (Randomized Evaluation of Long-Term Anticoagulation Therapy), dabigatran was non-inferior to warfarin in reducing incidence of stroke with atrial fibrillation [1]. Bleeding risk was less than that of warfarin with dabigatran dosed at 110 mg twice daily, but equivalent at 150 mg twice daily. We present a case of acute kidney injury (AKI) in the setting of conversion from warfarin to dabigatran that reveals a pathophysiological mechanism of injury that likely has relevance to warfarin nephropathy.
Case report
A 67-year-old Caucasian male, with previously normal kidney function [serum creatinine concentration (sCr) 1.0 mg/dL (88 μmol/L), eGFR 78 mL/min/1.73 m2 using the CKD-EPI equation, and no proteinuria on urinalysis], presented with AKI and 1 week of gross hematuria. The past medical history included coronary disease and atrial fibrillation. He denied family history of kidney disease, but did have one previous episode of ‘dark-colored urine’ 2 months prior that resolved spontaneously.
Medications included aspirin 81 mg daily, sotalol, omeprazole, lisinopril and dabigatran 150 mg twice daily. One week prior to presentation, warfarin had been held for 3 days and then dabigatran initiated, at request of the patient. Aspirin was continued for cardioprotection. His blood pressure was 140/76 mmHg. Physical examination was unremarkable. SCr was 5.5 mg/dL (420 μmol/L) with no evidence for systemic hemolysis. The international normalized ratio (INR) was 1.6 with a prothromboplastin time of 62 s. He was initially managed with i.v. fluids, and anticoagulants were held. A serological workup, including antineutrophilic cytoplasmic antibody (ANCA), cryoglobulins, C3/C4, serum/urine protein electrophoresis, and viral serologies, was negative. Because hematuria persisted and creatinine continued to rise, he underwent kidney biopsy.
The biopsy showed extensive, diffuse tubular injury with nuclear dropout, sloughing of brush border membrane and cytoplasmic reabsorption droplets containing eosinophilic proteinaceous material (Figure 1A). There were also several, large intratubular RBC casts. The glomeruli showed capillary congestion and mesangial matrix expansion and hypercellularity (Figure 1B). The vessels were unremarkable and a Prussian-Blue stain showed focal positivity in 1–2% of tubular profiles. Immunofluorescence studies showed 2–3+ mesangial positivity for IgA (Figure 1C) and 1+ mesangial positivity for C3. IgG, IgM and C1q stains were negative. There was no significant interstitial infiltrate. Ultrastructural evaluation showed segmental foot process effacement and large mesangial immune complex deposits (Figure 1D). The proximal tubular epithelial cells showed cytoplasmic reabsorption droplets containing heme-proteins and intratubular granular casts (Figure 1E). Immunohistochemistry revealed strong, diffuse positivity for CD163 (Figure 1F).
Fig. 1.
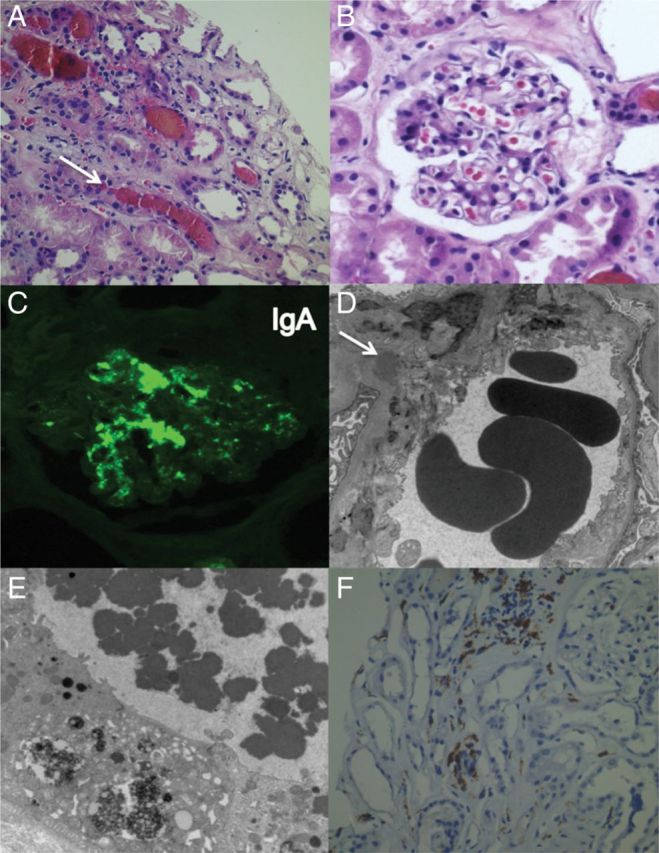
(A) Diffuse acute tubular injury with RBC casts and cytoplasmic reabsorption droplets (arrow) (H&E 400×). (B) Glomerulus with increased mesangial cellularity (H&E 400×). (C) IF stain for IgA with strong mesangial staining pattern (200×); (D) EM with mesangial immune complex deposits (arrow; ×8000). Basement membranes with normal thickness of 230 ± 60 nm; (E) EM of tubular epithelium with cytoplasmic reabsorption droplets containing heme proteins (arrow; 13 000×). (F) Immunohistochemistry stain for CD163 (×400).
The patient had previously had mild undiagnosed IgA nephropathy that was found on biopsy but in the setting of a high dose of dabigatran and aspirin he developed massive hematuria (likely predisposed by IgA nephropathy) and acute tubular necrosis from the direct toxic effect of heme pigment in the proximal tubular cells. The patient's creatinine stabilized at 6.0 mg/dL (530 μmol/L), and he was treated with normal saline to encourage diuresis. He was discharged home and his creatinine improved over a 3-month period to 1.8 mg/dL (160 μmol/L). He eventually resumed warfarin without evidence of hematuria or AKI.
Discussion: hematuria and acute kidney injury
AKI in the setting of IgA nephropathy may result from immune-mediated proliferative glomerulonephritis, glomerular bleeding leading to tubular injury or a process completely unrelated to the disease [2]. Previous case reports have shown that hematuria itself may lead to an acute decline in renal function [3]. Warfarin-related nephropathy (WRN) is seen in patients with or without chronic kidney disease on anticoagulation with INRs over three [4]. AKI during warfarin therapy is usually hemodynamic due to hemorrhage, interstitial nephritis or occlusive RBC casts [4–6]. These RBC casts form as a consequence of enhanced glomerular hemorrhage that might be augmented by drugs that increase glomerular filtration pressure. WRN is characterized by AKI and may accelerate progression of CKD [7]. Kidney disorders associated with hematuria, such as IgA nephropathy, may predispose the patient to warfarin-induced glomerular hemorrhage. However, <1.5% of patients with IgA nephropathy and gross hematuria present with AKI [8]. Therefore, the reason for the development of AKI in WRN is still unknown. Moreover, the kidney biopsies in patients with WRN show that the percentage of tubules with RBC casts is low (<20%) as reported in the previous cases [6]. These observations raise the question whether other additional mechanisms might be involved in the pathogenesis of tubular injury in WRN.
This patient's biopsy showed large RBC casts, extensive acute tubular injury and cytoplasmic protein reabsorption droplets in proximal tubular cells. Our patient did have underlying IgA nephropathy, discovered at the time of kidney biopsy, but the conversion to dabigatran in association with aspirin precipitated AKI. The IgA nephropathy was largely quiescent with normal kidney function and only one previous episode of ‘dark urine’. Moderate IgA staining on kidney biopsy, previous high eGFR and low-level proteinuria suggest mild pre-existing disease.
Our patient's biopsy showed RBC casts in only 10% of tubules, but extensive, diffuse cytoplasmic protein reabsorption droplets in most proximal tubular cells. There was focally positive iron staining and heme aggregates in both the proximal tubular lumens and in the cytoplasm of proximal tubular cells. High hemoglobin concentrations in the urine and tubular RBC casts lead to increased uptake of hemoglobin in the proximal tubule cells. This uptake occurs via the megalin–cubulin receptors and internalization through apical vacuoles that become protein absorption droplets [9, 10]. The heme molecule in its free form is tubulo-toxic through lysosomal overload, inflammation, apoptosis and oxygen-radical formation [11, 12]. Studies in isolated tubular segments have shown significant toxicity of heme proteins through oxidative damage [13]. Heme further induces significant mitochondrial damage and augments ischemia-induced lipid peroxidation, augmented by Fe2+ [13]. Exposure of tubular cells to high concentrations of heme proteins may increase synthesis of the vasoconstrictor peptide endothelin-1, leading to ischemic injury or monocyte migration [14]. A possible mechanism through which dabigatran might enhance heme-induced tubulotoxicity is by heme oxygenase-1 inhibition, thus preventing the breakdown of heme to biliverdin and iron. The expression of CD-163 positive macrophages suggests an immune role in the presence of direct tubular and interstitial damage through the scavenging of toxic heme compounds [15]. However, it is quite possible that the underlying hematuria and subsequent injury overwhelmed this compensatory mechanism.
This patient did have underlying IgA nephropathy, with mesangial immune complexes, but minimal proteinuria, suggesting mild disease. The AKI present on biopsy was not directly caused by the IgA nephropathy, but rather a result of significant anticoagulation with dabigatran and aspirin. It is quite possible that the combination of warfarin, dabigatran and aspirin during the transition period resulted in excessive anticoagulation. The INR was mildly elevated at 1.6 possibly suggesting persistent vitamin K antagonism and thereby anticoagulation from warfarin in addition to the direct thrombin inhibition of dabigatran. The progression of AKI most likely exacerbated the effects by elevating serum levels of dabigatran further (although levels were not measured). All anticoagulants, such as dabigatran or warfarin, may cause severe hematuria, leading to acute tubular injury.
In summary, excessive glomerular hemorrhage may lead to AKI via several pathological mechanisms (see Figure 2). Red blood cells may be endocytosed by proximal tubular cells, resulting in heme protein induced necrosis. The burden of these different injury mechanisms overcomes the regenerative capacity of the tubular cells perpetuating injury. We propose that the mechanism of injury in this patient was similar to WRN, but the patient was not taking warfarin. Therefore, this disease would more properly be renamed ‘anticoagulation-related nephropathy’.
Fig. 2.

Injury mechanisms of heme toxicity: 1. Heme-proteins increase endothelin-1 levels, causing vasoconstriction and ischemic injury. 2. Heme-proteins stimulate inflammatory infiltrate to the interstitium. 3. The tetrapyrrole heme molecule induces lysosomal overload and lysosomal cell injury. 4. Heme-proteins induce apoptosis and oxygen-radical formation. 5. Heme-proteins cause injury through oxidative damage to the mitochondria. 6. Heme augments ischemia-induced peroxidation.
Conflict of interest statement. The results presented in this paper have not been published previously in whole or part, except in abstract format. G.W.M.: nothing to disclose. R.L.L.: nothing to disclose. U.C.B.: nothing to disclose.
References
- 1.Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. NEJM. 2009;361:1139–1151. doi: 10.1056/NEJMoa0905561. doi:10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 2.Kincaid-Smith P, Bennett WM, Dowling JP, et al. Acute renal failure and tubular necrosis associated with hematuria due to glomerulonephritis. Clin Nephrol. 1983;19:206–210. [PubMed] [Google Scholar]
- 3.Wen YK, Chen ML. The spectrum of acute renal failure in IgA nephropathy. Ren Fail. 2010;32:428–433. doi: 10.3109/08860221003646345. doi:10.3109/08860221003646345. [DOI] [PubMed] [Google Scholar]
- 4.Brodsky SV, Nadasdy T, Rovin BH, et al. Warfarin-related nephropathy occurs in patients with and without chronic kidney disease and is associated with an increased mortality rate. Kidney Int. 2011;80:181–189. doi: 10.1038/ki.2011.44. doi:10.1038/ki.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kapoor KG, Bekaii-Saab T. Warfarin-induced allergic interstitial nephritis and leucocytoclastic vasculitis. Intern Med J. 2008;38:281–283. doi: 10.1111/j.1445-5994.2008.01646.x. doi:10.1111/j.1445-5994.2008.01646.x. [DOI] [PubMed] [Google Scholar]
- 6.Brodsky SV, Satoskar A, Chen J, et al. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of 9 cases. Am J Kidney Dis. 2009;54:1121–1126. doi: 10.1053/j.ajkd.2009.04.024. doi:10.1053/j.ajkd.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 7.Brodsky SV, Collins M, Park E, et al. Warfarin therapy that results in an international normalization ratio above the therapeutic range is associated with accelerated progression of chronic kidney disease. Nephron Clin Pract. 2010;115:c142–c146. doi: 10.1159/000312877. doi:10.1159/000312877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kveder R, Ales J, Kovac D, et al. Acute kidney injury in immunoglobulin A nephropathy: potential role of macroscopic hematuria and acute tubulointerstial injury. Ther Apher Dial. 2009;13:273–277. doi: 10.1111/j.1744-9987.2009.00723.x. doi:10.1111/j.1744-9987.2009.00723.x. [DOI] [PubMed] [Google Scholar]
- 9.Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int. 1996;49:314–326. doi: 10.1038/ki.1996.48. doi:10.1038/ki.1996.48. [DOI] [PubMed] [Google Scholar]
- 10.Miller F, Palade GE. Lytic activities in renal protein absorption droplets: an electron microscopical cytochemical study. J Cell Biol. 1964;23:519–552. doi: 10.1083/jcb.23.3.519. doi:10.1083/jcb.23.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zager RA, Gamelin LM. Pathogenetic mechanisms in experimental hemoglobinuric acute renal failure. Am J Physiol. 1989;256:F446–F455. doi: 10.1152/ajprenal.1989.256.3.F446. [DOI] [PubMed] [Google Scholar]
- 12.Lin HL, Xu XS, Lu HX, et al. Pathological mechanisms and dose dependency of erythrocyte-induced vulnerability of atherosclerotic plaques. J Mol Cell Cardiol. 2007;43:272–280. doi: 10.1016/j.yjmcc.2007.05.023. doi:10.1016/j.yjmcc.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Moussavian MR, Slotta JE, Kollmar O, et al. Hemoglobin induces cytotoxic damage of glycine-preserved renal tubules. Transpl Int. 2007;20:884–894. doi: 10.1111/j.1432-2277.2007.00538.x. doi:10.1111/j.1432-2277.2007.00538.x. [DOI] [PubMed] [Google Scholar]
- 14.Zoja C, Morigi M, Figliuzzi M, et al. Proximal tubular cell synthesis and secretion of endothelin-1 on challenge with albumin and other proteins. Am J Kidney Dis. 1995;26:934–941. doi: 10.1016/0272-6386(95)90058-6. doi:10.1016/0272-6386(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 15.Moreno JA, Matrin-Cleary M, Gutierrez E, et al. AKI associated with macroscopic hematuria: clinical and pathophysiologic consequences. CJASN. 2012;7:175–184. doi: 10.2215/CJN.01970211. [DOI] [PubMed] [Google Scholar]