

Abstract
Fibronectin glomerulopathy occurs between the second and fifth decades of life in most patients, and it is known to be slowly progressive with mild proteinuria leading to kidney failure. The case of a 78-year-old woman with a rapid course of nephrotic syndrome due to fibronectin glomerulopathy is reported. She had proteinuria that rapidly increased to 6.8 g/day in a month and microscopic haematuria. A renal biopsy specimen showed lobular glomerulopathy and membranoproliferative glomerulonephritis-like lesions on light microscopy. There was scanty staining for immunoglobulins and complement. Electron microscopy revealed granular deposits with fibril formation. Immunohistochemistry of the fibronectin showed intense staining in the mesangium and peripheral loop. Therefore, this case was diagnosed as fibronectin glomerulopathy. The kidney function was rapidly decreasing, necessitating haemodialysis 2 months after renal biopsy. It is important to consider fibronectin glomerulopathy in the differential diagnosis of nephrotic syndrome in older people.
Keywords: fibronectin glomerulopathy, haemodialysis, nephrotic syndrome
Background
Fibronectin glomerulopathy is an autosomal-dominant renal disease. Common clinical features are mild proteinuria and varying degrees of haematuria, hypertension and slow progression to end-stage renal disease over 15–20 years [1]. A diagnosis is made only by renal biopsy. Electron microscopy shows abnormal electron-dense deposits in the mesangium and subendothelial zone. The deposits may be finely granular or have a fibrillary substructure with randomly arranged, 12- to 16-nm fibrils. The diagnosis of fibronectin glomerulopathy is confirmed when the intense staining for the plasma isoform of fibronectin is found in the mesangium and along the capillary walls [2, 3]. Currently, there is no specific treatment for fibronectin glomerulopathy.
Case report
A 78-year-old Japanese woman was admitted because of systemic oedema. Approximately 11 months earlier, her serum creatinine level was within the normal range (Cre 38.1 µmol/L), and her urinalysis was also normal at a medical check-up. Eight months earlier, non-steroidal anti-imflammatory drugs (NSAIDs) were prescribed for arthralgia. Six weeks before hospitalization, she visited us for the first time. Her serum creatinine was 101 µmol/L. Urinalysis showed proteinuria of 0.65 g/day and haematuria of 30–49 red blood cells/high-power field. The NSAIDs were discontinued, and after 2 weeks, the serum creatinine decreased to 68.6 µmol/L, although the urinalysis findings did not improve.
One month later, she came to the outpatient clinic again because of systemic oedema. Urinalysis revealed proteinuria of 10.2 g/gCre (protein-creatinine ratio), and laboratory tests showed a serum total protein of 48 g/L, a serum albumin of 26 g/L and a serum creatinine of 101 µmol/L. She was diagnosed with nephrotic syndrome and was admitted. On admission, she had a blood pressure of 148/93 mmHg and her weight had increased 11 kg in a month. A percutaneous renal biopsy was performed for accurate diagnosis of her nephrotic syndrome.
Light microscopy contained 40 glomeruli. Haematoxylin and eosin-stained sections showed massive, homogeneous, eosinophilic deposits in the mesangial space with formation of a lobular shape (Figure 1A). No crescent formation was seen. Capillary walls had double contours, and almost all glomeruli had numerous periodic acid-Schiff (PAS)-positive deposits in the mesangial space and between the double-contour capillary walls (Figure 1B and C). Amyloid staining and Congo-red staining were both negative. Immunofluorescence studies showed slight intensity of IgG in a peripheral pattern. However, other immunofluorescence microscopy tests were all negative. Electron microscopy revealed granular deposits in the mesangial space (Figure 1D). The granular deposits showed focal fibril formation with higher magnifications (Figure 1E). The fibrils were randomly arranged, with no central core, and their diameter was 12–14 nm. Fibronectin glomerulopathy was suspected because of this fibril formation and size. Therefore, plasma fibronectin immunostaining was performed. Immunohistochemistry of the fibronectin clearly showed intense staining in the mesangial space and a peripheral pattern (Figure 1F). Thus, the case was diagnosed as having fibronectin glomerulopathy based on these histological findings.
Fig. 1.
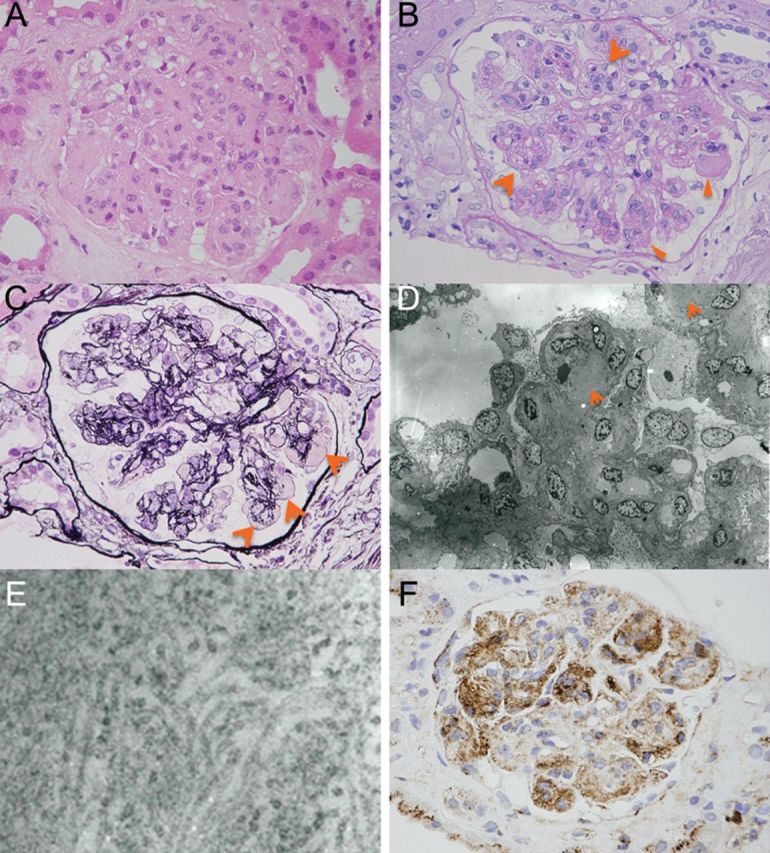
(A) Haematoxylin and eosin-stained section showed massive, homogeneous, eosinophilic deposits in the mesangium space with formation of a lobular shape (×300). (B) PAS stain. Capillary walls were with double contour (arrows) and almost all glomeruli had numerous PAS-positive deposits (arrowheads) in the mesangium space and between double contour capillary walls (×300). (C) Periodic acid silver methenamine stain. There were silver methenamine stain-negative and PAS-positive materials (arrows) in the mesangium space (×300). (D) Electron micrograph showed faint deposits in the mesangium space (arrows), not the typical electron-dense deposits, but granular deposits with focal fibril formation (×2800). (E) Electron micrograph at high magnification (×60 000). The deposits had fibrillary substructure, with randomly arranged, 12–14 nm fibrils. They had no central core. (F) Immunochemistry for plasma fibronectin showed intense staining in the mesangial and peripheral loop (×300).
She had progressive pre-renal acute kidney injury due to nephrotic syndrome. Two months after kidney biopsy, haemodialysis was started, and she became dialysis-dependent thereafter.
Discussion
A case of rapidly progressive nephrotic syndrome due to fibronectin glomerulopathy in an elderly woman that resulted in end-stage renal disease was described.
Fibronectin glomerulopathy is an autosomal-dominant renal disease. However, the present patient family history was negative for kidney disease. As reported by other groups, she could be a sporadic case of fibronectin glomerulopathy. Castelletti et al. [4] sequenced the gene encoding fibronectin-1 (FN1) and found three heterozygous missense mutations, Y973C, W1925R and L1974R, which co-segregated with the disease in 40% of pedigrees. However, other families (60%) affected by fibronectin glomerulopathy showed no causative gene mutations in FN1. FN1 was sequenced in the present patient, but she did not have these missense mutations. Although it was reported that fibronectin glomerulopathy is uncommonly caused by systemic lupus erythematosus, this patient was negative for systemic lupus erythematosus [5].
In fibronectin glomerulopathy, most patients have mild proteinuria, microscopic haematuria, hypertension and slowly decreasing renal function over 15–20 years (Table 1). Only a small subset present with nephrotic-range proteinuria, although it is a slowly progressive proteinuria [6]. However, in the present patient, nephrotic syndrome occurred rapidly, and proteinuria increased to 6.8 g/day in a month. To the best of our knowledge, such a rapid clinical course of nephrotic syndrome has not been previously reported in fibronectin glomerulopathy.
Table 1.
Past case reports of fibronectin glomerulopathy
| Reporter | Case numbers | Age at biopsy (years) | Pcr at biopsy (µmol/L) | Proteinuria (at biopsy) | Renal outcome (Follow-up years) |
|---|---|---|---|---|---|
| Strom et al. [6] | 23 patients (6 families) | 14–59 | 45.8–137 | 21 patients: positive (9 patients: nephrotic) | 4 patients: HD (2,7,10,13 years) |
| Gemperle et al. [1] | 9 patients (1 family) | 20–46 | Not reported | 7 patients: positive, (1 patient: nephrotic) | 2 patients: HD (14, 14), 2 patients: CAPD (15,21 years) |
| Castelletti et al. [4] | 8 patients (1 family) | 12–59 | 53.4–168 | 6 patients: positive (4 patients: nephrotic), 2 patients: unknown | HD(-) (max. follow-up: 23 years) |
| Uesugiet al. [7] | 5 patients (4 families) | 3–75 | 15.3–122 | 5 patients: positive (2 patients: nephrotic) | HD(-) (max. follow-up: 10 years) |
| Sato et al. [8] | 1 patient | 23 | 68.6 | 1–2 g/day | not reported |
| Niimi et al. [9] | 1 patient | 3 | 15.3 | Nephrotic | Pcr was stable (7 years) |
| Nadamuni et al. [10] | 1 patient | 50 | 61.0 | Nephrotic | Pcr was stable (1 year) |
| Baydar et al. [11] | 1 patient | 24 | 62.5 | 1 g/day | Pcr was stable (2.5 years) |
| This Case | 1 patient | 78 | 99.1 | 6.8 g/day | HD |
Pcr, Plasma creatinine.
Fibronectin glomerulopathy is usually diagnosed between the second and fifth decades of life [6], and the oldest patient was 75 years old [7]. This case is interesting because proteinuria occurred at 78 years of age, the oldest onset of fibronectin glomerulopathy reported to date.
Taken together, fibronectin gromerulopathy should be considered in the differential diagnosis of rapidly progressive nephrotic syndrome in elderly patients.
Conflict of interest statement. None declared.
References
- 1.Gemperle O, Neuweiler J, Reutter FW, et al. Familial glomerulopathy with giant fibrillar (fibronectin-positive) deposits: 15-year follow-up in a large kindred. Am J Kidney Dis. 1996;28:668–675. doi: 10.1016/s0272-6386(96)90247-4. [DOI] [PubMed] [Google Scholar]
- 2.Weiss M, Tzavella K, Müller-Höcker J, et al. Fibrillary glomerulonephritis. Case report for differential nephrotic syndrome diagnosis. Pathology. 1998;19:141–145. doi: 10.1007/s002920050266. [DOI] [PubMed] [Google Scholar]
- 3.Yong JL, Killingsworth MC, Spicer ST, et al. Fibronectin non-amyloid glomerulopathy. Int J Clin Exp Pathol. 2009;3:210–216. [PMC free article] [PubMed] [Google Scholar]
- 4.Castelletti F, Donadelli R, Banterla F, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci USA. 2008;105:2538–2543. doi: 10.1073/pnas.0707730105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonella S, Angelo P, Claudia F, et al. Fibronectin glomerulopathy: an uncommon cause of nephrotic syndorome in systemic erythematosus. NDT Plus. 2008;1:225–227. doi: 10.1093/ndtplus/sfn037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strøm EH, Banfi G, Krapf R, et al. Glomerulopathy associated with predominant fibronectin deposits: a newly recognized hereditary disease. Kidney Int. 1995;48:163–170. doi: 10.1038/ki.1995.280. [DOI] [PubMed] [Google Scholar]
- 7.Uesugi N, Katafuchi R, Taguchi H, et al. Clinicopathological and morphometrical analysis of 5 cases from 4 families of fibronectin glomerulopathy. Nihon Jinzo Gakkai Shi. 1999;41:49–59. [PubMed] [Google Scholar]
- 8.Sato H, Matsubara M, Marumo R, et al. Familial lobular glomerulopathy: first case report in Asia. Am J Kidney Dis. 1998;31:E3. doi: 10.1053/ajkd.1998.v31.pm10074583. [DOI] [PubMed] [Google Scholar]
- 9.Niimi K, Tsuru N, Uesugi N, et al. Fibronectin glomerulopathy with nephrotic syndrome in a 3-year-old male. Pediatr Nephrol. 2002;17:363–366. doi: 10.1007/s00467-002-0833-2. [DOI] [PubMed] [Google Scholar]
- 10.Nadamuni M, Piras R, Mazbar S, et al. Fibronectin glomerulopathy: an unusual cause of adult-onset nephrotic syndrome. Am J Kidney Dis. 2012;60:839–842. doi: 10.1053/j.ajkd.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 11.Ertoy Baydar D, Kutlugun AA, Bresin E, et al. A case of familial glomerulopathy with fibronectin deposits caused by the Y973C mutation in fibronectin. Am J Kidney Dis. 2013;61:514–518. doi: 10.1053/j.ajkd.2012.08.050. [DOI] [PubMed] [Google Scholar]