

Highlight
In comparing diploids with their isogenic synthetic autopolyploids of watermelon, we noted and characterized tissue-specific alternative splicing across the genome.
Key words: genome duplication, gene expression, RACE-PCR, RNA-seq, tetraploid, alternative splicing, watermelon (Citrullus lanatus).
Abstract
The exploitation of synthetic polyploids for producing seedless fruits is well known in watermelon. Tetraploid progenitors of triploid watermelon plants, compared with their diploid counterparts, exhibit wide phenotypic differences. Although many factors modulate alternative splicing (AS) in plants, the effects of autopolyploidization on AS are still unknown. In this study, we used tissues of leaf, stem, and fruit of diploid and tetraploid sweet watermelon to understand changes in gene expression and the occurrence of AS. RNA-sequencing analysis was performed along with reverse transcription quantitative PCR and rapid amplification of cDNA ends (RACE)-PCR to demonstrate changes in expression and splicing. All vegetative tissues except fruit showed an increased level of AS in the tetraploid watermelon throughout the growth period. The ploidy levels of diploids and the tetraploid were confirmed using a ploidy analyser. We identified 5362 and 1288 genes that were up- and downregulated, respectively, in tetraploid as compared with diploid plants. We further confirmed that 22 genes underwent AS events across tissues, indicating possibilities of generating different protein isoforms with altered functions of important transcription factors and transporters. Arginine biosynthesis, chlorophyllide synthesis, GDP mannose biosynthesis, trehalose biosynthesis, and starch and sucrose degradation pathways were upregulated in autotetraploids. Phloem protein 2, chloroplastic PGR5-like protein, zinc-finger protein, fructokinase-like 2, MYB transcription factor, and nodulin MtN21 showed AS in fruit tissues. These results should help in developing high-quality seedless watermelon and provide additional transcriptomic information related to other cucurbits.
Introduction
Watermelon [Citrullus lanatus var. lanatus (Thunb.) Matsum. & Nakai] is a member of the cucurbits that is widely cultivated for its sweet dessert fruit, which is also an important source of water, sucrose, and other phytochemicals. Commercial varieties of watermelon include seedless triploids produced by crossing female synthetic autotetraploids with a male diploid (Kirhara, 1951; Andrus et al., 1971; Ahmad et al., 2013). Watermelon breeding is unique in that it takes advantage of synthetic polyploids in breeding commercially important seedless triploids and uses heterosis because of the dosage effects resulting from various combinations of diploid and tetraploid genomes. Autotetraploids are maintained year after year as a parental line in triploid breeding, so investigating the genomic-level changes of neopolyploids is of interest.
Based on phenotypic, cytological, and molecular characterization of neopolyploids, polyploids will pass through a bottleneck of instability before they settle down for diploidization. An immediate consequence of polyploidy is the change in gametic and filial frequencies. Mendelian laws of inheritance still apply to ploidy but with an increase in the number of ‘factors’ to consider. This situation produces ratios that are deviant from those observed in diploids (Comai, 2005). A variety of genetic and epigenetic processes are initiated to modify or silence the action of duplicated genes that can lead to loss or retention of duplicates, often resulting in subfunctionalization or neofunctionalization (Langham et al., 2004; Zhang and Kishino, 2004; Comai, 2005; Maere et al., 2005; Nam et al., 2005; Rastogi and Liberles, 2005; Jackson and Chen, 2010). Some of these changes are rapid and fixed immediately after chromosome doubling, whereas others are stochastic and gradual (Jackson and Chen, 2010). Moreover, autopolyploids have to overcome the odds of random chromosome segregation due to complications of pairing during meiosis (Jackson and Chen, 2010), which explains why their triploid or aneuploid progeny are seedless.
Polyploidy gives rise to new allelic variants with wide gene-expression changes (Feldman and Levy, 2009; Wang et al., 2012; Zhang et al., 2014) and causes expansion of gene families (Zahn et al., 2005; Veron et al., 2007). Differential gene-expression patterns among diploids and tetraploids have been studied to investigate the effects of natural and synthetic polyploidy for novel organ development and resistance to abiotic stresses (Chen and Ni, 2006; Doyle et al., 2008). The molecular basis of evolutionary advantage could be related to gene expression changes, which have been demonstrated in resynthesized polyploids in Arabidopsis (Wang et al., 2006), Brassica (Gaeta et al., 2009), and Gossypium (Flagel et al., 2008). RNA sequencing (RNA-seq) revealed the differential expression of ~50% of paralogues among 18 000 duplicated genes in soybean; the genes showed subfunctionalization on testing seven different tissues (Roulin et al., 2013). Gene expression at transcriptome and proteome levels was investigated in soybean (Coate et al., 2014). Differential gene expression among the ploidy levels included non-additive expression, tissue-specific expression changes, and parent-dependent repression or activation, as documented in wheat, cotton, and sugarcane (Jackson and Chen, 2010). Analysis of proteomes from autopolyploid cabbage did not reveal major alterations related to ploidy change (Albertin et al., 2005). However, Arabidopsis showed high (~18%) and low (~6.8%) proteomic divergence in allopolyploid and autopolyploid genomes, respectively, compared with diploid genomes (Ng et al., 2012). This study indicated the presence of rapid alterations at both post-transcriptional and post-translational levels in polyploids. Heterosis causes polyploids to be more vigorous than their diploid progenitors, and gene redundancy shields polyploids from the deleterious effect of mutations (Comai, 2000).
More than 60% of intron-containing genes undergo alternative splicing (AS) in plants (Syed et al., 2012). This number will increase when AS is explored in different tissues, developmental stages, and environmental conditions. Although the functional impact of AS on protein complexity is still understudied in plants, recent examples demonstrate its importance in regulating plant processes (Syed et al., 2012). Exon skipping, alternative 5ʹ and 3ʹ splice sites, intron retention, mutually exclusive exons, alternative promoters/5ʹ transcription start sites (TSSs), and alternative 3ʹ poly(A) sites are the seven main types of known AS events (Grabowski, 2002; Keren et al., 2010). In addition to increasing transcript level, AS can also cause transcript degradation via nonsense-mediated decay, thus potentially reducing the level of gene expression (Dinesh-Kumar and Baker, 2000; Barbazuk et al., 2008). As a result, AS can modulate phenotypic changes related to developmental growth and physiological processes (Staiger and Brown, 2013).
Recently, AS patterns have been studied among natural and synthetic polyploids of Brassica napus. Two independently synthesized lines showed parallel loss of AS events after polyploidy (Zhou et al., 2011b ). This finding is intriguing because it shows that even two independent events of polyploidy, using the same species, results in an identical pattern of AS loss, pointing to a non-random response of the so-called ‘genomic shock’ after two genomes physically interact with each other (Syed et al., 2012). The same study showed 26–30% of the duplicated genes with changes in AS compared with the parents, with some showing organ specificity or response to abiotic stress (Zhou et al., 2011b ). Because many crop species are polyploids, AS has played an important role in the evolution and adaptation of cultivated crops to different environmental conditions and niches. This area will be of great interest in the future, particularly with the power of high-throughput sequencing (Syed et al., 2012).
Several factors are known to modulate splicing events in different organisms. For example, light can cause intron retention in Physcomitrella patens (Wu et al., 2014). AS plays a key role in plant growth and development and defence systems. Use of alternative 5ʹ TSSs produced multiple transcripts from an RNA-binding protein (LGD1) regulating rice architecture (Thangasamy et al., 2012). AS of a superfamily transporter (ZIFL1) resulted in full-length and truncated alternative protein isoforms with altered regulatory roles in vacuolar and plasma membranes, respectively (Remy et al., 2013). In tobacco, the NBS-LRR class of plant resistance genes underwent AS as a mechanism of resistance to tobacco mosaic virus (Dinesh-Kumar and Baker, 2000). In addition, AS increased the complexity of microRNA-based gene regulation in Arabidopsis (Yang et al., 2012).
Recently, the RNA-seq approach combined with whole-genome sequencing has offered new ways to map and quantify transcriptomes and locate alternative isoforms (Wang et al., 2009; Ilut et al., 2012; Walters et al., 2013; Vitulo et al., 2014). Autopolyploidy is the ideal system to study ploidy-dependent gene regulation because natural allopolyploidy introduces confounding factors arising from two similar but diverse genomes. In this research, we used a watermelon diploid and its isogenic autopolyploid derivative to investigate the regulation of AS. Since seedless watermelon fruits are harvested from triploids, the variation in size of various tissues among the ploids of watermelon motivated us to investigate the effects of gene expression at the whole-genome level and to scan for splice variants. In watermelon, seedless fruits are produced by crossing a tetraploid female with a diploid male; therefore, we characterized the ploidy-specific transcriptome and observed the effects of a duplicated genome on AS.
Materials and methods
Plant materials and growth conditions
Watermelon diploid cultivar Charleston Gray (CLD1; PI-635699, http://www.ars-grin.gov) and its tetraploid (CLT1; PI-635696) were selected from the F9 backcross progeny of a cross of Charleston Gray and Sugar Baby after colchicine treatment. We additionally used Sugar Baby (WM) as a diploid cultivar. The field plot technique consisted of three biological replications. Ten plants per genotype were grown in each replication using standard horticultural procedures.
Evaluation of phenotypic traits
All phenotypic traits were measured for three replications as suggested by Ahmad et al. (2013). Total soluble solids of flesh juice (i.e. Brix %) was measured by use of a hand refractometer (ATAGO, Minatoku, Tokyo, Japan). Rind pressure was measured with a FT 327 Penetrometer (QA Supplies LLC, Norfolk, VA, USA) by piercing through the rind before cutting, and flesh pressure was measured with the same penetrometer by piercing the flesh after cutting the fruit.
Preparation of nuclei and flow cytometry
The procedure for ploidy analyses was slightly modified from a previous protocol (Dolezel and Bartos, 2005). To release nuclei, 0.5cm2 of fresh young leaves was placed in a Petri dish containing 1ml ice-cold extraction buffer (0.1M citric acid containing 0.5% Tween 20). Leaf pieces were further chopped into fine pieces with use of a razor blade. The homogenate was then filtered through a 45-µm nylon filter to remove cell debris, and 1.5ml of 4ʹ,6-diamidino-2-phenylindole (DAPI) staining buffer was added. Flow cytometry involved the use of a ploidy analyser (Partec GmbH, Munich, Germany) 1min after staining buffer was added. Gain value was adjusted so that the G1 peak of nuclei isolated from the control diploid Sugar Baby channel was positioned on 50. Thereafter, peaks representing G1 nuclei from CLD1 and CLT1 were expected at channels 50 and 100, respectively. We kept the total nuclei count above 5000 in each experiment. Finally, the Partec software package (Partec GmbH) was used to analyse the coefficient of variation percentage (CV%) for peaks at 50 and 100 for CLD1, and at 100 and 200 for CLT1 tissues.
RNA extraction and RNA-seq
Total RNA was isolated from frozen leaves, stems (30-d-old), and young fruits (10 d after pollination) of plants for all three biological replicates after ploidy confirmation, and total RNA was stored in a –80°C freezer for downstream gene-expression experiments. An amount of 50mg of tissue was lysed in microvials with zirconia beads (BioSpec, Bartlesville, OK, USA) and TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in a MagNA lyser (Roche, Basel, Switzerland) at 7000rpm for two cycles. After lysis, RNA purification steps were followed as suggested by the manufacturer (Zymo Research, Irvine, CA, USA). On-column DNase digestion was also performed. RNA integrity was measured by using a Nanodrop 1100 (NanoDrop, Wilmington, DE, USA) and a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For RNA-seq of each genotype, total RNA from different tissues was pooled within a biological replication in equimolar concentrations. Two biological replications of each genotype (CLD1-1, CLD1-2; CLT1-1, CLT1-2; and WM1-1, WM1-2) were used for 50bp paired-end RNA-seq (Genomic Services Laboratory, Huntsville, AL, USA) with Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, USA).
RNA sequence data analysis for expression and AS
As a part of the quality-control procedure, poor-quality reads were discarded on the basis of Illumina’s chastity filter. To assess the quality of sequencing and identify splice junctions, RNA-seq reads were mapped to the watermelon reference genome (Guo et al., 2013) with the TopHat algorithm (Trapnell et al., 2009). The alignment results are reported in SAM/BAM format. Reads with an alignment (MAPQ) score of 255 were identified from the output SAM files. The mapping rates for various samples are summarized (Table 1). The unique mapping rate was 42.9%, on average, exceeding an acceptable representation of 30%. TopHat-generated read alignments along with transcript annotation information from the reference genome were used with Cufflinks software to quantify gene/transcript expression and detect novel transcripts. The expression estimates were reported in units of fragments per kilobase of exon per million fragments mapped (FPKM). For assessing the influence of ploidy on gene/transcript expression, the expression of CLT1 transcripts was compared with that of CLD1. Transcripts with minimum FPKM of ≥1 were considered for differential expression analysis by using Cufflinks (Trapnell et al., 2010). To draw a heat map, hierarchical clustering of transcripts involved the use of Cluster 3.0 (de Hoon et al., 2004) with Spearman’s rank correlation as the distance measure. JAVA TreeView (Saldanha, 2004) was used to visualize the comparative map.
Table 1.
Mapping rates for various samples from RNA-seq
| Samples | Total read counts | Total PF mate pairs | Uniquely mapped mate pairs | Singleton | Unique mapping rate (%) |
|---|---|---|---|---|---|
| WM-1 | 33518638 | 31246600 | 8038310 | 1862752 | 31.69 |
| WM-2 | 39978004 | 37219446 | 7040148 | 1871008 | 23.94 |
| CLD1-1 | 83902943 | 80286133 | 24374324 | 3076898 | 34.19 |
| CLD1-2 | 131410680 | 120798531 | 58623148 | 6081401 | 53.56 |
| CLT1-1 | 75698759 | 71353201 | 35889608 | 4051355 | 55.98 |
| CLT1-2 | 164895585 | 154193891 | 79143169 | 10226231 | 57.96 |
Gene prediction, functional annotation, and pathway analysis
Protein sequences for cucumber and Arabidopsis were aligned to watermelon contigs using TblastN (E-value 1e–5), and high-scoring pairs were grouped by using genBlastA. Next, alignments were refined by including 500bp from each side of the alignment and re-aligning the DNA sequence to the parental protein sequence using Genewise. Genscan, with parameters optimized for plant genomes, was chosen to predict gene models. The transcript sequences were mapped to watermelon unigene identifiers by sequence similarity. Wherever possible, the sequences were mapped to UNIPROT identifiers and annotated with gene ontology (GO) IDs using Biomart. The GO annotations were categorized into broader GO classes with plant-specific GO-Slim terms at the Categorizer web server (Hu et al., 2008). The pathway annotations (version 2) of watermelon unigenes were downloaded from the Cucurbit Genomics Database. Gene-set enrichment analysis involved use of the GeneTrail server (http://genetrail.bioinf.uni-sb.de/). Gene-set enrichment analysis was done to identify significantly affected pathways, which offer more-specific insights than the broader GO categories. The gene identifiers mapped to the pathway were used as the gene sets, and the differentially expressed gene lists in each of the comparisons were used as the test sets. A minimum of two genes in each category was the criterion for consideration. Significant pathways were identified as those passing a hypergeometric test with a false discovery rate-corrected P value of <0.05.
Splicing model, semi-quantitative reverse transcription (RT)-PCR, and quantitative RT-PCR (RT-qPCR)
To draw differentially spliced products, we used the RNA sequences matched to their respective locus number from the reference sequence database, and aligned the mRNA-to-genomic sequence by use of the mRNA-to-genomic alignment program Spidey (http://www.ncbi.nlm.nih.gov/spidey). Multiple spliced transcripts were aligned to each locus. The final alignment also showed the splicing donor/acceptor sites and mismatches. To show tissue-related expression patterns as well as transcripts with AS, we performed both semi-quantitative RT-PCR and RT-qPCR for all tissues of WM, CLD1, and CLT1. The DNase-treated RNA was used in RT reactions to synthesize cDNAs using SuperScript II reverse transcriptase (Invitrogen) as suggested by the manufacturer.
PCR for each transcript involved respective primer sets (see Supplementary Table S7 at JXB online) and GoTaq Green Master Mix (Promega, Madison, WI, USA) with the following conditions for 26–34 cycles: denaturation at 94°C for 10 s, annealing between 54 and 58°C for 30 s, and extension at 72°C for 30 s to 3min for different transcripts. Semi-quantitative RT-PCR was performed for three biological replications with Elongation Factor1 as an endogenous control. RT-qPCR of three biological replications involved a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and FastStart SYBR Green (Roche). The expression of selected genes was normalized to that of the 18S rRNA gene. Finally, the mean relative gene expression was calculated by the 2–ΔΔCt method (Livak and Schmittgen, 2001).
5ʹ and 3ʹ rapid amplification of cDNA ends (RACE)
To map the TSSs, alternative splice junctions and ends of transcripts [different poly(A) sites], we used FirstChoice RNA ligase-mediated RACE (Ambion, Austin, TX, USA) with specific primers (Supplementary Table S7). RACE-PCR was performed twice from two biological replications to confirm spliced transcripts along with the 5ʹ and 3ʹ ends of each gene. The RACE-PCR products were cloned and confirmed by sequencing. Finally, full-length cDNAs were aligned by use of the program Spidey to obtain variants with TSSs, intron–exon junctions, and poly(A)s.
Results
Phenotypic characterization of CLD1 and CLT1 lines
Throughout vegetative growth, plants were tested for ploidy levels to confirm the stability of ploidy. To accomplish this, the ploidy level of Sugar Baby, CLD1, and CLT1 was confirmed at multiple times at 30-d intervals after they were transplanted into the field. CLT1 had a ploidy peak that was twice that of the diploid (Fig. 1). We observed growth from the seedling to maturity stage and measured growth parameters including fruit traits. For all traits other than fruit size, mean values were higher for CLT1 than for CLD1 plants (Fig. 2A–D; Table 2); the fruit size of the diploid and tetraploid did not differ (Fig. 2E; Table 2).
Fig. 1.
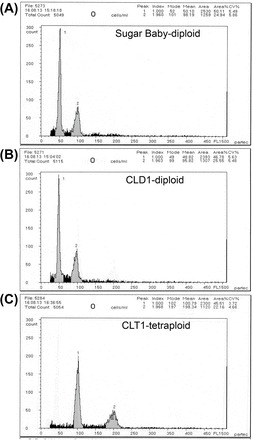
Quantification of genome size of watermelon ploidy by Partec ploidy analyser. Flow cytometry of the relative DNA content of DAPI-stained nuclei from young shoot tips. The x-axis and y-axis represent the relative signal intensity and number of nuclei, respectively. Sugar Baby (A) was used as a control standard diploid to compare the ploidy level of diploid (B) and tetraploid (C) plants. Experiments were repeated at least three times from three independent plants.
Fig. 2.

Morphological features of diploid (CLD1) and tetraploid (CLT1) plants. Detailed morphological features of diploid and tetraploid Charleston Gray field-grown watermelon. (A) Phenotype of field-grown plants 70 d after planting. (B) Morphology of leaves detached from mother plants. (C) Comparison of stems collected from the middle of the main stem showing variation in width and trichome morphology. (D) Phenotypic variation of staminate flowers collected during flowering. (E) Fruits showing no significant morphological difference between diploid and tetraploid. Bars, 5cm (B, E); 1cm (C, D).
Table 2.
Analysis of quantitative traits between CLD1 (diploid) and CLT1 (tetraploid) watermelon plants
| Leaf length (cm) | Leaf width (cm) | Stem thickness (mm) | Flower size (mm) | Fruit length (cm) | Fruit width (cm) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 |
| 15.70±0.61 | 19.94±1.01*** | 13.64±0.44 | 22.38±2.27*** | 5.40±0.32 | 7.40±0.47*** | 34.3±2.1 | 45.8±2.0*** | 16.84±0.69 | 17.16±0.54 | 17.16±0.68 | 17.02±0.79 |
| L/W ratio | Rind pressure (kg mm–1) | Rind thickness (mm) | Fruit weight (kg) | Flesh pressure (kg/mm) | Brix % (TSS) | ||||||
| CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 | CLD1 | CLT1 |
| 0.95±0.04 | 1.01±0.06 | 0.58±0.08 | 0.73±0.03** | 11.3±0.7 | 15.0±0.7** | 1.99±0.25 | 2.18±0.34 | 1.45±0.09 | 1.56±0.13 | 9.48±0.54 | 10.04±0.36 |
Asterisks indicate significant differences between CLD1 and CLT1 as analysed by Student’s t-test: **0.001<P< 0.01; ***P<0.001.
Effect of ploidy on global transcriptome
To understand the effect of ploidy on gene expression, we generated 529.5 million 50-nt paired-end reads of Sugar Baby, CLD1, and CLT1 using the Illumina HiSeq 2000 platform. A total of 495 million reads were obtained after trimming and pass filtering. Our average mapping rate was good: 42.9% compared with the basal 30%. However, the mapping rate was lower than normal for the second replication of Sugar Baby (Table 1). Overall, our results indicated that the quality and quantity of RNA-seq map reads met the acceptable range for downstream analyses.
The correlation (scatter) plots for expression of replicates of CLD1, CLT1, Sugar Baby, and between CLD1 and CLT1 are in Fig. 3A–D. The replicates showed a high degree of expression correlation (R 2>0.6). However, Sugar Baby replicates showed alow correlation (R 2=0.474). For assessing the effect of ploidy on expression, we compared the expression of CLT1, Sugar Baby, and the CLD1 transcriptome. Only transcripts with FPKM ≥1 were considered for differential expression analysis. All transcripts were assigned names based on mapping to watermelon reference genome annotations.
Fig. 3.
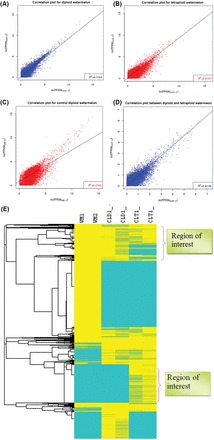
Scatter plot and heat map of RNA-seq expression analysis. Scatter plots of RNA-seq expression data for Sugar Baby, CLD1, and CLT1 replicates showing a high degree of correlation, as expected. Transcripts with ≥1 FPKM were used. The results showed correlation between replications of diploid (CLD1-1 vs CLD1-2) (A), tetraploid (CLT1-1 vs CLT1-2) (B), Sugar Baby (WM-1 vs WM-2) (C), and diploid and tetraploid (CLD1-2 vs CLT1-2) (D). The plot is on a natural log-transformed scale. In the heat map (E), transcript abundance was compared across samples (Sugar Baby 1, Sugar Baby 2, CLD1-1, CLD1-2, CLT1-1, CLT1-2). The yellow boxes represent transcripts with high expression and blue boxes transcripts with low expression.
To observe the transcriptomic difference between the diploid and tetraploid, we drew a heat map using two biological replications of all three samples (Fig. 3E). Two unique regions showed differential regulation between CLD1 and the tetraploid, whereby clusters of genes were up- or downregulated specific to the tetraploid. Of the total transcripts, 6650 were differentially expressed between CLD1 and the tetraploid; 5362 and 1288 genes were upregulated in the tetraploid CLT1 and CLD1, respectively (Fig. 3E, Table 3, and Supplementary Figs S1 and S2 at JXB online). In addition, 2259 genes were upregulated and 611 were downregulated in CLT1 compared with Sugar Baby. In this comparison, 295 transcripts were exclusive to CLT1 and 29 were specifically expressed in Sugar Baby (Fig. 3E and Supplementary Table S3 at JXB online). Our RNA-seq revealed 303 genes upregulated (Supplementary Table S1 at JXB online) and 348 downregulated (Supplementary Table S2 at JXB online) in the tetraploid compared with the diploid, with fold change >2.0 (P<0.001). Compared with CLT1 versus CLD1, CLT1 versus Sugar Baby showed 1287 upregulated and 491 downregulated genes (Supplementary Table S3). Among genes in the CLT1 versus CLD1 comparison, the top 20 upregulated genes in the tetraploid are given in Table 4. The genes included auxin response factor, serine/threonine protein kinase, fructokinase, aminotransferase, and calcium-transporting ATPase. In addition, the top 20 downregulated genes in CLT1 are given in Table 5. The gene list included indole-3-acetic acid-amido synthase, peroxidase, DNA-binding protein, nitrate transporter, WRKY transcription factor, and mitogen-activated protein kinase kinase. Genes such as tubulin β1 chain, pollen Ole e allergen and extensin, and two-component response regulator ARR11 were uniformly upregulated in CLT1 compared with the diploid CLD1 and Sugar Baby (Supplementary Table S4 at JXB online). Nucleoside phosphorylase, pathogenesis-related protein 4B, ABC transporter ATP-binding protein, aquaporin 2, and glycine-rich protein were important among genes downregulated in the tetraploid compared with Sugar Baby (Supplementary Table S5 at JXB online).
Table 3.
Number of genes expressed in diploid and tetraploid watermelon
| Category of genes | CLD1 | CLT1 |
|---|---|---|
| Total upregulated | 1288 | 5362 |
| Log2 fold change (>2.0 to <8.06) | 405 | 1508 |
| Significant P value (<0.001) | 376 | 1261 |
| Corrected Q value (<0.001) | 362 | 908 |
| Annotated | 348 | 302 |
| Expressed only in respective ploidy levels | 37 | 91 |
| Unique genes annotated | 20 | 31 |
Table 4.
List of top 20 upregulated transcripts in tetraploid watermelon and corresponding gene names
| Fold change (log 2) | P value | ICuGI Ref. ID | Gene name |
|---|---|---|---|
| 8.06069 | 0.000213 | Cla000557 | Auxin response factor |
| 6.45777 | 4.57E-11 | Cla001640 | Hydroxycinnamoyl-CoA shikimate/ quinate hydroxycinnamoyl transferase |
| 6.45171 | 3.61E-05 | Cla007846 | Myb-like transcription factor 1 |
| 6.40979 | 6.66E-16 | Cla010329 | S-Ribonuclease |
| 6.39404 | 0.000499 | Cla019574 | α/β-Hydrolase fold protein |
| 6.38678 | 2.02E-12 | Cla010060 | Carboxyl-terminal proteinase |
| 6.08616 | 6.22E-15 | Cla003043 | Pollen Ole e 1 allergen and extensin family protein |
| 5.73977 | 4.01E-11 | Cla016488 | Receptor-like kinase, serine/threonine protein kinase |
| 5.64255 | 0 | Cla016148 | Two-component response regulator ARR11, SHAQKYF class |
| 5.5681 | 5.39E-12 | Cla018530 | Serine/threonine protein kinase |
| 5.56251 | 2.12E-06 | Cla006646 | SAUR-like auxin-responsive protein |
| 5.29933 | 0.011601 | Cla019529 | GPI ethanolamine phosphate transferase, alkaline-phosphatase-like |
| 5.11315 | 3.47E-08 | Cla023265 | Aminotransferase class-III |
| 5.04522 | 1.99E-06 | Cla004401 | Dynamin-like protein |
| 4.99178 | 2.44E-07 | Cla008071 | Fructokinase-like 2, carbohydrate/purine kinase |
| 4.97748 | 6.93E-14 | Cla001615 | Tubulin α1 chain |
| 4.93461 | 0.001169 | Cla003358 | Origin recognition complex subunit 6 |
| 4.73379 | 0.010574 | Cla023303 | Myosin-like protein |
| 4.65835 | 0.011623 | Cla022638 | Calcium-transporting P-type ATPase |
| 4.52862 | 0.000135 | Cla000977 | Pantothenate kinase 2 |
Table 5.
List of top 20 downregulated transcripts in tetraploid watermelon and corresponding gene names
| Fold change (log2) | P value | ICuGI Ref. ID | Gene name |
|---|---|---|---|
| –10.0503 | 0.00386588 | Cla012877 | Enoyl-(acyl-carrier-protein) reductase II;2-nitropropane dioxygenase, NPD |
| –7.90913 | 0 | Cla013991 | Indole-3-acetic acid-amido synthetase |
| –7.49702 | 0 | Cla016287 | 1-Aminocyclopropane-1-carboxylate oxidase |
| –7.02036 | 0 | Cla007431 | Glyoxalase/bleomycin resistance protein/dioxygenase |
| –6.53801 | 0 | Cla003194 | Peroxidase |
| –5.70691 | 0 | Cla003993 | AT-hook DNA-binding protein |
| –5.65104 | 0 | Cla009607 | Aluminum-induced protein |
| –5.39458 | 0 | Cla015292 | Non-specific lipid-transfer protein |
| –5.30548 | 0 | Cla022102 | Phosphate-induced protein 1 |
| –5.16735 | 0 | Cla017623 | U-box domain-containing protein |
| –5.07383 | 0 | Cla012250 | Nitrate transporter |
| –5.04399 | 0 | Cla007389 | Auxin-responsive GH3-like |
| –5.02762 | 1.01E–05 | Cla014736 | Mitochondrial ubiquitin ligase activator of NFKB 1 |
| –5.00521 | 1.25E–11 | Cla004233 | WRKY transcription factor 27 |
| –4.99167 | 0 | Cla014725 | Leucine-rich repeat receptor-like kinase |
| –4.93805 | 1.85E–11 | Cla017001 | Ribonuclease |
| –4.93512 | 1.58E–12 | Cla013951 | Nodulin MtN21 family protein |
| –4.92981 | 1.54E–11 | Cla018437 | Mitogen-activated protein kinase kinase |
| –4.8747 | 3.34E–08 | Cla016627 | Auxin-induced SAUR-like protein |
| –4.86353 | 0 | Cla014161 | Inositol-3-phosphate synthase |
Finally, to evaluate the potential functions of genes with altered transcription levels from three different tissues of CLD1 and CLT1, we performed GO analysis. The genes with a >2-fold expression change in CLT1 and CLD1 were assigned different GO categories such as biological process, molecular function, and cellular location. We have shown only the top 10 categories (Fig. 4). For upregulated genes, biological processes included sucrose metabolism and starch metabolism, whereas for downregulated genes, starch metabolism preceded sucrose metabolism. For molecular function, ATP-binding-related genes from CLD1 and CLT1 were at the top. In the cellular component class, the cell-wall-located proteins were most prominent, followed by cytoplasmic-located proteins.
Fig. 4.
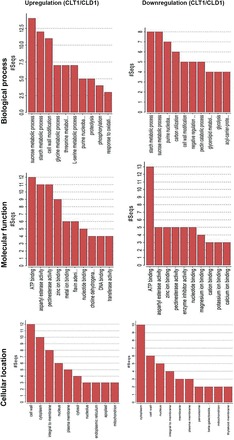
GO of up- and downregulated genes in tetraploid compared with diploid watermelon. Significantly regulated genes were classified by biological process, molecular function, and cellular location. Most of the genes are specific for proteins in the cytoplasm and cell wall for energy metabolism. (This figure is available in colour at JXB online.)
We then investigated the expression profile of genes involved in multiple signalling such as hormone, cell division, and storage, with the RNA-seq data supported by RT-qPCR. We focused on a few genes among both up- and downregulated genes in terms of expression in the tetraploid, and compared the expression of key genes across leaf, stem, and fruit tissues between the diploid and tetraploid (Table 6). The leaf showed higher upregulation of cell-division and growth-related genes (dynamin-like protein C and amine oxidase) and light receptors (chlorophyll a/b-binding protein). Similarly, genes related to iron storage, signal transduction, and aquaporin were upregulated in tetraploid stems. Most of the genes were downregulated in fruit tissues, especially GPI ethanolamine phosphate transferase. These results showed that groups of certain genes were uniquely up- or downregulated in particular tissues because of changes in ploidy level.
Table 6.
RT-qPCR analysis of genes that differentially expressed across tissues in tetraploid compared with diploid watermelon
| Gene name | ICuGI Ontology/Function | Leaf | Stem | Fruit |
|---|---|---|---|---|
| Origin recognition complex subunit | Origin of replication/Recruitment of Cdc6 | 3.93±1.57 | 6.74±1.38 | –5.72±0.06 |
| GPI ethanolamine phosphate transferase 2 | Anchoring to the cell membrane | 1.98±0.11 | 1.46±0.21 | –585.34±0.00 |
| Serine-threonine protein kinase | Signal transduction | 2.74±0.32 | 86.42±7.34 | –1.40±0.31 |
| Inositol-3-phosphate synthase | Inositol biosynthesis | –49.61±0.00 | 1.43±0.60 | –6.36±0.05 |
| Indole-3-acetic acid-amido synthetase | Auxin homeostasis/disease resistance | 1.49±0.10 | –2.89±0.06 | –5.07±0.01 |
| Non-specific lipid-transfer protein 8 | Membrane transfer of phospholipids | 1.91±0.32 | –3.39±0.05 | –5.49±0.02 |
| Ferritin-2 | Iron storage and availability | –8.78±0.02 | 38.58±13.73 | –84.90±0.00 |
| Amine oxidase family protein | Deamination of polyamines required for cell growth | 28.81±1.91 | 9.21±5.56 | –1.21±0.25 |
| Phloem filament protein | Major protein composing phloem filaments | 4.56±1.18 | –2.63±0.04 | –3.46±0.14 |
| Cellulose synthase | Production of cellulose | 1.63±0.23 | 61.97±7.62 | –1.33±0.18 |
| MLP-like protein 28 | Defence response | 1.36±0.14 | –2.97±0.02 | –8.00±0.04 |
| Chloroplastic chlorophyll a-b-binding protein P4 | Light receptors for photosystems | 20.34±1.04 | 3.33±0.86 | –10.45±0.04 |
| Aquaporin PIP2.1 | Petal expansion/regulates water channels | 2.44±0.07 | 41.58±18.74 | 1.04±0.16 |
| ABC transporter A family member 8 | Regulates pump activity in membranes | 1.50±0.18 | –10.31±0.08 | –3.84±0.01 |
| Ethylene-responsive proteinase inhibitor 1 | Inhibition of protease activity | 1.14±0.20 | –2.67±0.17 | –4.60±0.03 |
| Cellulose synthase-like protein H1-like isoform 1 | Cellulose production | 2.64±0.10 | 4.05±0.44 | –21.89±0.01 |
| Ubiquitin ligase protein cop1 | Polyubiquitination/protein degradation | –6.98±0.01 | –6.53±0.02 | –15.20±0.01 |
| Dynamin-like protein C | Cell division/membrane trafficking | 113.66±23.43 | –5.15±0.02 | –8.48±0.01 |
| Serine carboxypeptidase | Protein cleavage/protein turnover | –878.63±0.00 | 77.87±32.89 | –17.47±0.01 |
Data are means±standard deviation relative to internal control 18S rRNA. Bold indicates upregulated in CLT1; italic indicates downregulated in CLT1.
Our pathway analysis of CLT1/CLD1 and CLT1/Sugar Baby included argininosuccinate synthases, glutamate dehydrogenase, and ornithine aminotransferase of the arginine biosynthesis pathway; magnesium chelatase subunit, protoporphyrinogen IX oxidase, and protochlorophyllide reductase of chlorophyllide synthesis; mannose-6-phosphate isomerase, mannose-1-phosphate guanyltransferase, and glucose-6-phosphate isomerase of GDP mannose biosynthesis; and members of trehalose biosynthesis, and starch and sucrose degradation. In addition, mRNA levels of enzymes such as carbamoyl-phosphate synthase, argininosuccinate lyase, and argininosuccinate synthase of the arginine biosynthesis pathway, which is part of the key pathway of α-amino acid citrulline metabolism, were upregulated ~1.5–2.4 times. A few more genes in the ascorbate gluthathione cycle, which is part of ethylene biosynthesis for fruit ripening, were almost equally expressed in both ploids (Supplementary Table S6 at JXB online).
Genome duplication spatially regulates AS in watermelon
We identified transcripts with AS due to genome duplication. The effect of whole-genome duplication on AS with autopolyploid is not known, although a few studies have investigated allopolyploidy or duplication of few genes. To this end, we identified 22 genes with transcript isoforms from RNA-seq data analyses and tested leaf, stem, and fruit tissues between the diploid and tetraploid (Table 7). The sequences of all transcript variants of genes were first aligned manually with their corresponding genomic sequence using Spidey and then mapped (Fig. 5). For these 22 genes, few transcripts showed the exact spliced transcript given in the watermelon reference genome sequence, and the remaining transcript variants were new. This information will enrich the transcriptome data for watermelon and its close relatives. Among these spliced genes, phloem protein 2 (PP2), the most abundant and enigmatic protein in the phloem sap, can exert effects over long-distance translocation with its lectin activity or RNA-binding properties. Rhodanese domain protein is closely related to plant senescence-associated proteins. A few gene families such as copine and dynamin regulate multiple functions during growth and development. Copines are Ca2+-dependent phospholipid-binding proteins thought to be involved in membrane trafficking and may also be involved in cell division and growth. Dynamin is a microtubule-associated force-producing protein involved in the production of microtubule bundles and is able to bind and hydrolyse GTP; it is implicated in endocytic protein sorting and cell division.
Table 7.
List of AS genes showing spatial expression in diploid (CLD1) and tetraploid (CLT1) watermelon
| Transcript | Locus | Location on chromosome | Gene name | Splicing variation |
|---|---|---|---|---|
| TSS14582 | Cla001006 | Chr 1:25897061–25900419 | Phloem protein 2 | ES, IR |
| TSS72233 | Cla013509 | Chr 2:28638066–28642630 | Protein disulfide isomerase family | 5ʹ-TSS, 5ʹ-ASS, 3ʹ-PA |
| TSS76921 | Cla008582 | Chr 2:32886676–32892905 | Rhodanese domain protein | 5ʹ-TSS, 3ʹ-ASS, 3ʹ-PA |
| TSS95362 | Cla008232 | Chr 3:1324441–1327387 | PGR5-like protein 1B, chloroplastic | ES, IR |
| TSS93444 | Cla009531 | Chr 3:15383157–15393213 | Zinc finger CCCH domain-containing protein 16 | ES, 3ʹ-PA |
| TSS110401 | Cla008071 | Chr 4:2707053–2711710 | Fructokinase-like 2 | 3ʹ-PA |
| TSS150556 | Cla012250 | Chr 6:19655212–19659396 | Nitrate transporter | 5ʹ-TSS, IR, 3ʹ-PA |
| TSS153874 | Cla012247 | Chr 6:19680362–19683167 | Oxidoreductase zinc-containing alcohol dehydrogenase family | 5ʹ-TSS, IR, 3ʹ-PA |
| TSS151927 | Cla019245 | Chr 6:26359223–26361665 | Abhydrolase domain-containing protein FAM108B1 | 3ʹ-PA |
| TSS152633 | Cla006689 | Chr 6:3238334–3241279 | Unknown protein | IR |
| TSS168906 | Cla012755 | Chr 7:26133714–26138192 | Golgi SNAP receptor complex member 1 | 3ʹ-PA |
| TSS172092 | Cla012768 | Chr 7:26308321–26311730 | SEC14 cytosolic factor family protein | 5ʹ-TSS, IR, 3ʹ-PA |
| TSS171012 | Cla011845 | Chr 7:8984422–8997770 | MYB transcription factor | ES |
| TSS186970 | Cla013951 | Chr 8:14502524–14509196 | Nodulin MtN21 family protein | IR, 3ʹ-PA |
| TSS188951 | Cla022427 | Chr 8:23526156–23527378 | Gibberellin-regulated protein 1 | 5ʹ-TSS, IR, 3ʹ-PA |
| TSS203418 | Cla015387 | Chr 9:1996822–2002776 | LOC398447 protein | 5ʹ-TSS, ES, IR, 3ʹ-PA |
| TSS206432 | Cla004401 | Chr 9:26340026–26346617 | Dynamin-like protein | IR, ES, 3ʹ-PA |
| TSS206875 | Cla010408 | Chr9:31338176–31341053 | Cinnamoyl CoA reductase-like protein | IR, 3ʹ-PA |
| TSS207932 | Cla015518 | Chr 9:838662–848405 | Polyphosphoinositide phosphatase | IR |
| TSS54907 | Cla005769 | Chr 11:11671132–11676692 | Copine-3 | IR, 3ʹ-PA |
| TSS54151 | Cla002053 | Chr 11:475350–479230 | DNA-directed RNA polymerase II subunit 3 | IR, 3ʹ-PA |
| TSS50731 | Cla001640 | Chr 11:5645850–5652537 | Hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase | 5ʹ-TSS, IR, 3ʹ-PA |
Chr, chromosome; 5ʹ-TSS, 5ʹ transcription start site; ES, exon skipping; IR, intron retention; 5ʹ-ASS, 5ʹ alternative splice site; 3ʹ-PA, 3ʹ poly(A) sites, as revealed by RNA-seq.
Fig. 5.

Transcript variants of products with AS between diploid and tetraploid watermelon using Spidey. Differentially spliced transcripts from RNA-seq were aligned manually to the corresponding locus of the International Cucurbit Genomics Initiative (ICuGI) genome using Spidey. The darker lines indicate genomic regions of the respective locus based on the start and end of transcripts. All forms of splicing variation are mapped into corresponding loci. Primers used in this study (Supplementary Table S7) for semi-quantitative RT-PCR are marked. The ICuGI locus number is given above each gene.
* indicates the annotated mRNA transcript from the database identified from RNA-seq data.
To functionally validate and identify which tissues of the diploid and tetraploid had these specific isoforms, we performed semi-quantitative RT-PCR of the 22 genes with unique primer sets (Supplementary Table S7) that discriminated alternative forms (Figs 5 and 6). In general, we divided splice variants into three subcategories based on splicing modulation. The first group showed similar patterns across tissues and across ploidy. The second group exhibited splicing variation only across tissues (spatially), regardless of ploidy level. Finally, the third group was unique to the diploid or tetraploid for certain tissues. Splice variants of Cla012755 and Cla012768 existed in all tissues, with varied expression among tissues. However, spatial splicing differences occurred in selective tissues: the first transcript of Cla008071 and Cla012250, second transcript of Cla002053 and Cla006689 and three forms of Cla022427 expressed preferably in fruit tissue of both ploidy levels. Similarly, the first transcript of Cla012247 was preferentially spliced and expressed in leaves. Thus, AS occurred spatially among different tissues at both ploidy levels.
Fig. 6.

Validation of products with AS of selected genes by semi-quantitative RT-PCR with unique sets of primers. The amplified products were obtained by PCR using location-specific forward and reverse primers (see Supplementary Table S7 for primer sequences and Fig. 5 for locations) to differentiate splice variants. The lanes from left to right are marker (M), leaf (L), stem (S) and fruit (F) of the diploid (CLD1) and tetraploid (CLT1), and genomic DNA (gD). Amplicons were the products of 26–34 cycles for different genes. Elongation Factor1 (Cla004730) was an internal control. Arrows indicate faint PCR products as well as very close amplicons.
We found remarkable AS patterns across ploidy for the same tissues. The RT-PCR results indicated that ploidy regulated AS across different tissues (Fig. 6). We observed these unique differences for at least one or more of the transcripts of PP2, chloroplastic PGR5-like protein, zinc-finger protein, fructokinase-like 2, MYB transcription factor in fruit, and nodulin MtN21 in leaf and fruit tissues. Exon skipping of zinc-finger protein occurred in diploid stem and tetraploid fruit and chloroplastic PGR5-like protein in tetraploid fruit tissue. In addition, alternative poly(A) sites of the dynamin, nitrate transporter, nodulin MtN21, and abhydrolase genes were absent in tetraploid fruits, and RT-PCR confirmed the existence of alternative poly(A) sites (Fig. 6). These results further indicated that ploidy-specific AS exists mostly in fruit tissue. We also provided a tissue-related expression profile of genes with AS using RT-qPCR with unique primers (Fig. 7). RT-qPCR results were similar to RT-PCR results across the diploid and tetraploid when considering the cumulative expression of all transcripts. Because of its high sensitivity, RT-qPCR could be used with primers designed in designated exons to ascertain the reliability of transcripts with AS between the diploid and tetraploid. The RT-qPCR results agreed with the RT-PCR findings for most of the transcripts (Fig. 8). Some tissues did not give threshold cycle (C T) values, possibly because of a low level of expression or the absence of transcripts (Fig. 8, labelled ND).
Fig. 7.
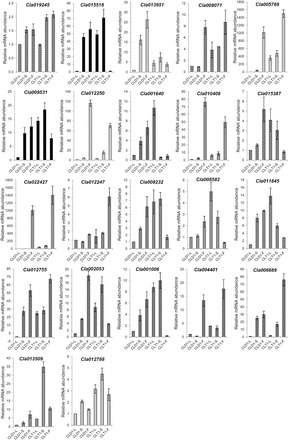
Cumulative expression of loci with AS between diploid and tetraploid watermelon quantified by RT-qPCR. Validation of RNA-seq data and semi-quantitative RT-PCR by RT-qPCR. Three technical and three biological replicates were performed for leaf (L), stem (S), and fruit (F) tissues from the diploid and tetraploid. Expression of the calibrator sample (diploid leaf; CLD1-L) was set to 1. The expression of each gene is the cumulative value of all transcripts per se, and unique primer sets were designed in common exon region. Data are means±standard deviation from three biological replicates.
Fig. 8.
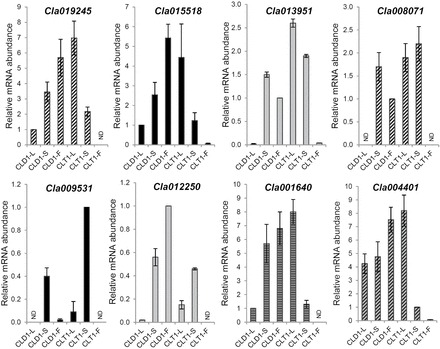
Quantitative expression of selected splicing variants of a few spliced loci of watermelon quantified by RT-qPCR. Validation of semi-quantitative RT-PCR results with use of RT-qPCR for splicing analysis. Three technical and three biological replicates were used for leaf (L), stem (S), fruit (F) tissues from diploid and tetraploid. A few genes for certain tissues lacked a C T value and are labelled as non-detectable (ND). The expression of any one of the samples was set to 1 for different genes. Primer sets were designed in exons with AS variants that were unique to certain transcripts. Data are means±standard deviation from three biological replicates.
Finally, we performed RACE-PCR to map the transcript ends in different tissues. To validate splicing variation, four genes namely, nitrate transporter (Cla012250), nodulin family protein (Cla013951), polyphosphoinositide phosphatase (Cla015518), and fructokinase zinc finger (Cla008071), were amplified with a RACE-specific adaptor and sequence-specific primers. Transcript variants of rhodanese domain protein, zinc-finger protein, and MYB transcription factor, showing exon skipping/intron retention/5ʹ-alternative splice site/3ʹ-alternative splice site (internal splicing variation) were amplified from RT-PCR directly. All amplicons from RT-PCR and RACE-PCR were sequenced and aligned to corresponding genomic loci. The translated proteins from these altered variants may lead to variation in protein isoforms with additional, missing, or changed amino acids. Together, our transcriptome results suggested that genes are expressed spatially in different tissues across ploidy, and that whole-genome duplication plays a crucial ploidy-dependent role in AS of at least a few genes in selected tissues. Spatially regulated AS in the watermelon genome depends on ploidy. This AS difference among tissues across ploidy might be due to differential expression, which has to be explored.
Discussion
Watermelon is one of the unique cultivars in which polyploid is used routinely to generate seedless fruit types. Ploidy levels alter plant architecture and fruit quality significantly, so studying differential gene expression and AS across various ploidy levels of watermelon is important. Knowledge of altered gene expression and AS has potential use in altering fruit quality known to occur among various ploidy levels.
In this study, we addressed the impact of gene duplication on AS and the transcriptome profile using a diploid and its tetraploid watermelon. Results from ploidy analysis (Fig. 1) confirmed that CLT1s are true tetraploids of CLD1. Despite the increased plant growth as well as fruit size that have been documented in tetraploid watermelon (Ahmad et al., 2013), we observed that the leaves, stems, and flowers were larger in the tetraploid than in the diploid but that fruit size was similar. Alteration of the transcriptome might somehow modify phenotypic variation, thus affecting overall plant growth and development.
The unique mapping rate of RNA-seq reads in the current study was 43%. This is an acceptable mapping rate for the following reasons: (i) the mapping was done against a diploid watermelon genome of an elite Chinese ecotype 97103 (Guo et al., 2013), which is different from the genome of the American tetraploid ecotypes used in this study; (ii) it is within the general range of RNA-seq mapping rates of between 34 and 60% in several other plant species (Severin et al., 2010; Chettoor et al., 2014; Liu et al., 2014; Thakare et al., 2014; Wang et al., 2014). We recently published a high-resolution map (Reddy et al., 2014) with 10 480 single-nucleotide polymorphisms made using a cross of other ecotypes and found that the map was not fully collinear with the existing whole-genome sequence. In addition, the tetraploid genotype does not have complete whole-genome assembly and annotation. However, given the large total number of reads sequenced per sample, the mapped reads selected for further downstream analyses were determined to be of sufficient quality and quantity (Table 1).
Studying gene expression is less complicated for autopolyploid than allopolyploid plants because allopolyploidy results in two homeologous genomes, such as in cotton (Udall et al., 2006). From our transcriptome data, two interesting regions from the heat map that belonged to up- and downregulated genes in the tetraploid compared with the diploid were found. Several genes were upregulated (with ≥4-fold change in expression) in the tetraploid (5362 genes) compared with CLD1 (1288 genes). Studies in a soybean allotetraploid showed a 1.4-fold larger transcriptome than its diploid progenitor, and the tetraploid had >87% of its genes duplicated (Coate and Doyle, 2010). The genes that were up- and downregulated agreed with their copy-number changes (Zhou et al., 2011a ), as occurred for most proteins in a linear fashion in maize ploidy (Yao et al., 2011). The overall increased expression pattern in tetraploids was more likely due to duplicated genes than single-copy genes (Gu et al., 2004). Of 34 members of the cytochrome P450 family, 29 were upregulated in CLT1 compared with CLD1, which explains their plausible involvement in multiple biochemical pathways such as biosynthesis of phenylpropanoids, terpenoids, alkaloids, and lipids (Chapple, 1998).
The levels of protein divergence between diploid and allotetraploid watermelon were relatively high compared with between diploids and autotetraploids in Arabidopsis (Ng et al., 2012). However, subsequent proteomic studies with our diploid and tetraploid tissues will show the diversity of the proteome including proteins due to AS in watermelon. A few genes such as UDP glucose pyrophosphorylase and glycogen/starch/α-glucan phosphorylase in the sucrose synthesis pathway were upregulated, whereas nitrate transporter, IAA-amido synthase, WRKY transcription factor, and nodulin MtN21 were downregulated in CLT1 compared with CLD1. The same set of genes that was highly expressed in Arabidopsis was repressed in the resynthesized allotetraploids (Chen, 2007). Comparative analysis of gene expression between CLT1/CLD1 and CLT1/Sugar Baby showed that most genes were upregulated, including the light-harvesting complex consisting of chlorophylls a and b and the chlorophyll a/b-binding proteins in tetraploids. Therefore, stomatal count from chloroplasts can be used as a rapid method to identify polyploids (Krishnaswami and Andal, 1978; Noh et al., 2012). In tetraploid watermelon, cell-cycle-related genes such as cell-division cycle protein 123-like and cell-division control proteins were downregulated, which might be due to an indirect relationship between genome size and mitotic cell division (Bennett, 1977; Cavalier-Smith, 2005).
A previous study showed that an increased number of chromosomes can result in a new type of epigenetic gene inactivation, thereby creating differences in gene-expression patterns (Mittelsten Scheid et al., 1996). In CLT1, chromatin remodelling subunit (Cla005113), a member of the SWI/SNF complex regulating FLOWERING LOCUS C in Arabidopsis (Jegu et al., 2014), was upregulated. The absence or low expression of some transcripts in CLT1 might have been caused by gene silencing or gene loss associated in part with cytosine methylation. Certain genes were expressed only in allopolyploids of Arabidopsis suecica and wheat, and were not detected in the parents (Kashkush et al., 2002; Madlung et al., 2002). Most of the genes that are involved in sucrose and starch metabolism and ATP-binding activity have cell-wall and cytoplasmic-specific cellular locations. Many critical pathways including those involved in amino acid biosynthesis were altered in the tetraploid watermelon compared with Sugar Baby and CLD1. Our study also explained differential expression patterns of several genes involved in energy-harvesting pathways. Several pathways including argininosuccinate synthases, glutamate dehydrogenase, and ornithine aminotransferase of the arginine biosynthesis pathway; magnesium chelatase subunit, protoporphyrinogen IX oxidase, and protochlorophyllide reductase of chlorophyllide synthesis; mannose-6-phosphate isomerase, mannose-1-phosphate guanyltransferase, and glucose-6-phosphate isomerase of GDP mannose biosynthesis; and members of trehalose biosynthesis, starch and sucrose degradation were altered in autotetraploids.
The ‘one gene to one protein’ paradigm was rewritten with the discovery of intervening sequences in eukaryotic genes and further splicing of introns (Berget et al., 1977). AS gave rise to multiple transcripts through seven types of cotranscriptional and post-transcriptional events. Different developmental cues and environmental signals could regulate AS in plants via many potential mechanisms (Reddy et al., 2013). AS plays critical roles in abiotic stress (Arabidopsis SAD1), biotic stress (tobacco Toll-interleukin1 receptor N gene, Arabidopsis RPS4), hormonal regulation (Arabidopsis Serine/Arginine-rich protein), development (Arabidopsis transporter ZIFL1, maize Vp1, Arabidopsis SUA), flowering time (Arabidopsis MAF1 and SVP), and the circadian clock response (Arabidopsis CCA1 and PRR9) in plants (Staiger and Brown, 2013).
Genome duplication along with AS could lead to proteome diversity in plants. However, little is known about the existence and significance of AS in autopolyploids. Our study showed that differential splicing occurred in 22 genes, including phloem protein2, zinc-finger CCCH domain protein, nodulin family protein, gibberellin-regulated protein, and nitrate transporter. Spatial regulation of these genes was confirmed among different tissues. Specifically, RT-qPCR analysis supported the existence of certain transcripts with AS in various tissues between the diploid and tetraploid (Fig. 8). We used RACE-PCR to further map the full-length transcripts of a few genes (nitrate transporter, zinc-finger CCCH domain protein, and fructokinase-like 2) between leaf and young fruit tissues. Most of the spliced variants distinguished in diploid and tetraploid plants were observed in fruit tissue rather than in the leaf and stem. Thus, AS between the diploid and tetraploid plants occurred tissue specifically. Studies of Brassica allopolyploid showed that allopolyploidy generated myriad qualitative and quantitative differences in AS events, and several thousand transcript forms with AS were gained or lost in polyploids (Tack et al., 2014).
In this study, we observed a few genes with tissue-specific splicing variants in synthetic autopolyploids. Deregulation of some important pre-mRNA splicing factors such as cleavage and polyadenylation specificity factor subunit, splicing factor subunit U2af, and pre-mRNA splicing factor (coiled-coil domain-containing protein Cwf18) might play roles in splicing. In addition, factors such as histone acetyl transferase and RNA polymerase II were involved in AS. However, most of these factors showed expression in the range of 1.3–2.0-fold change.
The response of the duplicated-genome dosage effect on expression per se has to be considered to accurately quantify the transcriptome in allopolyploid plants (Coate and Doyle, 2010). Previously, Guo et al. (1996) established a 1:1 dosage effect for the expression of many genes in maize autopolyploid with the exception of negative dosage effects for a few genes. Because our research focus was a synthetic autopolyploid, we validated read numbers from RNA-seq with RT-qPCR results. In addition, the strength of our transcriptome comparison between diploid and autotetraploids relied on the results of tissue-related AS. Our AS data qualitatively support the existence of splicing variants due to genome duplication in watermelon.
The critical question to answer is why we found no difference in fruit traits between the diploid and tetraploid. However, most of the spliced variants we observed showed differences among the fruit tissues between ploidy levels. Similarly, several transcripts were downregulated in autotetraploid fruits. Post-transcriptional regulation events and post-translational modifications in polyploids have been widely studied (Koh et al., 2012). This phenomenon of post-transcriptional or translational modifications in polyploids is termed transcriptomic shock (Buggs et al., 2009). Understanding why the whole-genome duplication did not alter fruit size would be of interest. Transcriptome analyses over the fruit development of diploid, triploid, and tetraploid watermelons from pollination to maturity along with detailed qualitative trait observation would help clarify this issue.
To conclude, we provide evidence that autotetraploidization can alter the transcriptome and modulate AS in watermelon. This finding might help polyploid-plant biologists in general and cucurbit researchers specifically to further explore gene regulation among the autopolyploids.
Supplementary data
Supplementary data are available at JXB online.
Supplementary Table S1. List of upregulated genes in CLT1 compared with CLD1.
Supplementary Table S2. List of downregulated genes in CLT1 compared with CLD1.
Supplementary Table S3. Transcripts up- and downregulated in CLT1 vs WM.
Supplementary Table S4. Top 20 genes upregulated in tetraploid CLT1 compared with Sugar Baby.
Supplementary Table S5. Top 20 genes downregulated in tetraploid CLT1 compared with Sugar Baby.
Supplementary Table S6. List of different pathways altered in tetraploid compared to diploid.
Supplementary Table S7. List of primers used for RT-PCR and RT-qPCR.
Acknowledgements
This project was supported by the USDA-NIFA (no. 2013-38821-21453), NSF-EPSCOR (no. 1003907), Gus R. Douglass Institute, and NIH (no. P20RR016477). The authors are grateful to R. Jarret, PGRCU, USDA-ARS (Griffin, GA, USA) for providing the seeds for the germplasm accessions.
Glossary
Abbreviations:
- AS
alternative splicing
- FPKM
fragments per kilobase of exon per million fragments mapped
- GO
gene ontology
- RACE
rapid amplification of cDNA ends
- RNA-seq
RNA-sequencing
- RT-qPCR
quantitative reverse transcription PCR
- TSS
transcription start site.
References
- Ahmad I, Hussain T, Nafees M, et al. 2013. Morphological dissimilarity between tetraploid and diploid watermelon (Citrullus lanatus Thunb.). World Applied Sciences Journal 21, 858–861. [Google Scholar]
- Albertin W, Brabant P, Catrice O, Eber F, Jenczewski E, Chevre AM, Thiellement H. 2005. Autopolyploidy in cabbage (Brassica oleracea L.) does not alter significantly the proteomes of green tissues. Proteomics 5, 2131–2139. [DOI] [PubMed] [Google Scholar]
- Andrus CF, Seshadri VS, Grimball PC. 1971. Production of seedless watermelons. Agricultural Research Service, Technical Bulletin , Vol. 1425: US Department of Agriculture. [Google Scholar]
- Barbazuk WB, Fu Y, McGinnis KM. 2008. Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Research 18, 1381–1392. [DOI] [PubMed] [Google Scholar]
- Bennett MD. 1977. The time and duration of meiosis. Philosophical Transactions of the Royal Society B: Biological Sciences 277, 201–226. [DOI] [PubMed] [Google Scholar]
- Berget SM, Moore C, Sharp PA. 1977. Spliced segments at the 5ʹ terminus of adenovirus 2 late mRNA. Proceedings of the National Academy of Sciences, USA 74, 3171–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggs RJA, Doust AN, Tate JA, Koh J, Soltis K, Feltus FA, Paterson AH, Soltis PS, Soltis DE. 2009. Gene loss and silencing in Tragopogon miscellus (Asteraceae): comparison of natural and synthetic allotetraploids. Heredity (Edinburgh) 103, 73–81. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. 2005. Economy, speed and size matter: evolutionary forces driving nuclear genome miniaturization and expansion. Annals of Botany 95, 147–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple C. 1998. Molecular-genetic analysis of plant cytochrome P450-dependent monooxygenases. Annual Review of Plant Physiology and Plant Molecular Biology 49, 311–343. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Ni Z. 2006. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays 28, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. 2007. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annual Review of Plant Biology 58, 377–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chettoor A, Givan S, Cole R, Coker C, Unger-Wallace E, Vejlupkova Z, Vollbrecht E, Fowler J, Evans M. 2014. Discovery of novel transcripts and gametophytic functions via RNA-seq analysis of maize gametophytic transcriptomes. Genome Biol 15, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate JE, Bar H, Doyle JJ. 2014. Extensive translational regulation of gene expression in an allopolyploid (Glycine dolichocarpa). Plant Cell 26, 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate JE, Doyle JJ. 2010. Quantifying whole transcriptome size, a prerequisite for understanding transcriptome evolution across species: an example from a plant allopolyploid. Genome Biology and Evolution 2, 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L. 2000. Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12, 1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L. 2005. The advantages and disadvantages of being polyploid. Nature Reviews Genetics 6, 836–846. [DOI] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S. 2004. Open source clustering software. Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Dinesh-Kumar SP, Baker BJ. 2000. Alternatively spliced N resistance gene transcripts: their possible role in tobacco mosaic virus resistance. Proceedings of the National Academy of Sciences, USA 97, 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezel J, Bartos J. 2005. Plant DNA flow cytometry and estimation of nuclear genome size. Annals of Botany 95, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, Wendel JF. 2008. Evolutionary genetics of genome merger and doubling in plants. Annual Review of Genetics 42, 443–461. [DOI] [PubMed] [Google Scholar]
- Feldman M, Levy AA. 2009. Genome evolution in allopolyploid wheat—a revolutionary reprogramming followed by gradual changes. Journal of Genetics and Genomics 36, 511–518. [DOI] [PubMed] [Google Scholar]
- Flagel L, Udall J, Nettleton D, Wendel J. 2008. Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biology 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeta RT, Yoo SY, Pires JC, Doerge RW, Chen ZJ, Osborn TC. 2009. Analysis of gene expression in resynthesized Brassica napus allopolyploids using Arabidopsis 70mer oligo microarrays. PLoS One 4, e4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski P. 2002. Alternative splicing in parallel. Nature Biotechnology 20, 346–347. [DOI] [PubMed] [Google Scholar]
- Gu Z, Rifkin SA, White KP, Li WH. 2004. Duplicate genes increase gene expression diversity within and between species. Nature Genetics 36, 577–579. [DOI] [PubMed] [Google Scholar]
- Guo M, Davis D, Birchler JA. 1996. Dosage effects on gene expression in a maize ploidy series. Genetics 142, 1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zhang J, Sun H, et al. 2013. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nature Genetics 45, 51–58. [DOI] [PubMed] [Google Scholar]
- Hu Z-L, Bao J, Reecy JM. 2008. CateGOrizer: A web-based program to batch analyze gene ontology classification categories. Online Journal of Bioinformatics 9, 108–112. [Google Scholar]
- Ilut DC, Coate JE, Luciano AK, Owens TG, May GD, Farmer A, Doyle JJ. 2012. A comparative transcriptomic study of an allotetraploid and its diploid progenitors illustrates the unique advantages and challenges of RNA-seq in plant species. American Journal of Botany 99, 383–396. [DOI] [PubMed] [Google Scholar]
- Jackson S, Chen ZJ. 2010. Genomic and expression plasticity of polyploidy. Current Opinion in Plant Biology 13, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegu T, Latrasse D, Delarue M, Hirt H, Domenichini S, Ariel F, Crespi M, Bergounioux C, Raynaud C, Benhamed M. 2014. The BAF60 subunit of the SWI/SNF chromatin-remodeling complex directly controls the formation of a gene loop at FLOWERING LOCUS C in Arabidopsis. Plant Cell 26, 538–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush K, Feldman M, Levy AA. 2002. Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 160, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nature Reviews Genetics 11, 345–355. [DOI] [PubMed] [Google Scholar]
- Kirhara H. 1951. Triploid watermelons. Proceedings of the American Society for Horticultural Science 58, 217–230. [Google Scholar]
- Koh J, Chen S, Zhu N, Yu F, Soltis PS, Soltis DE. 2012. Comparative proteomics of the recently and recurrently formed natural allopolyploid Tragopogon mirus (Asteraceae) and its parents. New Phytologist 196, 292–305. [DOI] [PubMed] [Google Scholar]
- Krishnaswami R, Andal R. 1978. Stomatal chloroplast number in diploids and polyploids of Gossypium . Proceedings of the Indian Academy of Sciences 87, 109–112. [Google Scholar]
- Langham RJ, Walsh J, Dunn M, Ko C, Goff SA, Freeling M. 2004. Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 166, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Missirian V, Zinkgraf M, Groover A, Filkov V. 2014. Evaluation of experimental design and computational parameter choices affecting analyses of ChIP-seq and RNA-seq data in undomesticated poplar trees. BMC Genomics 15, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔC T method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Madlung A, Masuelli RW, Watson B, Reynolds SH, Davison J, Comai L. 2002. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiology 129, 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, De Bodt S, Raes J, Casneuf T, Van Montagu M, Kuiper M, Van de Peer Y. 2005. Modeling gene and genome duplications in eukaryotes. Proceedings of the National Academy of Sciences, USA 102, 5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelsten Scheid O, Jakovleva L, Afsar K, Maluszynska J, Paszkowski J. 1996. A change of ploidy can modify epigenetic silencing. Proceedings of the National Academy of Sciences, USA 93, 7114–7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J, Kaufmann K, Theissen G, Nei M. 2005. A simple method for predicting the functional differentiation of duplicate genes and its application to MIKC-type MADS-box genes. Nucleic Acids Research 33, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DW, Zhang C, Miller M, Shen Z, Briggs SP, Chen ZJ. 2012. Proteomic divergence in Arabidopsis autopolyploids and allopolyploids and their progenitors. Heredity (Edinb) 108, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh J, Sheikh S, Chon HG, Seong MH, Lim JH, Lee SG, Jung GT, Kim JM, Ju H-J, Huh YC. 2012. Screening different methods of tetraploid induction in watermelon [Citrullus lanatus (thunb.) Manst. and Nakai]. Horticulture, Environment, and Biotechnology 53, 521–529. [Google Scholar]
- Rastogi S, Liberles DA. 2005. Subfunctionalization of duplicated genes as a transition state to neofunctionalization. BMC Evolutionary Biology 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AS, Marquez Y, Kalyna M, Barta A. 2013. Complexity of the alternative splicing landscape in plants. Plant Cell 25, 3657–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy UK, Nimmakayala P, Levi A, et al. 2014. High-resolution genetic map for understanding the effect of genome-wide recombination rate on nucleotide diversity in watermelon. G3 (Bethesda) pii: g3.114.012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy E, Cabrito TR, Baster P, Batista RA, Teixeira MC, Friml J, Sa-Correia I, Duque P. 2013. A major facilitator superfamily transporter plays a dual role in polar auxin transport and drought stress tolerance in Arabidopsis. Plant Cell 25, 901–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, Stacey G, Doerge RW, Jackson SA. 2013. The fate of duplicated genes in a polyploid plant genome. Plant Journal 73, 143–153. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ. 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Severin A, Woody J, Bolon Y-T, et al. 2010. RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 10, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger D, Brown JW. 2013. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25, 3640–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed NH, Kalyna M, Marquez Y, Barta A, Brown JWS. 2012. Alternative splicing in plants—coming of age. Trends in Plant Science 17, 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack D, Darracq A, Adams KL. 2014. Abstract W629: Reshaped patterns of alternative splicing after allopolyploidy in Brassica napus . In Plant and Animal Genome XXII , 11–15 January, San Diego, CA, USA. [Google Scholar]
- Thakare D, Yang R, Steffen JG, Zhan J, Wang D, Clark RM, Wang X, Yadegari R. 2014. RNA-Seq analysis of laser-capture microdissected cells of the developing central starchy endosperm of maize. Genomics Data 2, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangasamy S, Chen PW, Lai MH, Chen J, Jauh GY. 2012. Rice LGD1 containing RNA binding activity affects growth and development through alternative promoters. Plant Journal 71, 288–302. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udall JA, Swanson JM, Nettleton D, Percifield RJ, Wendel JF. 2006. A novel approach for characterizing expression levels of genes duplicated by polyploidy. Genetics 173, 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veron AS, Kaufmann K, Bornberg-Bauer E. 2007. Evidence of interaction network evolution by whole-genome duplications: a case study in MADS-box proteins. Molecular Biology and Evolution 24, 670–678. [DOI] [PubMed] [Google Scholar]
- Vitulo N, Forcato C, Carpinelli EC, et al. 2014. A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype. BMC Plant Biology 14, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters B, Lum G, Sablok G, Min XJ. 2013. Genome-wide landscape of alternative splicing events in Brachypodium distachyon . DNA Research 20, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tian L, Lee HS, et al. 2006. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cao C, Ma Q, et al. 2014. RNA-seq analyses of multiple meristems of soybean: novel and alternative transcripts, evolutionary and functional implications. BMC Plant Biology 14, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang X, Paterson AH. 2012. Genome and gene duplications and gene expression divergence: a view from plants. Annals of the New York Academy of Sciences 1256, 1–14. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HP, Su YS, Chen HC, Chen YR, Wu CC, Lin WD, Tu SL. 2014. Genome-wide analysis of light-regulated alternative splicing mediated by photoreceptors in Physcomitrella patens . Genome Biology 15, R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang H, Li L. 2012. Alternative mRNA processing increases the complexity of microRNA-based gene regulation in Arabidopsis. Plant Journal 70, 421–431. [DOI] [PubMed] [Google Scholar]
- Yao H, Kato A, Mooney B, Birchler J. 2011. Phenotypic and gene expression analyses of a ploidy series of maize inbred Oh43. Plant Molecular Biology 75, 237–251. [DOI] [PubMed] [Google Scholar]
- Zahn LM, Kong H, Leebens-Mack JH, Kim S, Soltis PS, Landherr LL, Soltis DE, Depamphilis CW, Ma H. 2005. The evolution of the SEPALLATA subfamily of MADS-box genes: a preangiosperm origin with multiple duplications throughout angiosperm history. Genetics 169, 2209–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Deng M, Fan G. 2014. Differential transcriptome analysis between Paulownia fortunei and its synthesized autopolyploid. International Journal of Molecular Sciences 15, 5079–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kishino H. 2004. Genomic background predicts the fate of duplicated genes: evidence from the yeast genome. Genetics 166, 1995–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lemos B, Dopman EB, Hartl DL. 2011a. Copy-number variation: the balance between gene dosage and expression in Drosophila melanogaster . Genome Biology and Evolution 3, 1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Moshgabadi N, Adams KL. 2011b. Extensive changes to alternative splicing patterns following allopolyploidy in natural and resynthesized polyploids. Proceedings of the National Academy of Sciences, USA 108, 16122–16127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.