

Abstract
The aim of this review is to explore the role of mitochondria in regulating macrophage sterol homeostasis and inflammatory responses within the aetiology of atherosclerosis. Macrophage generation of oxysterol activators of liver X receptors (LXRs), via sterol 27-hydroxylase, is regulated by the rate of flux of cholesterol to the inner mitochondrial membrane, via a complex of cholesterol trafficking proteins. Oxysterols are key signalling molecules, regulating the transcriptional activity of LXRs which coordinate macrophage sterol metabolism and cytokine production, key features influencing the impact of these cells within atherosclerotic lesions. The precise identity of the complex of proteins mediating mitochondrial cholesterol trafficking in macrophages remains a matter of debate, but may include steroidogenic acute regulatory protein and translocator protein. There is clear evidence that targeting either of these proteins enhances removal of cholesterol via LXRα-dependent induction of ATP binding cassette transporters (ABCA1, ABCG1) and limits the production of inflammatory cytokines; interventions which influence mitochondrial structure and bioenergetics also impact on removal of cholesterol from macrophages. Thus, molecules which can sustain or improve mitochondrial structure, the function of the electron transport chain, or increase the activity of components of the protein complex involved in cholesterol transfer, may therefore have utility in limiting or regressing atheroma development, reducing the incidence of coronary heart disease and myocardial infarction.
Keywords: Atherosclerosis, Macrophage, Cholesterol, High density lipoproteins, Apolipoproteins, ATP binding cassette transporters, Scavenger receptor B1, Mitochondria (dys)function, Sterol 27-hydroxylase, Liver X receptors
Core tip: Mitochondrial cholesterol trafficking to CYP27A1 located on the inner mitochondrial membrane regulates the formation of oxysterol ligands for liver X receptors (LXRs) in sterol-laden macrophage “foam” cells. In turn, ligation of LXRα has profound implications for sterol removal and inflammatory responses in macrophage “foam” cells, both factors which may contribute to the effective resolution of atherosclerotic lesions and reductions in the incidence of coronary heart disease and its sequelae.
INTRODUCTION
Coronary heart disease (CHD) is the major cause of morbidity and mortality worldwide, and the single largest cause of disease burden, determined according to disability-adjusted life years, the sum of life lost and years lived with disability[1,2]. Genetic factors contribute to coronary heart disease, fuelled by behavioural (smoking, physical inactivity, unhealthy diet, excess alcohol intake), metabolic (hypertension, diabetes, elevated serum cholesterol, overweight and obesity) and environmental (poverty, stress, educational status) factors[1-3].
Atherosclerosis is the primary cause of coronary heart disease characterised by chronic and unresolved inflammatory responses at sites of perturbed laminar blood flow in large and medium-sized arteries[4-6]. Activation of the arterial endothelial layer allows the accumulation of low density lipoprotein (LDL) within the intima of the vessel, where it can become modified via oxidation or crosslinking, triggering the recruitment of monocytes, neutrophils, lymphocytes and circulating stem cells to sites of inflammation[4-6]. Within this complex microenvironment, monocytes differentiate into macrophages which lie within a broad phenotypic spectrum, ranging from pro- (M1) to anti-inflammatory (M2)[6].
Arterial macrophages become laden with excess cholesterol and cholesteryl esters, part via the unregulated uptake of modified LDL by scavenger receptors (e.g., CD36, CD68, LOX-1 and SR-AI/AII), and by phagocytosis of apoptotic cells, resulting in formation of “foam cells”, a hallmark of early “fatty streak”, developing, and unstable atherosclerotic lesions[7-10]. During the early phase of lesion development, this process may represent a protective mechanism; however, in more advanced lesions, cholesterol-laden macrophages, by releasing inflammatory cytokines and matrix metalloproteinases, contribute to chronic unresolved inflammation[10], accelerating the disease process and acute thrombotic events such as cerebrovascular stroke or myocardial infarction.
Thus, removal of cholesterol from macrophage “foam cells” may achieve successful regression and stabilisation of atheroma, and the importance of this pathway in protecting against CHD is supported by epidemiological studies in humans, and in genetically modified mice in which components of this pathway have been overexpressed or deleted. For example, HDL-cholesterol (HDL-C) emerged as an independent risk factor for cardiovascular disease in the Framingham Heart Study, offering a risk reduction of 2%-3% for each 1 mg/dL increase in HDL-C concentration[11,12]. HDL particles also possess antioxidant, anti-thrombotic and pro-fibrinolytic properties, and can counteract the chronic inflammation[13-16], proliferation of haematopoietic stem cells[17] and leucocytosis[10,18] which promote atherosclerosis. However, increasing the level of HDL-C, with niacin[19,20], fibrates[20] or dolcetrapib (dal-OUTCOMES III trial)[20,21], does not necessarily confer protection against CHD[19-21] and in patients with systemic inflammation, coronary heart disease, chronic renal disease or diabetes, the protective properties of HDL are lost, and the particles transformed into those with pro-atherogenic potential[22-24]. Thus, it is not just the level, but the quality, composition (including levels of cargo molecules such as sphingosine-1-phosphate)[25] and function of HDL particles that are important.
Some, but not all, of the beneficial effects associated with HDL are mediated via the interaction of ATP binding cassette (ABC) transporters, such as ABCA1, ABCG1 and ABCG4, with apolipoproteins and HDL (Figure 1). While ABCA1 promotes efflux of cholesterol and phospholipids to lipid-poor apolipoproteins, such as apoA-I and apoE[13], ABCG1 and ABCG4 promote efflux of cholesterol, oxysterols and desmosterol to HDL[26]. Thus, these transporters together in a sequential manner to generate nascent HDL, which can then mature to HDL3 and HDL2 within the reverse cholesterol transport pathway in the bloodstream[25].
Figure 1.
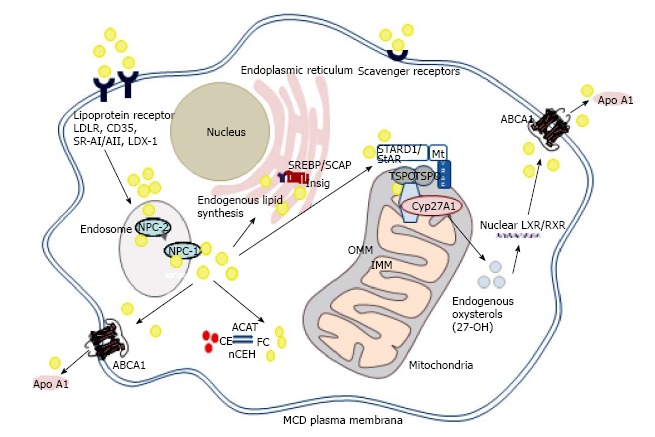
The role of mitochondrial cholesterol trafficking in regulation of macrophage sterol metabolism. Increased expression of steroidogenic acute regulatory protein (StAR, STARD1) or 18 kDa translocator protein (TSPO) drive cholesterol trafficking to mitochondrial sterol 27-hydroxylase (CYP27A1), enhancing endogenous production of oxysterols (24-, 25- and 27-hydroxycholesterol), in turn activating liver X receptors (LXR) and enhancing cholesterol efflux to apolipoprotein A-I (Apo AI) via ATP binding cassette transporter A1 (ABCA1). One current model for cholesterol transfer from the outer (OMM) to inner (IMM) mitochondrial membrane, derived from studies in steroidogenic cells, involves a complex of proteins, including StAR, TSPO, voltage-dependent anion channel (VDAC), regulatory subunits of protein kinase A (PKA-R1α), acyl CoA binding domains-1 and -3, ATPase family AAA domain-containing protein 3A (ASTAD3A) and optic atrophy type 1 proteins. Exogenous cholesterol delivered to the endocytic pathway via lipoprotein or scavenger receptors is transported either to the plasma membrane, enhancing cholesterol efflux via ABCA1, to lipid poor acceptors such as Apo AI or Apo E, or delivered to the endoplasmic reticulum (ER), retaining the Sterol Regulatory Element Binding Protein (SREBP)/SREBP-cleavage activating protein (SCAP) complex, in turn reducing cholesterol biosynthesis. Oxysterols enhance this process by binding to Insig-1/2 (insulin-induced gene-1 or -2). Excess cholesterol is esterified via Acyl CoA: Cholesterol Acyltransferase-1 (ACAT-1), and stored in lipid droplets within the cytoplasm as “foamy” droplets. nCEH: Neutral cholesteryl ester hydrolase; FC: Free cholesterol; CE: Cholesteryl ester: NPC-1/NPC-2: Niemann-Pick C1/C2 protein; StAR: Steroidogenic acute regulatory protein: RXR: Retinoic acid receptor.
Both rare and common genetic variations in ABCA1 influence the levels of HDL-C[26] and risk of ischaemic heart disease (IHD). However, the association between ABCA1 variants and coronary disease seem to be independent of the plasma level of HDL-C[27]. Instead, cholesterol efflux from macrophages is strongly linked to atherosclerosis and provides a novel way of assessing cardiovascular risk that provides a greater level of prediction than HDL-C[28]. Thus the expression and activity of the ABCA1 protein, and the quality and functionality of the nascent HDL generated, may prove valuable discriminants of the risk of cardiovascular disease[29].
Importantly, macrophage ABCA1 expression and cholesterol accumulation are intrinsically linked to the inflammatory status of these cells. Excess cholesterol proves cytotoxic and pro-inflammatory if recycling via ABCA1 is disrupted in macrophages[30-33]. Enhanced Toll-like receptor signalling is noted in ABCA1/ABCG1 null macrophages, resulting in increased expression of pro-inflammatory genes, and free cholesterol accumulation[34], while activation of Toll-like receptors 3 and 4 represses induction of ABCA1 and reduces macrophage cholesterol efflux[35]. Conversely, interleukin-6 (IL-6) attenuates pro-inflammatory responses and stimulates efflux of cholesterol via ABCA1 in human macrophages[36]. In good agreement with this integrated paradigm, macrophage ABCA1 limits inflammatory responses via ApoA-I dependent activation of the Jak2/Stat3 pathway[37,38], while macrophage sterol accumulation activates Liver X Receptor nuclear (LXR) transcription factors, achieving induction of ABCA1 and ABCG1 and repression of inflammation (below)[39,40].
MACROPHAGE LIPID METABOLISM AND INFLAMMATION ARE REGULATED BY LIVER X RECEPTORS
Activation of nuclear LXRs (LXRα/β) is marshals cellular responses to increasing levels of sterol, promoting cholesterol efflux (above)[39-43]. Liver X receptors form heterodimeric complexes with retinoic acid receptors (RXRs), and bind to imperfect direct repeats of the nuclear receptor half-site TGACCT[39-43]. Ligand binding dissociates co-repressor proteins, destined for ubiquitination and proteasomal degradation, and engages co-activator proteins such as histone demethylases and G-protein pathway suppressor-2 (GPS2), stimulating target gene transcription[44].
Activation of LXRα also represses cholesterol biosynthesis via novel negative LXR DNA-response elements within the promoter region of squalene synthase and lanosterol 14α-demethylase and suppresses uptake of LDL[45,46]. Oxysterols also bind to Insig-1/2, facilitating sequestration of sterol-regulatory element binding proteins (SREBPs) at the endoplasmic reticulum, ensuring repression of cholesterol biosynthesis and uptake[45]. Deletions of LXRα and LXRβ in murine models of atheroma cause lipid accumulation within the aortic root, even in the absence of an atherogenic diet[47,48].
It is also evident that LXRs modulate innate and adaptive immune responses mediated by macrophages, neutrophils, lymphocytes, neutrophils and dendritic cells[45], decreasing cytokine-mediated expression of a range of pro-inflammatory genes. This is achieved via a mechanism involving nuclear receptor co-repressor (NCoR), silencing mediator of retinoid and thyroid receptors (SMRT) and inhibition of nuclear factor kappa B (NFκB) signalling[45,48,49]. Activation of LXRs is also achieved by phagocytosis of apoptotic cells by macrophages increasing expression of receptor tyrosine kinase (Mertk), amplifying phagocytosis and cell clearance, and reducing production of inflammatory mediators[50]. Absence of LXR signalling enhances the apoptosis of macrophages challenged with Listeria monocytogenes, Escherichia coli or Salmonella typhinurium, via loss of the anti-apoptotic factor AIM/Spα[51,52].
MACROPHAGE GENERATION OF OXYSTEROL LIGANDS FOR LIVER X RECEPTORS
High levels of mitochondrial sterol 27-hydroxylase (CYP27A1) are found in human macrophages, and this enzyme can produce modified sterols, proven to act as LXR ligands in vitro and in vivo[53-56]. Loss of CYP27A1 leads to the lipid storage disease, cerebrotendinous xanthomatosis (CTX), which triggers accumulation of cholesterol and cholestanol in brain and tendons, progressive neurological deterioration, xanthomas and, as a secondary complication, accelerated atherosclerosis[57,58].
The rate-limiting step controlling CYP27A1 activity is the flux of cholesterol from the outer to the inner mitochondrial membrane, via a mitochondrial cholesterol trafficking complex (discussed below)[59]. Mitochondrial oxysterols therefore act as key cell signalling molecules, the levels of which can be moderated by sulfation (SULT2B1b), esterification (ACAT-1) or metabolism to soluble bile acid derivatives[53]. Conceivably, this process could be “uncoupled” by accumulation of free cholesterol at the interface between endoplasmic reticulum (ER) and mitochondrial membranes, triggering ER stress and proteasomal degradation of ABCA1, and opening of the permeability transition pore in mitochondria[53]. Esterification of excess oxysterols may then result: over 85% of the 27-hydroxycholesterol in human atherosclerotic lesions is esterified and incapable of activating LXRα and its downstream pathways[60,61]. Loss of this protective pathway predicates mitochondrial damage, apoptosis and cytotoxicity, features associated with addition of exogenous atheroma-relevant oxysterols (≥ 20 μmol/L) to cultured cells[62].
Thus, it is clear that the biological impact of oxysterols are not solely restricted to LXR activation[63-67]. For example, oxysterols also serve as endogenous ligands for G-protein coupled receptor 183 (Epstein-Barr virus-induced gene 2, EBI2)[63], function as selective estrogen receptor modulators[64], bind to the Smoothened molecule to modulate Hedgehog signalling[65], while CYP27A1-derived 7α and 7β, 27-hydroxycholesterol modify innate and adaptive immune responses by acting as agonists of retinoic acid-related (RAR) orphan receptor gamma t (ROPRγτ)[66].
Acute exposure of macrophages to exogenous oxysterols induce rapid (< second) oscillations in cytoplasmic [Ca2+] triggered by influx from the extracellular medium, followed by sustained increases in [Ca2+] mediated by translocation of TRPC1 (transient receptor potential, canonical) channels into lipid rafts in the plasma membrane[68]. Calcium transfer between ER and mitochondria is facilitated by mitochondria-associated membranes, which act as a hub for lipid transfer, regulation of mitochondrial morphology (fission, fusion and trafficking), apoptosis, autophagy and ER stress[69], although the role of endogenously generated oxysterols in these processes remains unknown at present. Certainly, chronic exposure to exogenous oxysterol congeners can activate calcium release from the ER, increasing dephosphorylation of Bcl-2 antagonist of cell death by the calcium-dependent phosphatase calcineurin, and promoting apoptosis[68].
TARGETING PROTEIN CONSTITUENTS OF THE MITOCHONDRIAL CHOLESTEROL TRAFFICKING COMPLEX: IMPACT ON MACROPHAGE STEROL METABOLISM AND INFLAMMATION
Despite intensive investigations in steroidogenic cells and tissues, the nature of the mitochondrial cholesterol trafficking complex remains a matter of debate. One recent model suggests a basal complex, forming contact sites between the outer and inner mitochondrial membranes, composed of the 18 kDa translocator protein (TSPO), adenine nucleotide transporter (ANT) and voltage-dependent anion channel (VDAC)[70-72]. In hormone-stimulated steroidogenic tissues, a “transduceosome” complex is formed, involving recruitment of the regulatory subunits of protein kinase A (PKA-R1α) and acyl CoA binding domain proteins-1 and -3. Elevated levels of cyclic adenosine monophosphate (cAMP) release PKA catalytic subunits to phosphorylate 37 kDa steroidogenic acute regulatory protein at the outer mitochondrial membrane; import of both StAR and cholesterol into the inner mitochondrial membrane and matrix facilitate both proteolytic processing of StAR to its 30 kDa form, and conversion of cholesterol into pregnenolone by CYP11A1[70-72]. However, a dynamic 800 kDa bioactive protein complex in steroidogenic cells has also been described, which does not involve ANT, but is composed of TSPO, VDAC, CYP11A1, ATPase family AAA domain-containing protein 3A (ASTAD3A) and optic atrophy type 1 proteins[73]; in this model, StAR facilitated binding of cholesterol to the 800 kDa complex, enhancing steroidogenesis.
Importantly, there is a growing realisation that key mitochondrial cholesterol trafficking proteins, such as StAR, play an important role in non-steroidogenic tissues[74]. This, combined with conflicting results regarding the impact of genetic deletion of TSPO on steroidogenesis and viability in mice[75-78], may lead to increased consideration of alternate functions for these proteins[74]. For example, StAR is expressed in endothelial cells, monocytes and macrophages[79-82], albeit at levels far lower than those found in adrenal or gonadal tissues[74]. By contrast, other components of the mitochondrial trafficking complex, such as TSPO, are widely expressed in a variety of tissues, including macrophages[78,81].
Importantly, both StAR and TSPO appear to impact on macrophage lipid and inflammatory phenotype, in part via the pathway involving sterol 27-hydroxylase, activation of LXRα and upregulation of ABCA1/ABCG1 mRNA and protein[81-83], arguing a functional role for these proteins in mediating cholesterol supply to CYP27A1. Overexpression of StAR decreased macrophage lipid content[82,83], repressed inflammation[82] and apoptosis[84] and increased macrophage cholesterol efflux[82,83], while a viral vector expressing StAR reduced aortic lipids and atheroma in apoE-/- mice[85]. However, exploiting any putative anti-atherogenic properties of StAR could prove problematic, due to the associated induction of lipogenesis in macrophages[83,86], presumably via LXRα dependent induction of Srebp1c[87].
This led to focus on other components of the mitochondrial cholesterol trafficking complex and, in particular, TSPO[81]. Transient overexpression of TSPO in human (THP-1) macrophages increased the levels of ABCA1 mRNA and protein, and enhanced efflux of cholesterol to apoA-I, HDL and human serum, a finding reversed by gene knockdown of TSPO. Small molecule TSPO ligands also increased cholesterol efflux, an effect that was amplified in macrophages genetically engineered to overexpress TSPO[81]. Notably, TSPO overexpression caused a decline in macrophage total neutral lipid mass, without induction of lipogenesis, and effectively prevented “foam cell” formation following exposure to modified LDL[81]. These effects were associated with induction of both LXRα and PPARα the latter providing a plausible mechanism for the observed reductions in macrophage lipid mass[81]. Notably, overexpression of some of the other proposed components of the mitochondrial cholesterol trafficking complex, such as VDAC, ANT and ACBD1, discussed above, exerted minimal effects on the macrophage cholesterol efflux pathway[81].
Expression of TSPO is upregulated by exposure to modified LDL in human macrophages[81], and TSPO ligands have been used to image vascular inflammation in CD68 positive macrophages at sites of disturbed flow in murine carotid arteries[88], and macrophage burden[89] and intraplaque inflammation[90] within human carotid atherosclerotic lesions. Despite this evident association with inflammation, it appears that upregulation of TSPO, or signalling via this protein, may represent an adaptive mechanism designed to limit tissue damage. Overexpression of TSPO in microglia decreased production of pro-inflammatory cytokines, reflected in increased expression of alternately activated M2 stage-related genes and mediated via repression of NF-κB activation[91]. Similarly, TSPO ligands inhibited the proliferation of retinal microglial cells, and repressed the output of reactive oxygen species and TNFα[92]. In good agreement, levels of TSPO are higher in dystrophic murine retina, and in microglia treated with LPS, while TSPO ligand XBD173 repressed the expression of chemokine (C-C motif) ligand 2 (CCL2), IL-6 and iNOS[93]. The TSPO ligand, PK11195 has proved effective in ameliorating the severity of disease in an experimental murine model of multiple sclerosis, by reducing inflammatory responses and promoting oligodendroglial regeneration[94]. TSPO has also been posited as a novel target for Alzheimer’s disease[95], anxiety, psychiatric and neurologic disorders[96-99], pain[100], cancer[101] and vascular dysfunction[88-90,102]. At present, it is not known how many of these effects are related to the cholesterol trafficking function of TSPO, although LXRs influence expression of an array of genes involved in cholesterol homeostasis, glucose metabolism, inflammation and Alzheimer’s disease[103]. It is also clear that some of the reported effects of TSPO and its ligands may require re-evaluation, given the lack of phenotype recently reported in healthy TSPO -/- mice[75,76].
MITOCHONDRIAL STRUCTURE AND BIOENERGETICS: IMPACT ON CHOLESTEROL HOMEOSTASIS
Mitochondria exhibit constant movement, fusion and fission[104]. The mitochondrial membrane protein mitofusin (Mfn2) is involved in maintaining mitochondrial morphology, energy provision, and cellular growth and apoptosis[105-107]. Recently, Mfn2 has emerged as a regulator of macrophage cholesterol efflux, via upregulation of peroxisome proliferator activated receptor-γ (PPARγ) ABCA1, ABCG1 and scavenger receptor-B1 (SR-B1), reflected in marked reductions in cholesterol mass[107]. Overexpression of Mfn2 attenuates the formation of atherosclerotic lesions in rabbit carotid arteries, and levels of Mfn2 are progressively reduced during lesion formation in apoE-/- mice during atherogenesis; levels of Mfn2 are also reduced in atherosclerotic, compared with non-atherosclerotic, human arteries[107].
Remodelling of the inner mitochondrial membrane by optic atrophy 1 (OPA1) also alters the efficiency of mitochondrial cholesterol trafficking, at least in steroidogenic cells[108,109]. Increased steroidogenesis is reported in trophoblasts undergoing syncytilisation, which express increased levels of the pro-fission mitochondrial shaping protein Drp1 increased, and decreased levels of Opa1 and mitofusin. An inverse relationship between levels of Opa1 and steroidogenesis were also evidenced in cells genetically manipulated to express higher levels of Opa1, while accumulation of cholesterol at the inner mitochondrial membrane was observed in mitochondria lacking Opa1[108,109].
Finally, it is self-evident that ATP is needed to mount an effective non-adaptive immune response, and to fuel cholesterol biosynthesis and the activity of ABC transporters that determine the rate of macrophage cholesterol efflux. However, more subtle changes in mitochondrial function or loss of bioenergetic capacity, the emerging concept of the Bioenergetic Health Index (BHI)[110], have been shown to reduce the efficiency of mitochondrial cholesterol trafficking and hormone biosynthesis in steroidogenic tissues[111,112]. Dissipation of the mitochondrial membrane potential (Δψm using carbonyl cyanide m-chlorophenylhydrazone), inhibition of electron transport at complex III (using antimycin), reduction of pH (nigericin) and inhibition of ATP synthase (oligomycin) blocked the formation of progesterone and synthesis or import of StAR protein in Leydig cells[111,112].
A parallel study in macrophages supports the notion that acute loss of mitochondrial function is also associated with dysregulated cholesterol homeostasis[113]. Cholesterol efflux was inhibited by nigericin and oligomycin in RAW 264.7 macrophages; levels of ABCA1 protein decreased in response to oligomycin treatment, despite paradoxical increases in Abca1 mRNA[113,114], reflecting findings in carotid atherosclerotic lesions[114] Further, while oligomycin treatment did not alter cholesterol biosynthesis, cholesterol esterification was significantly inhibited, promoting apoptosis. Oligomycin induced expression of genes involved in cholesterol efflux (Abca1, Abcg4, Stard1) and cholesterol biosynthesis (Hmgr, Mvk, Scap, Srepb2) arguing that loss of coordinated regulation of sterol homeostasis is caused by loss of mitochondrial ATP generation[113]. In turn, accumulation of free cholesterol or fatty acids can trigger mitochondrial dysfunction, which could promote inflammation via loss of LXRα-dependent repression of NF-κB (above) and upregulation of cytokine expression, but also by NLRP3 inflammasome-dependent and –independent pathways[115].
QUESTIONS FOR THE FUTURE
This review summarizes the current evidence that, in part, macrophage sterol homeostasis, and inflammatory responses, can be linked to mitochondrial cholesterol trafficking, and mitochondrial structure and bioenergetics. Whether proteins involved in mitochondrial structure, fission, fusion or organelle dynamics can also impact on these processes is currently uninvestigated and an area of keen interest. More particularly, mitochondria-mediated hormetic effects in aging[116,117] suggest a retrograde signalling pathway by which mitochondrial dysfunction in a single distinct tissue elicits the mitochondrial stress response in some (but not all) distal tissues. In turn, this suggests that loss of effective mitochondrial function, such as that caused by hepatic insulin resistance for example, may be transmitted via “mitokines” to peripheral tissues, promoting vascular dysfunction and cardiovascular disease. These exciting findings offer some intriguing possibilities for therapeutic strategies aimed at sustaining or improving mitochondrial function.
Footnotes
P- Reviewer: Atamer A S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: An international patent application (PCT/GB2014/052585) has been filed on behalf of Glasgow Caledonian University, relating to cholesterol modulation. No other conflicting interests, commercial, personal, political, religious, intellectual or otherwise, exist relating to this manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 14, 2014
First decision: January 8, 2015
Article in press: March 18, 2015
References
- 1.Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization in collaboration with the World Heart Federation and the World Stroke Organization. Global Atlas on cardiovascular disease prevention and control. Available from: http: //whqlibdoc.who.int/publications/2011/9789241564373eng.pdf.
- 3.Dawber TR, Moore FE, Mann GV. Coronary heart disease in the Framingham study. Am J Public Health Nations Health. 1957;47:4–24. doi: 10.2105/ajph.47.4_pt_2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 5.Woollard KJ. Immunological aspects of atherosclerosis. Clin Sci (Lond) 2013;125:221–235. doi: 10.1042/CS20120576. [DOI] [PubMed] [Google Scholar]
- 6.Wolfs IM, Donners MM, de Winther MP. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb Haemost. 2011;106:763–771. doi: 10.1160/TH11-05-0320. [DOI] [PubMed] [Google Scholar]
- 7.Gerrity RG. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol. 1981;103:181–190. [PMC free article] [PubMed] [Google Scholar]
- 8.Sawamura T, Kakino A, Fujita Y. LOX-1: a multiligand receptor at the crossroads of response to danger signals. Curr Opin Lipidol. 2012;23:439–445. doi: 10.1097/MOL.0b013e32835688e4. [DOI] [PubMed] [Google Scholar]
- 9.Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. 2012;217:492–502. doi: 10.1016/j.imbio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Andrés V, Pello OM, Silvestre-Roig C. Macrophage proliferation and apoptosis in atherosclerosis. Curr Opin Lipidol. 2012;23:429–438. doi: 10.1097/MOL.0b013e328357a379. [DOI] [PubMed] [Google Scholar]
- 11.Gordon DJ, Knoke J, Probstfield JL, Superko R, Tyroler HA. High-density lipoprotein cholesterol and coronary heart disease in hypercholesterolemic men: the Lipid Research Clinics Coronary Primary Prevention Trial. Circulation. 1986;74:1217–1225. doi: 10.1161/01.cir.74.6.1217. [DOI] [PubMed] [Google Scholar]
- 12.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 13.Rosenson RS, Brewer HB, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. 2012;111:1079–1090. doi: 10.1161/CIRCRESAHA.111.258673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Annema W, von Eckardstein A. High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J. 2013;77:2432–2448. doi: 10.1253/circj.cj-13-1025. [DOI] [PubMed] [Google Scholar]
- 16.Scanu AM, Edelstein C. HDL: bridging past and present with a look at the future. FASEB J. 2008;22:4044–4054. doi: 10.1096/fj.08-117150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013;24:129–136. doi: 10.1016/j.tem.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy AJ, Westerterp M, Yvan-Charvet L, Tall AR. Anti-atherogenic mechanisms of high density lipoprotein: effects on myeloid cells. Biochim Biophys Acta. 2012;1821:513–521. doi: 10.1016/j.bbalip.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 20.Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379. doi: 10.1136/bmj.g4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 22.Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–155. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balder JW, Staels B, Kuivenhoven JA. Pharmacological interventions in human HDL metabolism. Curr Opin Lipidol. 2013;24:500–509. doi: 10.1097/MOL.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 24.Rye KA, Barter PJ. Predictive value of different HDL particles for the protection against or risk of coronary heart disease. Biochim Biophys Acta. 2012;1821:473–480. doi: 10.1016/j.bbalip.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Egom EE, Mamas MA, Soran H. HDL quality or cholesterol cargo: what really matters--spotlight on sphingosine-1-phosphate-rich HDL. Curr Opin Lipidol. 2013;24:351–356. doi: 10.1097/MOL.0b013e328361f822. [DOI] [PubMed] [Google Scholar]
- 26.Hellerstein M, Turner S. Reverse cholesterol transport fluxes. Curr Opin Lipidol. 2014;25:40–47. doi: 10.1097/MOL.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 27.Brunham LR, Singaraja RR, Hayden MR. Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu Rev Nutr. 2006;26:105–129. doi: 10.1146/annurev.nutr.26.061505.111214. [DOI] [PubMed] [Google Scholar]
- 28.Frikke-Schmidt R. Genetic variation in the ABCA1 gene, HDL cholesterol, and risk of ischemic heart disease in the general population. Atherosclerosis. 2010;208:305–316. doi: 10.1016/j.atherosclerosis.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hafiane A, Genest J. HDL, Atherosclerosis, and Emerging Therapies. Cholesterol. 2013;2013:891403. doi: 10.1155/2013/891403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86:1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francone OL, Royer L, Boucher G, Haghpassand M, Freeman A, Brees D, Aiello RJ. Increased cholesterol deposition, expression of scavenger receptors, and response to chemotactic factors in Abca1-deficient macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1198–1205. doi: 10.1161/01.ATV.0000166522.69552.99. [DOI] [PubMed] [Google Scholar]
- 33.Koseki M, Hirano K, Masuda D, Ikegami C, Tanaka M, Ota A, Sandoval JC, Nakagawa-Toyama Y, Sato SB, Kobayashi T, et al. Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-alpha secretion in Abca1-deficient macrophages. J Lipid Res. 2007;48:299–306. doi: 10.1194/jlr.M600428-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 37.Frisdal E, Lesnik P, Olivier M, Robillard P, Chapman MJ, Huby T, Guerin M, Le Goff W. Interleukin-6 protects human macrophages from cellular cholesterol accumulation and attenuates the proinflammatory response. J Biol Chem. 2011;286:30926–30936. doi: 10.1074/jbc.M111.264325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin K, Deng X, Mo ZC, Zhao GJ, Jiang J, Cui LB, Tan CZ, Wen GB, Fu Y, Tang CK. Tristetraprolin-dependent post-transcriptional regulation of inflammatory cytokine mRNA expression by apolipoprotein A-I: role of ATP-binding membrane cassette transporter A1 and signal transducer and activator of transcription 3. J Biol Chem. 2011;286:13834–13845. doi: 10.1074/jbc.M110.202275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 41.Im SS, Osborne TF. Liver x receptors in atherosclerosis and inflammation. Circ Res. 2011;108:996–1001. doi: 10.1161/CIRCRESAHA.110.226878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci USA. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci USA. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Traversari C, Russo V. Control of the immune system by oxysterols and cancer development. Curr Opin Pharmacol. 2012;12:729–735. doi: 10.1016/j.coph.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Rogers PM, Su C, Varga G, Stayrook KR, Burris TP. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem. 2008;283:26332–26339. doi: 10.1074/jbc.M804808200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shibata N, Glass CK. Macrophages, oxysterols and atherosclerosis. Circ J. 2010;74:2045–2051. doi: 10.1253/circj.cj-10-0860. [DOI] [PubMed] [Google Scholar]
- 49.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O’connell RM, Cheng G, Saez E, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 52.Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci USA. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen AM, Taylor JM, Graham A. Mitochondrial (dys)function and regulation of macrophage cholesterol efflux. Clin Sci (Lond) 2013;124:509–515. doi: 10.1042/CS20120358. [DOI] [PubMed] [Google Scholar]
- 54.Lund E, Björkhem I, Furster C, Wikvall K. 24-, 25- and 27-hydroxylation of cholesterol by a purified preparation of 27-hydroxylase from pig liver. Biochim Biophys Acta. 1993;1166:177–182. doi: 10.1016/0005-2760(93)90094-p. [DOI] [PubMed] [Google Scholar]
- 55.Song C, Liao S. Cholestenoic acid is a naturally occurring ligand for liver X receptor alpha. Endocrinology. 2000;141:4180–4184. doi: 10.1210/endo.141.11.7772. [DOI] [PubMed] [Google Scholar]
- 56.Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 2007;5:73–79. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Björkhem I. Cerebrotendinous xanthomatosis. Curr Opin Lipidol. 2013;24:283–287. doi: 10.1097/MOL.0b013e328362df13. [DOI] [PubMed] [Google Scholar]
- 58.Valdivielso P, Calandra S, Durán JC, Garuti R, Herrera E, González P. Coronary heart disease in a patient with cerebrotendinous xanthomatosis. J Intern Med. 2004;255:680–683. doi: 10.1111/j.1365-2796.2004.01316.x. [DOI] [PubMed] [Google Scholar]
- 59.Pandak WM, Ren S, Marques D, Hall E, Redford K, Mallonee D, Bohdan P, Heuman D, Gil G, Hylemon P. Transport of cholesterol into mitochondria is rate-limiting for bile acid synthesis via the alternative pathway in primary rat hepatocytes. J Biol Chem. 2002;277:48158–48164. doi: 10.1074/jbc.M205244200. [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Cruset S, Carpenter KL, Guardiola F, Stein BK, Mitchinson MJ. Oxysterol profiles of normal human arteries, fatty streaks and advanced lesions. Free Radic Res. 2001;35:31–41. doi: 10.1080/10715760100300571. [DOI] [PubMed] [Google Scholar]
- 61.Vaya J, Aviram M, Mahmood S, Hayek T, Grenadir E, Hoffman A, Milo S. Selective distribution of oxysterols in atherosclerotic lesions and human plasma lipoproteins. Free Radic Res. 2001;34:485–497. doi: 10.1080/10715760100300431. [DOI] [PubMed] [Google Scholar]
- 62.Larsson DA, Baird S, Nyhalah JD, Yuan XM, Li W. Oxysterol mixtures, in atheroma-relevant proportions, display synergistic and proapoptotic effects. Free Radic Biol Med. 2006;41:902–910. doi: 10.1016/j.freeradbiomed.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 63.Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, Guerini D, Baumgarten BU, Roggo S, Wen B, et al. Oxysterols direct immune cell migration via EBI2. Nature. 2011;475:524–527. doi: 10.1038/nature10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Umetani M, Shaul PW. 27-Hydroxycholesterol: the first identified endogenous SERM. Trends Endocrinol Metab. 2011;22:130–135. doi: 10.1016/j.tem.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, Rohatgi R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol. 2012;8:211–220. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, Yu J, Nelen MI, Liu X, Castro G, et al. Oxysterols are agonist ligands of RORγt and drive Th17 cell differentiation. Proc Natl Acad Sci USA. 2014;111:12163–12168. doi: 10.1073/pnas.1322807111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Traversari C, Sozzani S, Steffensen KR, Russo V. LXR-dependent and -independent effects of oxysterols on immunity and tumor growth. Eur J Immunol. 2014;44:1896–1903. doi: 10.1002/eji.201344292. [DOI] [PubMed] [Google Scholar]
- 68.Mackrill JJ. Oxysterols and calcium signal transduction. Chem Phys Lipids. 2011;164:488–495. doi: 10.1016/j.chemphyslip.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 69.van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843:2253–2262. doi: 10.1016/j.bbamcr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 70.Rone MB, Fan J, Papadopoulos V. Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim Biophys Acta. 2009;1791:646–658. doi: 10.1016/j.bbalip.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manna PR, Dyson MT, Stocco DM. Regulation of the steroidogenic acute regulatory protein gene expression: present and future perspectives. Mol Hum Reprod. 2009;15:321–333. doi: 10.1093/molehr/gap025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res. 2011;52:2111–2135. doi: 10.1194/jlr.R016675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rone MB, Midzak AS, Issop L, Rammouz G, Jagannathan S, Fan J, Ye X, Blonder J, Veenstra T, Papadopoulos V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol. 2012;26:1868–1882. doi: 10.1210/me.2012-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anuka E, Gal M, Stocco DM, Orly J. Expression and roles of steroidogenic acute regulatory (StAR) protein in ‘non-classical’, extra-adrenal and extra-gonadal cells and tissues. Mol Cell Endocrinol. 2013;371:47–61. doi: 10.1016/j.mce.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 75.Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology. 2014;155:89–97. doi: 10.1210/en.2013-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, Selvaraj V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem. 2014;289:27444–27454. doi: 10.1074/jbc.M114.578286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Papadopoulos V. On the role of the translocator protein (18-kDa) TSPO in steroid hormone biosynthesis. Endocrinology. 2014;155:15–20. doi: 10.1210/en.2013-2033. [DOI] [PubMed] [Google Scholar]
- 78.Wang HJ, Fan J, Papadopoulos V. Translocator protein (Tspo) gene promoter-driven green fluorescent protein synthesis in transgenic mice: an in vivo model to study Tspo transcription. Cell Tissue Res. 2012;350:261–275. doi: 10.1007/s00441-012-1478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma Y, Ren S, Pandak WM, Li X, Ning Y, Lu C, Zhao F, Yin L. The effects of inflammatory cytokines on steroidogenic acute regulatory protein expression in macrophages. Inflamm Res. 2007;56:495–501. doi: 10.1007/s00011-007-6133-3. [DOI] [PubMed] [Google Scholar]
- 80.Borthwick F, Taylor JM, Bartholomew C, Graham A. Differential regulation of the STARD1 subfamily of START lipid trafficking proteins in human macrophages. FEBS Lett. 2009;583:1147–1153. doi: 10.1016/j.febslet.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 81.Taylor JM, Allen AM, Graham A. Targeting mitochondrial 18 kDa translocator protein (TSPO) regulates macrophage cholesterol efflux and lipid phenotype. Clin Sci (Lond) 2014;127:603–613. doi: 10.1042/CS20140047. [DOI] [PubMed] [Google Scholar]
- 82.Ning Y, Bai Q, Lu H, Li X, Pandak WM, Zhao F, Chen S, Ren S, Yin L. Overexpression of mitochondrial cholesterol delivery protein, StAR, decreases intracellular lipids and inflammatory factors secretion in macrophages. Atherosclerosis. 2009;204:114–120. doi: 10.1016/j.atherosclerosis.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taylor JM, Borthwick F, Bartholomew C, Graham A. Overexpression of steroidogenic acute regulatory protein increases macrophage cholesterol efflux to apolipoprotein AI. Cardiovasc Res. 2010;86:526–534. doi: 10.1093/cvr/cvq015. [DOI] [PubMed] [Google Scholar]
- 84.Bai Q, Li X, Ning Y, Zhao F, Yin L. Mitochondrial cholesterol transporter, StAR, inhibits human THP-1 monocyte-derived macrophage apoptosis. Lipids. 2010;45:29–36. doi: 10.1007/s11745-009-3375-6. [DOI] [PubMed] [Google Scholar]
- 85.Ning Y, Xu L, Ren S, Pandak WM, Chen S, Yin L. StAR overexpression decreases serum and tissue lipids in apolipoprotein E-deficient mice. Lipids. 2009;44:511–519. doi: 10.1007/s11745-009-3299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Graham A, Borthwick F, Taylor J. Steroidogenic acute regulatory protein (StAR) and atherogenesis. In: Clark BJ, Stocco DM, editors. Cholesterol transporters of the START domain protein family in health and disease. New York: Springer Science; 2014. pp. 99–117. [Google Scholar]
- 87.Pawar A, Botolin D, Mangelsdorf DJ, Jump DB. The role of liver X receptor-alpha in the fatty acid regulation of hepatic gene expression. J Biol Chem. 2003;278:40736–40743. doi: 10.1074/jbc.M307973200. [DOI] [PubMed] [Google Scholar]
- 88.Cuhlmann S, Gsell W, Van der Heiden K, Habib J, Tremoleda JL, Khalil M, Turkheimer F, Meens MJ, Kwak BR, Bird J, et al. In vivo mapping of vascular inflammation using the translocator protein tracer 18F-FEDAA1106. Mol Imaging. 2014:13. doi: 10.2310/7290.2014.00014. [DOI] [PubMed] [Google Scholar]
- 89.Bird JL, Izquierdo-Garcia D, Davies JR, Rudd JH, Probst KC, Figg N, Clark JC, Weissberg PL, Davenport AP, Warburton EA. Evaluation of translocator protein quantification as a tool for characterising macrophage burden in human carotid atherosclerosis. Atherosclerosis. 2010;210:388–391. doi: 10.1016/j.atherosclerosis.2009.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gaemperli O, Shalhoub J, Owen DR, Lamare F, Johansson S, Fouladi N, Davies AH, Rimoldi OE, Camici PG. Imaging intraplaque inflammation in carotid atherosclerosis with 11C-PK11195 positron emission tomography/computed tomography. Eur Heart J. 2012;33:1902–1910. doi: 10.1093/eurheartj/ehr367. [DOI] [PubMed] [Google Scholar]
- 91.Bae KR, Shim HJ, Balu D, Kim SR, Yu SW. Translocator protein 18 kDa negatively regulates inflammation in microglia. J Neuroimmune Pharmacol. 2014;9:424–437. doi: 10.1007/s11481-014-9540-6. [DOI] [PubMed] [Google Scholar]
- 92.Wang M, Wang X, Zhao L, Ma W, Rodriguez IR, Fariss RN, Wong WT. Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J Neurosci. 2014;34:3793–3806. doi: 10.1523/JNEUROSCI.3153-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karlstetter M, Nothdurfter C, Aslanidis A, Moeller K, Horn F, Scholz R, Neumann H, Weber BH, Rupprecht R, Langmann T. Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J Neuroinflammation. 2014;11:3. doi: 10.1186/1742-2094-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Daugherty DJ, Selvaraj V, Chechneva OV, Liu XB, Pleasure DE, Deng W. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med. 2013;5:891–903. doi: 10.1002/emmm.201202124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chua SW, Kassiou M, Ittner LM. The translocator protein as a drug target in Alzheimer’s disease. Expert Rev Neurother. 2014;14:439–448. doi: 10.1586/14737175.2014.896201. [DOI] [PubMed] [Google Scholar]
- 96.Nothdurfter C, Baghai TC, Schüle C, Rupprecht R. Translocator protein (18 kDa) (TSPO) as a therapeutic target for anxiety and neurologic disorders. Eur Arch Psychiatry Clin Neurosci. 2012;262 Suppl 2:S107–S112. doi: 10.1007/s00406-012-0352-5. [DOI] [PubMed] [Google Scholar]
- 97.Girard C, Liu S, Adams D, Lacroix C, Sinéus M, Boucher C, Papadopoulos V, Rupprecht R, Schumacher M, Groyer G. Axonal regeneration and neuroinflammation: roles for the translocator protein 18 kDa. J Neuroendocrinol. 2012;24:71–81. doi: 10.1111/j.1365-2826.2011.02215.x. [DOI] [PubMed] [Google Scholar]
- 98.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 99.Papadopoulos V, Lecanu L. Translocator protein (18 kDa) TSPO: an emerging therapeutic target in neurotrauma. Exp Neurol. 2009;219:53–57. doi: 10.1016/j.expneurol.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hernstadt H, Wang S, Lim G, Mao J. Spinal translocator protein (TSPO) modulates pain behavior in rats with CFA-induced monoarthritis. Brain Res. 2009;1286:42–52. doi: 10.1016/j.brainres.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Austin CJ, Kahlert J, Kassiou M, Rendina LM. The translocator protein (TSPO): a novel target for cancer chemotherapy. Int J Biochem Cell Biol. 2013;45:1212–1216. doi: 10.1016/j.biocel.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 102.Lazzarini R, Sakai M, Costa-Pinto FA, Palermo-Neto J. Diazepam decreases leukocyte-endothelium interactions in situ. Immunopharmacol Immunotoxicol. 2010;32:402–409. doi: 10.3109/08923970903468821. [DOI] [PubMed] [Google Scholar]
- 103.Cao G, Liang Y, Jiang XC, Eacho PI. Liver X receptors as potential therapeutic targets for multiple diseases. Drug News Perspect. 2004;17:35–41. doi: 10.1358/dnp.2004.17.1.829024. [DOI] [PubMed] [Google Scholar]
- 104.Liu R, Jin P, LiqunYu Y, Han L, Shi T, Li X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One. 2014;9:e92810. doi: 10.1371/journal.pone.0092810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu C, Ge B, He C, Zhang Y, Liu X, Liu K, Qian C, Zhang Y, Peng W, Guo X. Mitofusin 2 decreases intracellular lipids in macrophages by regulating peroxisome proliferator-activated receptor-γ. Biochem Biophys Res Commun. 2014;450:500–506. doi: 10.1016/j.bbrc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 106.Gan KX, Wang C, Chen JH, Zhu CJ, Song GY. Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J Gastroenterol. 2013;19:1572–1581. doi: 10.3748/wjg.v19.i10.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abhijit S, Bhaskaran R, Narayanasamy A, Chakroborty A, Manickam N, Dixit M, Mohan V, Balasubramanyam M. Hyperinsulinemia-induced vascular smooth muscle cell (VSMC) migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol Cell Biochem. 2013;373:95–105. doi: 10.1007/s11010-012-1478-5. [DOI] [PubMed] [Google Scholar]
- 108.Fülöp L, Rajki A, Katona D, Szanda G, Spät A. Extramitochondrial OPA1 and adrenocortical function. Mol Cell Endocrinol. 2013;381:70–79. doi: 10.1016/j.mce.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 109.Wasilewski M, Semenzato M, Rafelski SM, Robbins J, Bakardjiev AI, Scorrano L. Optic atrophy 1-dependent mitochondrial remodeling controls steroidogenesis in trophoblasts. Curr Biol. 2012;22:1228–1234. doi: 10.1016/j.cub.2012.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chacko BK, Kramer PA, Ravi S, Benavides GA, Mitchell T, Dranka BP, Ferrick D, Singal AK, Ballinger SW, Bailey SM, et al. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond) 2014;127:367–373. doi: 10.1042/CS20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hales DB, Allen JA, Shankara T, Janus P, Buck S, Diemer T, Hales KH. Mitochondrial function in Leydig cell steroidogenesis. Ann N Y Acad Sci. 2005;1061:120–134. doi: 10.1196/annals.1336.014. [DOI] [PubMed] [Google Scholar]
- 112.Allen JA, Shankara T, Janus P, Buck S, Diemer T, Hales KH, Hales DB. Energized, polarized, and actively respiring mitochondria are required for acute Leydig cell steroidogenesis. Endocrinology. 2006;147:3924–3935. doi: 10.1210/en.2005-1204. [DOI] [PubMed] [Google Scholar]
- 113.Allen AM, Graham A. Mitochondrial function is involved in regulation of cholesterol efflux to apolipoprotein (apo)A-I from murine RAW 264.7 macrophages. Lipids Health Dis. 2012;11:169. doi: 10.1186/1476-511X-11-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Albrecht C, Soumian S, Amey JS, Sardini A, Higgins CF, Davies AH, Gibbs RG. ABCA1 expression in carotid atherosclerotic plaques. Stroke. 2004;35:2801–2806. doi: 10.1161/01.STR.0000147036.07307.93. [DOI] [PubMed] [Google Scholar]
- 115.Lawlor KE, Vince JE. Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta. 2014;1840:1433–1440. doi: 10.1016/j.bbagen.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 116.Woo DK, Shadel GS. Mitochondrial stress signals revise an old aging theory. Cell. 2011;144:11–12. doi: 10.1016/j.cell.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 117.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]