

Abstract
Fungi that have the enzymes cyanase and carbonic anhydrase show a limited capacity to detoxify cyanate, a fungicide employed by both plants and humans. Here, we describe a novel two-gene cluster that comprises duplicated cyanase and carbonic anhydrase copies, which we name the CCA gene cluster, trace its evolution across Ascomycetes, and examine the evolutionary dynamics of its spread among lineages of the Fusarium oxysporum species complex (hereafter referred to as the FOSC), a cosmopolitan clade of purportedly clonal vascular wilt plant pathogens. Phylogenetic analysis of fungal cyanase and carbonic anhydrase genes reveals that the CCA gene cluster arose independently at least twice and is now present in three lineages, namely Cochliobolus lunatus, Oidiodendron maius, and the FOSC. Genome-wide surveys within the FOSC indicate that the CCA gene cluster varies in copy number across isolates, is always located on accessory chromosomes, and is absent in FOSC’s closest relatives. Phylogenetic reconstruction of the CCA gene cluster in 163 FOSC strains from a wide variety of hosts suggests a recent history of rampant transfers between isolates. We hypothesize that the independent formation of the CCA gene cluster in different fungal lineages and its spread across FOSC strains may be associated with resistance to plant-produced cyanates or to use of cyanate fungicides in agriculture.
Keywords: Fusarium oxysporum, horizontal gene transfer, metabolic gene cluster, convergent evolution, gene duplication, fungicide resistance
Introduction
Cyanate (CNO−) is a toxic defense compound produced by a wide range of bacterial, plant, and fungal species and is currently commonly used in agriculture as a general purpose pesticide and fungicide. Like the closely related compound cyanide (CN−), cyanate inhibits cytochrome C (Jain and Kassner 1984), thereby shutting down oxidative phosphorylation and starving cells of ATP (Albaum et al. 1946). The widespread use of toxic cyanate compounds in nature is reflected in the broad taxonomic distribution of the enzyme cyanase, which detoxifies cyanate by converting it to ammonia (NH4+) and carbon dioxide (CO2). Found throughout plants, bacteria, and nematodes (Wilson et al. 2009), cyanase is nearly ubiquitous in the Basidiomycota, the Ascomycota (other than the yeasts, where it is absent), as well as in other fungal phyla (http://supfam.cs.bris.ac.uk/SUPERFAMILY/cgi-bin/taxviz.cgi?sf=55234, last accessed February 23, 2014; Wilson et al. 2009).
Cyanase consumes bicarbonate (H2CO3), which is supplied through the conversion of water (H2O) and carbon dioxide (CO2) by the enzyme carbonic anhydrase (Ebbs 2004) (fig. 1A). Carbonic anhydrase, which plays an important role in numerous processes, including respiration, pH homeostasis, and bicarbonate-dependent carboxylation reactions (Elleuche and Pöggeler 2010), has been found in all cellular organism genomes except for a single syntrophic bacterium that grows on carbon dioxide provided by other bacteria (Nishida et al. 2009). Interestingly, cyanate also inhibits carbonic anhydrase (fig. 1A), potentially slowing cyanate detoxification by reducing bicarbonate production (Peng et al. 1993).
Fig. 1.—
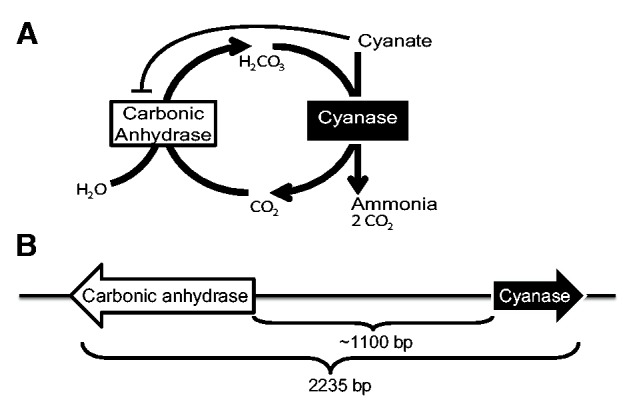
(A) Cyanase and carbonic anhydrase enzymes are both required to detoxify cyanate. Cyanase converts the toxic compound cyanate into nontoxic ammonia and carbon dioxide. Carbonic anhydrase converts carbon dioxide and water into bicarbonate, which cyanase requires in order to convert cyanate to ammonia and carbon dioxide. (B) A novel clustered form of the genes (i.e., CCA gene cluster) encoding these enzymes may increase the capacity for cyanate detoxification. In several members of the FOSC, the genes are divergently transcribed and separated on the chromosome by a highly variable intergenic region that is approximately 1,100 bp in length.
Although both cyanase and carbonic anhydrase co-occur in the majority of fungal genomes, they are not known to be physically colocalized. For example, in members of the Fusarium oxysporum species complex (FOSC), the native cyanase gene is located on chromosome 4 whereas the native carbonic anhydrase gene is on chromosome 8 (Ma et al. 2010). In the course of our studies on the evolution of fungal metabolism (McGary et al. 2013), we discovered, but did not report, a novel gene cluster (CCA gene cluster) that comprises cyanase and carbonic anhydrase gene copies on accessory chromosomes in whole-genome sequence from several members of the FOSC (fig. 1B). In this study, our analysis of 234 fungal genomes spanning the diversity of the fungal kingdom revealed that duplicates of the same genes are also clustered in Oidiodendron maius, a facultative endomycorrhizal fungus, and Cochliobolus lunatus, a pathogen of several monocots. Phylogenetic analysis of all cyanase and carbonic anhydrase gene copies shows that the CCA gene cluster originated independently at least twice, suggesting that clustering is likely a response to a shared selection pressure. Furthermore, molecular screening of 163 FOSC isolates identified 73 CCA gene cluster-containing isolates from 6 continents, 41 different hosts, diverse sequence types (STs) and forma specialis (following O’Donnell et al. 2009), as well as from several different vegetative compatibility groups (VCGs). The gene cluster’s pattern of presence and absence in FOSC isolates from the same VCG, forma specialis, and ST group across multiple continents suggests that the FOSC CCA gene cluster might also reflect a response to a recent widespread selection pressure.
Materials and Methods
Discovering the CCA Gene Cluster
The cyanase–carbonic anhydrase gene cluster was first identified through a screen of 234 fungal genomes using previously reported methods to identify metabolically linked genes that are colocated on a chromosome (Slot and Rokas 2010, 2011; Campbell et al. 2012, 2013; Zhang et al. 2012; McGary et al. 2013; Greene et al. 2014).
Phylogenetic Analysis of the CCA Gene Cluster
Cyanase sequences from F. oxysporum f. sp. lycopersici 4287 (NRRL 34936) (http://www.broadinstitute.org/, second annotation, last accessed March 1, 2012), O. maius Zn version 1.0 (http://www.jgi.doe.gov/, last updated May 24, 2011, last accessed March 1, 2012), and C. lunatus m118 version 2.0 (http://www.jgi.doe.gov/, last updated February 13, 2012, last accessed March 1, 2012) and other Pezizomycotina cyanase sequences of EC 4.2.1.104 (Kanehisa et al. 2004) were aligned with MAFFT version 6.923 b using the E-INS-I strategy (Katoh and Toh 2008) and quality trimmed with trimAl version 1.4.rev11 using the automated1 strategy (Capella-Gutierrez et al. 2009). The optimal model of sequence evolution was determined using ProtTest v2.4 (Abascal et al. 2005). Maximum-likelihood (ML) trees were built in RAxML version 7.2.8 using the PROTGAMMALGF model and clade support was assessed by 100 bootstrap pseudoreplicates (Stamatakis and Alachiotis 2010). Tests of monophyly were performed in RAxML using the log-likelihood Shimodaira–Hasegawa (SH) test to compare the unconstrained best tree and the best tree given a constrained topology (Shimodaira and Hasegawa 1999).
Identifying CCA Gene Clusters in FOSC Genomes
Cluster sequences were identified in the ten FOSC genomes available through the BROAD Institute Fusarium Comparative Database (http://www.broadinstitute.org/annotation/genome/fusarium_group/MultiHome.html, last accessed May 30, 2012; Ma et al. 2010) and the FOSC, O. maius, and C. lunatus genomes available through the Joint Genome Institute (Grigoriev et al. 2012), using the nucleotide sequence of the cluster originally identified in F. oxysporum f. sp. lycopersici 4287 as the query sequence in a tBLASTn search through the NCBI (National Center for Biotechnology Information) BLAST webpage (Altschul et al. 1997, 2005).
Phylogenetic Analysis of Clusters in Published FOSC Genomes
Nucleotide sequences of CCA gene clusters identified in published genomes were aligned in ClustalW2 (Larkin et al. 2007) and revised manually. An ML phylogeny was generated using RAxML version 7.2.6, using the GTRGAMMA model of sequence evolution with 100 bootstrap pseudoreplicates (Stamatakis and Alachiotis 2010).
Selecting FOSC Isolates
Isolates were selected from a previously reported collection of 850 FOSC strains (O’Donnell et al. 2009), which are available upon request from the ARS culture collection (NRRL, http://nrrl.ncaur.usda.gov/TheCollection/Requests.html, last accessed February 23, 2015), National Center for Agricultural Utilization Research, Peoria, Illinois. To minimize redundant screening of closely related strains, isolates were selected for screening based on ST groups, that is groups of isolates that shared identical sequences at the conserved transcription elongation factor 1-α (EF1-α) and nuclear ribosomal intergenic spacer region (IGS rDNA) loci (O’Donnell et al. 2009). In total, 163 isolates were selected to maximize coverage of the species complex and increase the likelihood that clusters would be detected using the following criteria:
Known hosts: Sampling focused on isolates found on hosts (tomato, muskmelon, cotton, and pea) of the four fully sequenced isolates known to contain the CCA gene cluster. For each ST group including isolates found on tomato, cotton, melon, or pea, one isolate was selected from this host and one more from each additional host in the ST group. In total, 110 isolates were chosen by these criteria.
Likely hosts: Sampling included hosts thought more likely to have isolates regularly exposed to cyanate: Potato and banana because they require higher doses of metam sodium (NCAP 2006), and Brassicaceae because they emit isothiocyanate compounds that function as a natural antifungal defense. We included carnation because diverse Fusarium spp. infect this host (their leaves are commonly used to induce Fusarium spp. to produce sporodochia in culture) (Leslie and Summerell 2006). For ST groups that did not contain isolates from any of the “known hosts,” one isolate from a “likely host” was selected. In total, 52 isolates were selected under this criterion.
Coverage: One additional isolate (NRRL 38296 F. oxysporum f. sp. betae) was selected so that the published EF1-α phylogeny of the species complex (O’Donnell et al. 2009) was well-covered.
Outgroup: Two isolates of F. foetens (NRRL 31852 and 38302) were selected because this species is sister to the FOSC (O’Donnell et al. 2009). Genomic DNA was extracted using a cetyltrimethyl ammonium bromide (CTAB) protocol (Stewart and Via 1993).
Primer Design
Nucleotide sequences of the cyanase and carbonic anhydrase CCA gene cluster were sufficiently divergent from native copies that the cluster could be selectively amplified. To genotype the selected isolates for the presence or absence of the gene cluster, we designed polymerase chain reaction (PCR) primers A and B, which target the 3′-ends of carbonic anhydrase and cyanase, respectively (supplementary table S5, Supplementary Material online). This primer pair amplified a 2,173-bp fragment that covered 100% of the two genes and the intergenic region in the reference genome F. oxysporum f. sp. lycopersici 4287 (NRRL 34946). Genomic DNA from F. oxysporum f. sp. lycopersici MN25 (NRRL 54003) and F. oxysporum f. sp. raphani PHW815 (NRRL 54005), whose sequenced genomes do not contain the CCA gene cluster, were used as negative controls.
In order to detect partial CCA gene clusters, PCR primers were designed to target each gene’s open reading frame (ORF) using nucleotides that were invariant in CCA gene clusters from the sequenced FOSC genomes but divergent from the native gene copies. Primer F was designed to bind near the 5′-end of the carbonic anhydrase ORF and Primer E near the 5′-end of the cyanase ORF. Primers were tested with PCR and gel electrophoresis, which revealed that they were effective in gene cluster-specific amplification. Primers G, C, and D were designed to provide greater flexibility to the PCR amplification and eventual sequencing of the cluster. Primer testing revealed that some of the interior primers did not work in every strain in which the A ⇔ B fragment was amplified.
Screening FOSC Isolates for CCA Gene Cluster
Screening consisted of two rounds of PCR and gel electrophoresis for each isolate. In all, 163 isolates were screened for the presence of clusters using primers A and B in a long distance PCR protocol with the following temperature profile: 3 min at 95 °C, followed by six cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min plus 30 s for each additional cycle, followed by 29 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 5 min. In total, 73 isolates yielded detectable bands of the appropriate size in both rounds and were thus considered positive for the CCA gene cluster.
Reverse Transcription PCR to Assess Expression of the Cluster in the FOSC
Erlenmeyer flasks (300 ml) containing 50 ml of yeast malt broth (O’Donnell et al. 2009) adjusted to pH 6.6 with potassium phosphate buffer were inoculated separately with 5 × 105 conidia of F. oxysporum f. sp. lycopersici NRRL 34936, F. oxysporum f. sp. vasinfectum NRRL 25433, F. oxysporum f. sp. melonis NRRL 26046 or F. oxysporum f. sp. pisi NRRL 37622 and then grown at 25 °C on a rotary shaker at 100 rpm for 48 h, which was designated the 0 h time point. After1-ml aliquots were removed from each flask, 1.5 ml of a 1 M sodium cyanate (product # 185086, Sigma-Aldrich, St. Louis, MO) stock was added to each flask to set the concentration at 30 mM cyanate/ml. Subsequently, 1-ml aliquots were removed from each flask at 4, 8, 24, and 48 h time points. Samples taken at the five time points were immediately placed in 1.5-ml eppendorf tubes and spun at 12,000 g in a tabletop centrifuge for 10 min to obtain a mycelial pellet. Following removal of the supernatant, the pellets were pulverized with plastic pestles in 750 ml of TRIzol reagent, then spun at 12,000 g for 3 min, after which 600 μl supernatant was recovered for use in a Direct-zol RNA extraction kit (Zymo Research Corp., Irvine, CA) that included in-column DNase treatment. Extracted RNA was quantified with a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific, Wilmington, DE) and sample quality confirmed with an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA). Reverse transcription (RT)-PCR was performed with an Agilent Easy-A One Tube RT-PCR kit, using 2 μl of RNA, an annealing temperature of 55 °C, and the following primers: Cyanate-RT: GGGTTGGAAGGGAGCCATTTG and Primer H: CTTTCGAAGGACGACTTGC. Standard PCR was then performed using the RT-PCR product as the template with the following temperature profile: 94 °C for 90 s, 40 cycles of 94 °C for 30 s, 45 s at 52 °C, 2 min at 68 °C, a final extension of 5 min at 68 °C, and ending with an indefinite 4 °C soak. PCR products were purified by vacuum filtration using Montage PCR96 filter plates (Millipore Corp., Billerica, MA) and sequenced to verify that they were transcripts of the cyanate cluster. A negative control, with water in place of the RNA template, yielded no bands during the RT-PCR or standard PCR steps.
Assessing Phylogenetic Distribution of the CCA Gene Cluster
In order to assess phylogenetic signal in the distribution of the cluster relative to the EF1-α phylogeny, we used Fritz and Purvis’s D statistic for binary character dispersion on a phylogeny (Fritz and Purvis 2010). D is given by the formula: , where is the observed sum of estimated sister-clade differences in the binary trait across the entire tree, db is the expected sum of estimated sister-clade differences in the binary trait across the entire tree under a Brownian model of evolution, and is the expected sum of estimated sister-clade differences in the binary trait across the entire tree under a random model, that is, shuffling the trait labels at the tips of the tree. D = 1 if the observed trait has a phylogenetically random distribution across the tips of the phylogeny and D > 1 if the trait has an overdispersed distribution. D = 0 if the trait has a clumped distribution according to a Brownian motion model.
Sequencing
Sanger sequencing of the entire gene cluster of each of the 73 cluster-positive isolates was performed for three fragments: A, G/C, and B. Primer G gave better coverage, so C was only used when G failed to amplify in a particular isolate. Primers G and C failed to amplify in six isolates, so only primers A and B were used in these cases. After the A ⇔ B PCR product from the 73 cluster-positive strains was sequenced at GENEWIZ, Inc. the chromatogram files were edited with PeakScanner software v1.0 (Applied Biosystems) to insure they were error free. Sequences are available in GenBank: Accessions KJ950372–KJ950435.
Assembly, Editing, and Alignment of Sequences
Isolates with amplicons that failed to sequence and those with less than 50% coverage of the region were excluded from the analysis, bringing the total number of isolates represented in the alignment to 59. Fragments that were sequenced with the A, G/C, and B primers were assembled by profile alignment in the software program SeaView against a known gene cluster and then manually spliced into one composite sequence for each isolate (Gouy et al. 2010). Assembled sequences were aligned with the global alignment software MAFFT version 6.847 (Katoh and Toh 2008) and then trimmed to remove poorly aligned nucleotide sites with TrimAl software using the “gappyout” option (Capella-Gutierrez et al. 2009). EF1-α sequences for all of the isolates included in the alignment were downloaded from the FUSARIUM-ID version 1.0 database (http://isolate.fusariumdb.org/, last accessed February 23, 2015; Geiser et al. 2004) and aligned with MAFFT (Katoh and Toh 2008). All alignments are available through TreeBASE, submission 15573.
Constructing Phylogenies
Phylogenies were constructed with RAxML software version 7.2.6, using the GTRGAMMA model of sequence evolution with 100 bootstrap pseudoreplicates (Stamatakis and Alachiotis 2010). Separate phylogenies were inferred for the whole cluster, carbonic anhydrase, cyanase, the concatenated genes, IGS rDNA, and EF1-α partitions. The CCA sequence from F. oxysporum f. sp. melonis NRRL 26406 was removed because it was missing a large portion of the carbonic anhydrase ORF and thereby distorting the phylogenies through long-branch attraction. Consensus phylogenies were then constructed using DENDROPY software with a “majority rules” threshold bootstrap support frequency of 0.50 at each bipartition (Sukumaran and Holder 2010). The best ML trees were compared visually using the MirrorTree MTSERVER (http://csbg.cnb.csic.es/mtserver/, last accessed February 23, 2015; Ochoa and Pazos 2010).
Results
The CCA Gene Cluster Evolved Independently at Least Twice in Fungi
During previous work (McGary et al. 2013), we serendipitously noted the physical linkage of the genes encoding cyanase and carbonic anhydrase (a CCA gene cluster). Here, we report that screening of 234 fungal genomes (supplementary table S1, Supplementary Material online) for CCA gene clusters revealed their presence in three genomes, those of the facultative endomycorrhizal fungus O. maius (Leotiomycetes), the fungal plant pathogen C. lunatus (Dothideomycetes) and the FOSC (Sordariomycetes), as represented by F. oxysporum f. sp. lycopersici NRRL 34936. In each of these genomes, the CCA cluster genes are separated by approximately 600–2,400 bp and divergently oriented, that is, the genes are immediately adjacent and transcribed in opposite directions, which is also commonly referred to as a head-to-head arrangement. Examination of the ten genes on either side of each CCA gene cluster (or to the end of the assembly scaffold) revealed no shared synteny, and BLAST2Seq pairwise searches of the intergenic regions revealed no meaningful similarity between the clusters from each species with the longest hit limited to 14 bp and the lowest Expect value only 0.045.
The cyanase genes from the CCA gene clusters of the FOSC, C. lunatus, and O. maius do not group together in the ML phylogeny, suggesting that the CCA gene cluster originated independently in these three taxa (fig. 2). Even though average bootstrap support in the cyanase gene phylogeny was low (66.1%), the bootstrap values supporting the independent origins of the CCA gene cluster in the three taxa were 100% (for O. maius vs. C. lunatus), 100% (for the FOSC vs. C. lunatus), and 83% (for the FOSC vs. O. maius). Furthermore, an SH test, which compared the ML-supported phylogeny with a tree in which all clustered cyanase genes were constrained to form a monophyletic group, rejected the hypothesis of a single origin for the CCA gene cluster (P-value threshold for all SH tests ≤0.05; supplementary table S2, Supplementary Material online).
Fig. 2.—

ML tree of cyanase (EC 4.2.1.104) in the Pezizomycotina. Numbers above branches correspond to bootstrap support ≥50. Species containing a CCA gene cluster are shown in bold, and letters in parentheses indicate whether the gene copies are part of a CCA gene cluster (C) or are unclustered (U). SH tests support at least two independent origins of the CCA gene cluster (P ≤ 0.05; supplementary table S2, Supplementary Material online).
SH tests were unable to reject the monophyly of clustered cyanase genes from the FOSC and O. maius, suggesting at least two independent origins of the CCA gene cluster, once for the cluster in C. lunatus and once for the cluster in the FOSC and O. maius (supplementary table S2, Supplementary Material online). However, if the CCA gene cluster present in the FOSC and O. maius had the same origin, explaining its presence in these two species would seem to imply an additional, recent horizontal transfer between the FOSC and O. maius. Finally, the SH tests from a second phylogenetic analysis using increased taxon sampling from 163 Pezizomycotina genomes showed similar results, finding support for at least two separate origins of the CCA gene cluster (supplementary fig. S1 and table S3, Supplementary Material online).
In contrast to the cyanase gene phylogeny, the anhydrase gene phylogeny had much lower average bootstrap support (55.5%) and was much less informative about taxonomic relationships (supplementary fig. S2, Supplementary Material online). Unsurprisingly, SH tests were unable to reject most of the topological constraints imposed (supplementary table S2, Supplementary Material online).
Characterizing the CCA Gene Cluster within the FOSC
To further understand the evolution of the CCA gene cluster in the FOSC, we first examined the sequenced genomes of two related Fusarium species, F. graminearum and F. verticillioides. Orthologous CCA cluster genes were not detected in the genomes of these two species. Analyses of ten fully sequenced FOSC genomes revealed that four contained between one and three copies of the CCA gene cluster, and the other six lacked the cluster. Specifically, F. oxysporum f. sp. lycopersici NRRL 34936 possessed three nearly identical copies of the CCA gene cluster, F. oxysporum f. sp. vasinfectum NRRL 25433 two copies, and F. oxysporum f. sp. pisi HDV247 (NRRL 37622) and F. oxysporum f. sp. melonis NRRL 26406 one copy each (fig. 3). In contrast to the conserved locations of the native cyanase and carbonic anhydrase copies on the core chromosomes, all copies of the CCA gene cluster in the four cluster-containing isolates occupied different positions on multiple accessory chromosomes.
Fig. 3.—

Phylogenic analysis of native and clustered forms of cyanase and carbonic anhydrase supports a single origin of the cluster within the FOSC. The phylogeny revealed that multiple clusters in F. oxysporum f. sp. lycopersici 34936 and F. oxysporum f. sp. vasinfectum 25433 are copy number polymorphisms, most likely the result of very recent intragenomic duplication. For example, in F. oxysporum f. sp. lycopersici 34936, two of the three copies are identical, and a third differs from these two by only four SNPs, indicating an extremely recent duplication. The single copy native genes of F. graminearum and F. verticillioides were added to root the tree. Strains listed only by NRRL number and host are members of the FOSC.
To further characterize the CCA gene cluster within the FOSC, we screened 163 representative isolates from the ARS Culture Collection (NRRL) (supplementary table S4, Supplementary Material online). Approximately 45% of the isolates (73/163) contained the CCA gene cluster (supplementary table S4, Supplementary Material online). To examine whether the CCA gene cluster was functional, RT-PCR and sequencing were used to assess whether the genes were expressed in the presence and absence of cyanate. This experiment revealed that the genes of the cluster were expressed in the four cluster-containing isolates tested (table 1). Intriguingly, three of the isolates appeared to express the cluster genes constitutively, whereas expression was induced in F. oxysporum f. sp. lycopersici NRRL 34936 only after exposure to 30 mM cyanate.
Table 1.
Expression of the CR Genes in Four Cluster-Positive Strains over 2 Days of Growth in Liquid Culture
| Strain | 0 h | 4 h | 8 h | 24 h | 48 h |
|---|---|---|---|---|---|
| Fol NRRL 34936 | − | + | + | + | + |
| Fov NRRL 25433 | + | + | + | + | + |
| Fom NRRL 26406 | + | + | + | + | + |
| Fop NRRL 37622 | + | + | + | + | + |
Note.—Shaded cells indicate RT-PCR results were confirmed by sequencing. Note that three of the four strains appeared to express the cluster genes constitutively prior to the addition of cyanate at the zero time point. However, expression was only detected in Fusarium oxysporum f. sp. lycopersici NRRL 34936 after exposure to 30 mM cyanate. Fol, F. oxysporum f. sp. lycopersici; Fov, F. oxysporum f. sp. vasinfectum; Fom, F. oxysporum f. sp. melonis; Fop, F. oxysporum f. sp. pisi.
Distribution of the CCA Gene Cluster in the FOSC Does not Correlate with Geography, Genetic Structure, or Host
The 163 isolates screened for the presence of the CCA gene cluster were selected to represent the full range of diversity within the FOSC; they were from 6 different continents, included members of 6 VCGs and 20 formae speciales and were isolated from 41 different hosts. Thus, this screen allowed us to examine whether the presence of the CCA gene cluster was correlated with geography, host, or genetic structure. The CCA gene cluster was identified over a very broad geographical range in isolates collected on six different continents (fig. 4). Moreover, the six VCGs that contained isolates with the CCA gene cluster also contained isolates that lacked it. With the exception of isolates obtained from human hosts, the CCA gene cluster was detected in at least one isolate from every host that was represented by more than one isolate. Furthermore, in every instance where two or more isolates from the same host were screened, the CCA gene cluster was never found to be present in all isolates from the same host.
Fig. 4.—

Cladogram of EF1-α nucleotide sequences including the 163 FOSC isolates screened for the presence of a CCA gene cluster. Cluster-containing isolates, indicated by the solid black circles, are labeled with the host and continent from which they were isolated. Af, Africa; As, Asia; Au, Australia; Eu, Europe; NA, North America; SA, South America; Un, unknown.
Rapid Spread of the CCA Gene Cluster among the FOSC
Given the lack of any pattern to the distribution of the CCA gene cluster by geography, genetic structure, or host, we next investigated the evolutionary dynamics of the CCA gene cluster. Using the Fritz–Purvis D statistic, which measures the phylogenetic signal of a binary trait, we evaluated how well the CCA gene cluster’s distribution across the 163 screened isolates corresponded to the isolates’ phylogeny (Fritz and Purvis 2010). The distribution of the CCA gene cluster was found to only weakly resemble the phylogeny of the isolates (estimated D value [ED] = 0.83, P(no phylogenetic structure) = 0.013; P(Brownian motion) = 0.0), suggesting that the distribution of the CCA gene cluster was not primarily determined by vertical descent. At least, two scenarios are compatible with this result. In the first, the distribution of the cluster lacks a phylogenetic signal because the cluster has been rampantly duplicated and rapidly lost from many strains in a phylogenetically random manner. In the second, the distribution of the cluster is due to very recent, rapid horizontal spread of the cluster between strains by the exchange of accessory chromosomes.
Given that the distribution of the CCA gene cluster contained little phylogenetic signal, it was not surprising that our direct comparison of the CCA gene cluster tree to the species phylogeny inferred from EF1-α (O’Donnell et al. 2009) showed no discernible pattern of relationship between the two trees (fig. 5, supplementary figs. S3 and S4, Supplementary Material online). Consider, for example, that the CCA tree indicates a clade with 96% bootstrap support that includes the isolates 34936, 38446, and 38435 (fig. 5, in bold); however, the isolate are well dispersed in the EF1-α tree in separate clades with high bootstrap support (93%, 100%, and 100%, respectively). This discordance goes both ways; in the EF1-α tree the clade containing isolates 38446 and 38364 has 100% support, but in the CCA cluster tree the isolates are separated by several branches including one with 100% support.
Fig. 5.—

Mirrortree comparison of EF1-α (left) and whole CCA gene cluster (right) phylogenies. Lines connecting the same isolate indicate the two loci have very different evolutionary histories. Examples discussed in the text are highlighted with bold lines and labels.
The discrepancy between the inferred histories of CCA cluster and the isolates is also clear from inspection of their sequence divergences. For example, closely related isolates often had very distantly related copies of the CCA gene cluster, for example, isolate NRRL 38302 and isolate NRRL 38364 shared 97.5% nucleotide identity in EF1-α but 88% nucleotide identity in the CCA gene cluster. EF1-α is a highly conserved protein that is expected to evolve far more slowly than the CCA gene cluster, which includes an intergenic sequence, so the converse comparison is even more convincing: Distantly related isolates may have nearly identical CCA gene clusters, for example, isolate 38302 and isolate 45908 have 97.8% nucleotide identity in EF1-α and 98.8% nucleotide identity in the CCA gene cluster. In fact, of a total of 12 isolates that shared a nearly identical CCA gene cluster sequence with one or more other isolates (at 99% average nucleotide identity among groups), only two, isolates 34115 and 25609, also shared identical EF1-α sequences. Together, these results argue that the CCA gene cluster spread recently and rapidly through nonvertical means.
Discussion
Our investigation reveals a complex history for the CCA gene cluster with more than one possible explanation. Nevertheless, in evaluating the evidence, we consider one scenario the most likely (fig. 6): The CCA gene cluster emerged independently in three different classes of Pezizomycotina: C. lunatus in the Dothidiomycetes, the FOSC in the Sordariomycetes, and O. maius in the Leotiomycetes. In C. lunatus and O. maius, the CCA gene cluster appears to have formed by rearrangement of cyanase and carbonic anhydrase paralogs of the native genes in these species. In the FOSC, the CCA gene cluster probably arose by the same process of duplication and rearrangement of native genes in a single isolate, but on an accessory chromosome, which subsequently spread into other isolates by some combination of gene duplication and horizontal transfer (both processes are likely facilitated by accessory chromosomes). Alternatively, the CCA gene cluster may have originated only twice, once in C. lunatus and once in either O. maius or the FOSC, after which it spread to the other species through horizontal transfer. In either case, the distribution and inferred phylogeny of the CCA gene cluster in the purportedly clonal FOSC suggest widespread transfer independent of the genetic structure of the lineages inferred from VCGs. The most likely molecular mechanism of CCA gene cluster spread is through the exchange of accessory chromosomes, which are known to transfer between isolates (Ma et al. 2010).
Fig. 6.—

A model for the independent origin, through similar mechanisms, of the CCA gene clusters in C. lunatus, O. maius, and the FOSC. (1) The initial step to form a CCA gene cluster is the duplication of both the native cyanase and carbonic anhydrase genes, possibly in response to selection pressure imposed from increased levels of cyanate in the environment. (2) The CCA gene cluster forms when the duplicated cyanase or carbonic anhydrase relocates to the chromosomal location of the complementary duplicate gene. Alternatively, only one of the two genes may be duplicated initially and the CCA cluster forms in place simultaneously with the duplication of the complementary gene. (3) In the FOSC, the CCA gene cluster is consistently located on accessory chromosomes where the entire cluster is frequently duplicated and transferred to additional accessory chromosomes, which appears to facilitate transfer among clonal lineages within the FOSC and may contribute to substantial copy number variation between isolates. Large gray arrows represent the carbonic anhydrase ORF and large black arrows the cyanase ORF, with a thick black line indicating the chromosome. An alternative model in which the CCA gene cluster originated only twice, once in C. lunatus and once in either O. maius or the FOSC, after which it spread to the other species through horizontal transfer, is also consistent with our data.
The independent evolution of the CCA gene cluster in species from at least two and possibly three distinct classes (Leotiomycetes, Sordariomycetes and Dothidiomycetes; fig. 2) from varying ecological niches suggests that they have independently converged on a similar response to a similar selection pressure. In each class, the CCA gene cluster is found only in a single species and comprises paralogous copies that are not highly diverged from the native genes copies, which together suggest that the origins of the CCA gene clusters in all three lineages are relatively recent. The presence of the CCA cluster in FOSC isolates from six continents and nearly every host and forma specialis sampled further indicates that the cluster arose in response to a widespread, recent selection pressure. The fact that all CCA gene cluster copies in the four fully sequenced isolates occupy nonhomologous chromosomal locations suggests that it is selection for the CCA gene cluster itself that is driving its spread, rather than mere hitchhiking on the accessory chromosomes. The location of all CCA gene cluster copies on accessory chromosomes, which shuffle their contents rapidly through TE-mediated ectopic recombination and a breakage-fusion-bridge mechanism (Croll et al. 2013), also offers a plausible mechanism for the rapid independent spread of the CCA gene cluster.
Fungi are likely to be regularly exposed to naturally produced cyanates, particularly from plants. Thus, it is possible that these three CCA gene clusters arose in response to selection pressure from naturally occurring cyanate compounds. If that is the case, the fact that similar CCA gene clusters are absent from most fungal genomes that have both CA and cyanase suggests that this pressure is either quite specific or quite recent. Indeed, C. lunatus infects sorghum, a plant known to poison animals with cyanide when grown in stressful conditions, particularly drought (Gillingham et al. 1969). Alternatively, clustering of these two genes may be the signature of a genomic response to the global popularity of cyanate fungicides in agriculture over the past four decades. As Fusarium infections are commonly treated with cyanate-based fungicides (AMVAC Chemical Corporation 2013), the rapid nonvertical spread of the CCA gene cluster in the FOSC across multiple continents is consistent with this hypothesis. Although many other fungal species regularly exposed to cyanate fungicides do not have the CCA gene cluster, the incomplete spread of the CCA gene cluster through the FOSC, despite the accessory chromosomes that provide a mechanism for rapid transfer, implies that clustering of these two genes may simply not have had opportunity to arise and spread through the population of many fungal species.
The third species with the CCA cluster, O. maius, is not a pathogen, so it is not specifically targeted for fungicide treatment. However, as a facultative endomycorrhizal symbiont of cultivated plants including blueberry, azalea and rhododendron, O. maius is likely exposed to cyanate fungicides during treatment of other infections (Hagan and Cobb 1996), although it is possible that some of its hosts produce cyanates as well. Regardless of cyanate source, our discovery of the CCA gene cluster in three unrelated fungal species raises the hypothesis that this remarkable case of convergent evolution represents an adaptive response to exposure to natural or agricultural cyanate.
Supplementary Material
Supplementary data file S1, figures S1–S4, and tables S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Nathane Orwig for running some of the DNA sequences in the NCAUR DNA Core Facility. The mention of firm names or trade products does not imply that they are endorsed or recommended by the US Department of Agriculture over other firms or similar products not mentioned. The USDA is an equal opportunity provider and employer. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville, TN. This work was supported by funds provided by a Vanderbilt Undergraduate Summer Research Program Fellowship to M.H.E. and the National Science Foundation (2013170866 to M.H.E. and DEB-0844968 and DEB-1442113 to A.R.).
Literature Cited
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Albaum HG, Tepperman J, Bodansky O. The in vivo inactivation by cyanide of brain cytochrome oxidase and its effect on glycolysis and on the high energy phosphorus compounds in brain. J Biol Chem. 1946;164:45–51. [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMVAC Chemical Corporation. 2013. VAPAM® HL: A Soil Fumigant Solution for all Crops. Available from: http://www.amvac-chemical.com/products/documents/Vapam%20TB.sprd.c21%20copy.pdf. [Google Scholar]
- Campbell MA, Rokas A, Slot JC. Horizontal transfer and death of a fungal secondary metabolic gene cluster. Genome Biol Evol. 2012;4:289–293. doi: 10.1093/gbe/evs011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MA, Staats M, Kan JV, Rokas A, Slot JC. Repeated loss of an anciently horizontally transferred gene cluster in Botrytis. Mycologia. 2013;105:1126–1134. doi: 10.3852/12-390. [DOI] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll D, Zala M, McDonald BA. Breakage-fusion-bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet. 2013;9:e1003567. doi: 10.1371/journal.pgen.1003567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbs S. Biological degradation of cyanide compounds. Curr Opin Biotechnol. 2004;15:231–236. doi: 10.1016/j.copbio.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Elleuche S, Pöggeler S. Carbonic anhydrases in fungi. Microbiology. 2010;156:23–29. doi: 10.1099/mic.0.032581-0. [DOI] [PubMed] [Google Scholar]
- Fritz SA, Purvis A. Selectivity in mammalian extinction risk and threat types: a new measure of phylogenetic signal strength in binary traits. Conserv Biol. 2010;24:1042–1051. doi: 10.1111/j.1523-1739.2010.01455.x. [DOI] [PubMed] [Google Scholar]
- Geiser DM, et al. FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. Eur J Plant Pathol. 2004;110:473–479. [Google Scholar]
- Gillingham JT, Shirer MM, Starnes JJ, Page NR, McClain EF. Relative occurrence of toxic concentrations of cyanide and nitrate in varieties of sudangrass and sorghum-sudangrass hybrids. Agron J. 1969;61:727. [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Greene GH, McGary KL, Rokas A, Slot JC. Ecology drives the distribution of specialized tyrosine metabolism modules in fungi. Genome Biol Evol. 2014;6:121–132. doi: 10.1093/gbe/evt208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev IV, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012;40:D26–D32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan AK, Cobb PP. 1996. Controlling insects and diseases on azaleas and rhododendrons. Available from: http://www.agrisk.umn.edu/cache/arl01681.htm. [Google Scholar]
- Jain A, Kassner RJ. Cyanate binding to the ferric heme octapeptide from cytochrome c. A model for anion binding to high spin ferric hemoproteins. J Biol Chem. 1984;259:10309–10314. [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Leslie JF, Summerell BA, editors. The Fusarium laboratory manual. Blackwell Publishing; 2006. Media—recipes and preparation; pp. 3–14. [cited 30 Sep 2014]. Ames, IA: Blackwell Publishing. [Google Scholar]
- Ma L-J, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary KL, Slot JC, Rokas A. Physical linkage of metabolic genes in fungi is an adaptation against the accumulation of toxic intermediate compounds. Proc Natl Acad Sci U S A. 2013;110:11481–11486. doi: 10.1073/pnas.1304461110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCAP Northwest Coalition for Alternatives to Pesticides. Metam sodium facts sheet. J Pestic Ref. 2006;26:12–16. [Google Scholar]
- Nishida H, Beppu T, Ueda K. Symbiobacterium lost carbonic anhydrase in the course of evolution. J Mol Evol. 2009;68:90–96. doi: 10.1007/s00239-008-9191-4. [DOI] [PubMed] [Google Scholar]
- Ochoa D, Pazos F. Studying the co-evolution of protein families with the Mirrortree web server. Bioinformatics. 2010;26:1370–1371. doi: 10.1093/bioinformatics/btq137. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, et al. A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genet Biol. 2009;46:936–948. doi: 10.1016/j.fgb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Peng Z, Merz KM, Banci L. Binding of cyanide, cyanate, and thiocyanate to human carbonic anhydrase II. Proteins. 1993;17:203–216. doi: 10.1002/prot.340170209. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–1116. [Google Scholar]
- Slot JC, Rokas A. Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. Proc Natl Acad Sci U S A. 2010;107:10136–10141. doi: 10.1073/pnas.0914418107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JC, Rokas A. Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr Biol. 2011;21:134–139. doi: 10.1016/j.cub.2010.12.020. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Alachiotis N. Time and memory efficient likelihood-based tree searches on phylogenomic alignments with missing data. Bioinformatics. 2010;26:i132–i139. doi: 10.1093/bioinformatics/btq205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CN, Via LE. A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques. 1993;14:748–750. [PubMed] [Google Scholar]
- Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26:1569–1571. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- Wilson D, et al. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009;37:D380–D386. doi: 10.1093/nar/gkn762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Rokas A, Slot JC. Two different secondary metabolism gene clusters occupied the same ancestral locus in fungal dermatophytes of the Arthrodermataceae. PLoS One. 2012;7:e41903. doi: 10.1371/journal.pone.0041903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.