

Abstract
Introduction
Pediatric adamantinomatous craniopharyngioma (ACP) is a histologically benign but clinically aggressive brain tumor that arises from the sellar/suprasellar region. Despite a high survival rate with current surgical and radiation therapy (75–95 % at 10 years), ACP is associated with debilitating visual, endocrine, neurocognitive and psychological morbidity, resulting in excheptionally poor quality of life for survivors. Identification of an effective pharmacological therapy could drastically decrease morbidity and improve long term outcomes for children with ACP.
Results
Using mRNA microarray gene expression analysis of 15 ACP patient samples, we have found several pharmaceutical targets that are significantly and consistently overexpressed in our panel of ACP relative to other pediatric brain tumors, pituitary tumors, normal pituitary and normal brain tissue. Among the most highly expressed are several targets of the kinase inhibitor dasatinib – LCK, EPHA2 and SRC; EGFR pathway targets – AREG, EGFR and ERBB3; and other potentially actionable cancer targets – SHH, MMP9 and MMP12. We confirm by western blot that a subset of these targets is highly expressed in ACP primary tumor samples.
Conclusions
We report here the first published transcriptome for ACP and the identification of targets for rational therapy. Experimental drugs targeting each of these gene products are currently being tested clinically and pre-clinically for the treatment of other tumor types. This study provides a rationale for further pre-clinical and clinical studies of novel pharmacological treatments for ACP. Development of mouse and cell culture models for ACP will further enable the translation of these targets from the lab to the clinic, potentially ushering in a new era in the treatment of ACP.
Introduction
Adamantinomatous craniopharyngioma (ACP) is the most common non-neural brain tumor with an incidence of approximately 1.9 cases/million patient-years in children [1–3]. Due to its sensitive sellar/suprasellar location and propensity to form large cysts, ACP often compresses and damages vital structures of the pituitary, hypothalamus and visual apparatus. Although seemingly well-demarcated on neuroimaging studies, histology reveals finger-like protrusions extending into neighboring visual and hypothalamic structures, eliciting tissue damage and gliosis [4]. This propensity to invade adjacent structures, in addition to the difficult surgical location, often precludes total resection in order to avoid the significantly increased risk of visual and hypothalamic damage associated with attempts to completely remove the tumor [5–8]. The current standard of subtotal resection followed by radiation reduces some of the morbidity, however, it makes recurrence relatively common, even after apparently successful primary therapy. Outcomes after recurrence are poorer, with significantly higher mortality and morbidity than after primary treatment [9–11].
While conservative surgery and radiation confer low mortality, the morbidity for survivors is still unacceptably high. Variable morbidities are associated with ACP but include endocrine, neurological, vascular, psychological and visual deficits [12]. As a result, ACP has been associated with the lowest quality of life (QoL) scores of any pediatric brain tumor [13]. Lifelong care is necessary for most childhood craniopharyngioma patients and ACP and is considered by many to be a chronic disease [14]. The introduction of rational therapy to treat craniopharyngioma could drastically reduce the morbidity associated with both the primary disease and current treatments by reducing the extent of resection and/or reducing or eliminating the need for subsequent radiation. Such a paradigm change in ACP treatment is critical to improving long term QoL for patients with this debilitating disease.
Progress regarding our understanding of the biological drivers of ACP growth has been slowed by the relative rarity of the tumor and the recalcitrance of ACP cells to laboratory growth. Lack of knowledge of the underlying biology, combined with the clinical complexity of ACP have led to an absence of standard systemic antitumor therapies. Few attempts to remedy this deficit have been made, in part because current therapy has acceptable survival outcomes. Nevertheless, substantial progress has been made recently through tissue banking collaborations and “omics” approaches.
Virtually all craniopharyngiomas in childhood are of the adamantinomatous type (ACP), contrasting with adults in whom up to 10 % of craniopharyngiomas are papillary, and are now known to be driven by BRAF V600E mutations [15]. The only known genetic alterations in adamatinomatous craniopharyngioma (ACP) are point mutations in exon 3 of CTNNB1 that lead to β-catenin accumulation and upregulation of downstream target gene expression. While the reported frequency of CTNNB1 sequence alterations ranges from 16–100 % [16–19], Brastianos and colleagues [15] recently used whole exome sequencing and mass spectrometric genotyping to identify CTNNB1 mutations in 92–96 % of ACP. It is likely, however, that genetic, epigenetic or other biological factors in addition to CTNNB1 mutation contribute to the pathogenesis of ACP. For instance, Larkin and colleagues [20] described 2 tumors that harbored alterations in both CTNNB1 and BRAF. Furthermore, ACP tumors with CTNNB1 mutation contain cells that do not demonstrate intranuclear β-catenin accumulation [21] and it has been suggested that some of the cells that comprise the tumor may not actually be CTNNB1 mutant “tumor” cells at all [22]. EGFR pathway activation has also recently been identified as a driver of migration and growth using in-vitro and xenotransplant models of ACP, supporting the testing of EGFR targeted therapies [23, 24]. In addition, through an embryonic mouse model of human ACP, the role of pituitary stem cells in ACP tumorigenesis is being explored [22, 25, 26].
The recent identification of BRAF mutations in papillary craniopharyngioma changes the paradigm in treating this (primarily adult) tumor because of the availability of BRAF V600E-specific inhibitors. By contrast, the identification of β-catenin/Wnt signaling as a driver of adamantinomatous craniopharyngioma (ACP) is of little use in guiding therapy because inhibitors of Wnt signaling downstream of β-catenin/TCF/LEF are not yet clinically viable [27]. Global gene expression analysis is therefore critical for determining the epigenetic effect of aberrant β-catenin driven transcription in ACP in order to find targets for rational therapy [22, 28].
Materials and methods
Tumor samples
A total of 15 ACP tumor samples were included in this study. Eleven specimens were from patients who underwent surgical procedures at Children’s Hospital Colorado, from 1995 through 2014. Tumor samples were collected at the time of surgery and snap frozen in liquid nitrogen or fixed in formalin and paraffin embedded. Additional specimens were contributed by the University of Alabama, Columbia University and Phoenix Children’s Hospital. The median age of this cohort was 7 years (range 0 to 18 years) (Table 1). Purity of ACP tumor samples was determined by histological analysis using hematoxylin and eosin staining in addition to immunostaining for β-catenin. A further 176 samples of other primary tumors and a variety of normal cerebral tissues were used for comparative purposes. This cohort included samples from the spectrum of pediatric and adult brain tumor types (20 atypical teratoid/rhabdoid tumor (AT/RT), 5 choroid plexus papilloma (CPP), 46 ependymoma (EPN), 12 glioblastoma (GBM), 22 medulloblastoma (MED), 9 meningioma (MEN), 15 pilocytic astrocytoma (PA), 13 primitive neuroepithelial tumor (PNET)) and other peripheral pediatric solid tumors (6 malignant peripheral nerve sheath tumors (MPNST), 8 rhabdomyosarcoma (RMS)). Specimens were classified according to WHO international histological tumor classification. Normal pediatric brain samples from a variety of anatomic sites were obtained during routine epilepsy surgery or autopsy at Children’s Hospital Colorado. All samples were obtained in compliance with internal review board regulations (COMIRB #95-500 and #09-0906).
Table 1.
β-catenin and BRAF mutational status of tumor and age of 15 ACP patient cohort used in transcriptome study
| UPN | β-Catenin | BRAF | Age at Dx |
|---|---|---|---|
| 411 | S33F | WT | 2 |
| 463 | S37F | WT | 4 |
| 598 | S37C | WT | 18 |
| 646 | D32N | WT | 7 |
| 673 | S37F | WT | 2 |
| 740 | S33F | WT | 12 |
| 802 | WT | WT | 7 |
| 883 | WT | WT | 6 |
| 956 | S37C | WT | 9 |
| 980 | D32N | WT | 13 |
| 1000 | S45F | WT | 0 |
| 9109 | WT | WT | 9 |
| 9201 | T41I | WT | - |
| 9202 | T41I | WT | - |
| 9302 | WT | WT | - |
Nucleic acid extraction, amplification and microarray preparation
RNA from all surgical specimens was extracted, amplified, labeled and hybridized to Affymetrix HG-U113 plus 2 microarray chips (Affymetrix, Santa Clara, CA, USA) according to manufacturer’s instructions and as described previously [29]. SNaPshot analysis for CTNNB1 and BRAF mutations was performed at the University of Colorado Pathology Core per manufacturer’s instructions and as described previously [22, 30]. Patient characteristics, including presence or absence of CTNNB1 mutations by SNaPshot analysis, are shown in Table 1. SNaPshot analysis was also used to examine BRAF mutational status (Table 1), specifically BRAF V600E, which was recently identified in papillary craniopharyngioma [20]. This mutation was not found in any ACP sample tested.
Microarray data analysis
Data analysis was performed in R (http://www.r-project.org), using packages publicly available through Bioconductor (http://www.bioconductor.org). As a first step, the scanned microarray data were background corrected and normalized using the gcRMA algorithm [31] resulting in log 2 gene expression values. Publically available microarray CEL file data were obtained for 9 normal pituitary and 14 pituitary adenoma samples from GEO (GSE26966); these samples were chosen because they were processed in the same manner as ours and through the same microarray core lab [32]. These were combined with the ACP, other tumor, and normal brain cohort as detailed above. Multiple probesets for a gene were then collapsed to 1 entry per gene, based on the mean best-expressed probeset for that gene. Hierarchical clustering was performed using the normalized gene expression data. Distances based on Spearman correlations were calculated for input to an agglomerative algorithm with use of complete linkage, as implemented in the Bioconductor hclust function. These microarray data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database [33] and are publicly accessible through GEO Series accession number GSE68015 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68015).
Differential gene expression between ACP and multiple other tumor and normal tissue types was calculated using the Bioconductor limma function [34]. Data were filtered before input to eliminate genes not expressed in any samples or that showed only limited variance across samples. The limma function performs pair-wise comparisons between a target group and each of the other user-defined groups in the dataset. It uses an Empirical Bayes approach to calculate a moderated t-statistic and calculates a false discovery rate (FDR) that accounts for multiple testing both within and across groups. The unique molecular signature of ACP was defined as genes that showed significant differential expression in all of the pair-wise comparisons (FDR ≤0.1 and mean fold difference ≥1.5) and for which the mean difference was in the same direction when compared with all of the other clusters (i.e., either all upregulated or all downregulated in all of the comparisons with respect to ACP).
In further analyses, individual tumor gene expression of select genes involved in putative ACP development/biology were extracted from the normalized dataset. For these comparisons, normalized hybridization intensity values for a selected gene for individual ACP samples were presented as fold-difference relative to the average of all other tumor and normal samples.
Data analysis using hierarchical clustering, NIH Database for Annotation, Visualization, and Integrated Discovery (DAVID) bioinformatics tools has been previously described [29]. Functional analysis of genes was performed with DAVID using the Gene Ontology, Protein Analysis Through Evolutionary Relationships (PANTHER) Biological Process and KEGG databases [35–37]. ACP upregulated genes were compared to known targets of FDA approved and other oncology drugs that had reached clinical trial stage, as published in the literature and compiled in Ingenuity’s KnowledgeBase (Ingenuity Systems, www.ingenuity.com). Oncology drug target gene expression in individual ACP samples (n = 15) was compared to all other tumor and normal tissue combined and assigned percentile scores. Drug target genes that were consistently highly expressed (>66th percentile) in all ACP samples were identified, and then ranked according to the highest average percentile expression.
Western blot validation of functional isoforms of putative drug targets
Protein levels of a selection of putative drug target genes in ACP were examined by Western blot analysis to validate the results of microarray analysis. Snap frozen tumor samples were homogenized in RIPA buffer (Sigma) supplemented with protease and phosphatase inhibitors (Roche). This study utilized 6 ACP samples and 3 each of AT/RT, EPN, GBM, MED, PA and normal brain (obtained from autopsy material). Proteins samples (30 μg) were resolved on a 26 well Criterion Gel (BioRad) and transferred to Immobilon PVDF membrane (Millipore). Membranes were probed with antibodies to SHH (Millipore #06-1106; 1:1000), MMP9 (Cell Signaling #3852; 1:1000), MMP12 (R&D Systems #AF917; 1:1000) and Actin (Cell Signaling #12262; 1:10,000).
Results
Identification of potential drug targets and ACP signature genes
The β-catenin and BRAF mutational status of each tumor sample is presented in Table 1. To explore the clinical relevance of our human pediatric ACP transcriptomic data, we screened the ACP signature for upregulation of genes associated with potential oncological drug targets for the treatment of these tumors. Genes with high expression in ACP samples (n = 15) versus all other normal and neoplastic tissue samples (n = 195) were compared to known targets of oncology drugs as published in the literature and compiled in IPA KnowledgeBase. This approach identified 13 drug target transcripts that were consistently overexpressed greater than the 66th percentile in ACP (Table 2). Three of the 13 consistently elevated drug target genes, LCK (lymphocyte-specific protein tyrosine kinase) (5.5 fold change (FC) in ACP versus all other samples combined, p = 8.2 × 10−16), EPHA2 (ephrin type-A receptor 2) (FC = 16.6, p = 6.8 × 10−16) and SRC (SRC proto-oncogene, non-receptor tyrosine kinase) (FC = 2.5, p = 3.7 × 10−7), are targets of a single FDA-approved drug, dasatinib (Fig. 1a). Other combinations of genes overexpressed by ACP that could be targeted by a single drug were identified. SHH (sonic hedgehog homolog) was shown to be highly expressed (FC = 96.9, p = 1.5 × 10−48) by ACP in this study (Fig. 1b), which is consistent with a recently developed mouse model of ACP [25] and is the focus of oncology drug development for a number of tumor types. SHH is translated as an inactive precursor 45 kDa protein that is cleaved to generate an active 19 kDa isoform. As a putative drug target in ACP, we measured the levels of precursor and active isoforms for SHH by Western blot analysis. Isoform levels were compared to a panel of other pediatric brain tumor types and normal brain. This revealed that although the inactive precursor is expressed broadly across all tumor types, the active isoform is present at high levels only in ACP (Fig. 2). ACP was also found to overexpress MMP9 (FC = 41.0, p = 9.8 × 10−14) and, more strikingly, MMP12 (FC = 819.7, p = 3.0 × 10−49) (Fig. 1c), which are both inhibited by AZD1236, a drug originally tested as a treatment for chronic obstructive pulmonary disease, but more recently investigated as an antitumor agent. Like SHH, MMP9 and 12 are translated as inactive precursor proteins (92 and 55 kDa respectively) that are cleaved to generate active isoforms (84 and 43/22 kDa, respectively). MMP isoform levels were compared to a panel of other pediatric brain tumor types and normal brain. The latent precursor of MMP9 was identified in all tumor types but not normal brain, whereas the active isoform was only observed in ACP (Fig. 2). Both the active and inactive isoforms of MMP12 were restricted to ACP (Fig. 2). ACP additionally overexpressed AREG (FC = 20.9, p = 4.8 × 10−17), EGFR (FC = 7.6, p = 6.0 × 10−5) and ERBB3 (FC = 22.8, p = 4.2 × 10−8) (Fig. 1d). Each of these is a target of the numerous EGFR/ERBB pathway inhibiting drugs (cetuximab, erlotinib, lapatinib).
Table 2.
Top 20 therapeutic target genes overexpressed in ACP compared to normal brain tissues and a variety of CNS and peripheral malignancies
| Symbol | Gene name | Average percentile | # of ACP >66th %-ile | Therapeutic agents | Fold | p-Value |
|---|---|---|---|---|---|---|
| MMP12 | matrix metallopeptidase 12 | 99.4 | 15 | AZD1236 | 820 | 3.03E-49 |
| SHH | sonic hedgehog homolog | 98.9 | 15 | Erismodegib, Vismodegib | 96.8 | 1.48E-48 |
| IL2RB | interleukin 2 receptor, beta | 96.7 | 15 | Denileukin diftitox | 12.4 | 4.87E-23 |
| LCK | lymphocyte-specific protein tyrosine kinase | 95.2 | 15 | Dasatinib, Pazopanib | 5.50 | 8.24E-16 |
| EPHA2 | EPH receptor A2 | 95.1 | 15 | Dasatinib, Regorafenib | 16.6 | 6.8E-16 |
| AREG | Amphiregulin | 93.9 | 15 | Cetuximab | 20.8 | 4.82E-17 |
| PIK3CD | phosphoinositide-3-kinase, catalytic, delta | 93.8 | 15 | Idelalisib | 6.79 | 3.55E-10 |
| IL6R | interleukin 6 receptor | 91.9 | 15 | Siltuximab | 6.13 | 7.06E-8 |
| MMP9 | matrix metallopeptidase 9 | 91.6 | 15 | AZD1236 | 41.0 | 9.79E-14 |
| EPCAM | epithelial cell adhesion molecule | 90.7 | 15 | Tucotuzumab celmoleukin | 87.4 | 2.01E-11 |
| SRC | v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog | 89.9 | 15 | Bosutinib, Dasatinib | 2.47 | 3.71e-7 |
| MUC1 | mucin 1 | 89.3 | 15 | HuHMFG1 | 6.30 | 1.27E-7 |
| ERBB3 | v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 | 86.7 | 15 | Lapatinib | 22.8 | 4.19E-08 |
| TNFSF11 | tumor necrosis factor (ligand) superfamily, member 11 | 91.7 | 14 | Denosumab | 5.17 | 4.28E-8 |
| MAPK14 | mitogen-activated protein kinase 14 | 91.3 | 14 | Regorafenib | 2.65 | 2.49E-8 |
| PTGS2 | prostaglandin-endoperoxide synthase 2 | 85.6 | 14 | Lenalidomide | 10.5 | 9.23E-7 |
| RRAS | related RAS viral (r-ras) oncogene homolog | 85.4 | 14 | Sorafenib | 3.61 | 2.32E-5 |
| PSMB1 | proteasome (prosome, macropain) subunit, beta type, 1 | 83.8 | 14 | Carfilzomib | 1.45 | 5.85E-5 |
| EGFR | epidermal growth factor receptor | 83.4 | 14 | Cetuximab, Erlotinib, Gefitinib, Lapatinib, etc. | 7.57 | 5.99E-05 |
| CD52 | CD52 molecule | 86.3 | 13 | Alemtuzumab | 8.85 | 4.51E-12 |
Fig. 1.
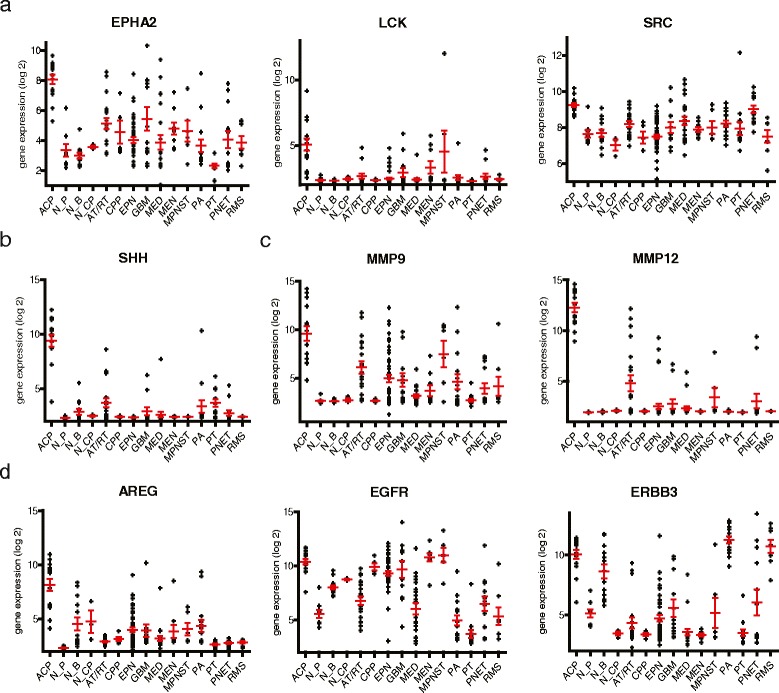
Gene expression of potential targets for therapeutic intervention in ACP. Expression of the indicated genes in ACP relative to a broad range of pediatric and adult brain tumor types: atypical teratoid/rhabdoid tumor (AT/RT), choroid plexus papilloma (CPP), ependymoma (EPN), glioblastoma multiforme (GBM), medulloblastoma (MED), meningioma (MEN), pilocytic astrocytoma (PA), primitive neuroectodermal tumor (PNET); peripheral pediatric solid tumors: malignant peripheral nerve sheath tumors (MPNST), rhabdomyosarcoma (RMS); in addition to malignant (pituitary adenoma) and normal pituitary (PT and N_P respectively); and normal brain and choroid plexus (N_B and N_CP respectively). Dasatinib targets, LCK (lymphocyte-specific protein tyrosine kinase), EPHA2 (ephrin type-A receptor 2) and SRC (SRC proto-oncogene, non-receptor tyrosine kinase) (a). Sonic hedgehog homolog (b). Matrix metalloproteases 9 & 12 (c). EGF pathway genes (d). Values are expressed as log2 gene expression. Horizontal red bars represent the mean, and error bars represent standard error of the mean (SEM)
Fig. 2.

Overexpression of active protein isoforms for SHH, MMP9 and MMP12 in ACP relative to other common pediatric brain tumors and normal brain. Western blot analysis was used to determine latent preforms and cleaved active isoforms of the indicated proteins. (Abbr: AT/RT, atypical teratoid/rhabdoid tumor; EPN, ependymoma; GBM, glioblastoma; MED, medulloblastoma; PA, pilocytic astrocytoma; Norm, normal brain)
Transcriptome microarray clustering analyses
Unbiased hierarchical clustering of ACP gene expression data with data from the panel of normal and neoplastic CNS samples and some non-CNS pediatric tumor types (as used above) afforded us further insights into the biology of ACP. ACP samples did not group with the cluster containing both normal and neoplastic pituitary (Fig. 3). Surprisingly, ACP formed a distinct cluster within a larger cluster containing MEN, MPNST and RMS, a histologically heterogeneous group of tumors derived from various tissue types. While it is perhaps not unexpected that a non-neural tumor like ACP clusters within this group of non-neural tumors, the fact that this cluster lies within the larger neuro-epithelial tumor and normal brain cluster and not with the pituitary tissues is difficult to interpret. This raises the possibility that ACP may have a completely different origin than has been hypothesized; it is more likely however that it has differentiated in such a way that it shares a convergent expression profile in common with these tumors not due to a common tissue of origin. Further analysis of the gene expression signatures responsible for these groupings as well as comparisons with papillary craniopharyngioma and head and neck cancers may give insight into the nature of these groupings.
Fig. 3.

Transcriptome cluster analysis reveals similarities between ACP, meningioma and rhabdomyosarcoma, with no relationship to adult pituitary or pituitary adenoma. Unbiased hierarchical clustering analysis of a panel of craniopharyngioma tumor samples compared to other pediatric tumors, normal brain tissue, pituitary tissue and adult pituitary adenomas. The top 30 % most variant genes, were used to generate the clustering dendrogram above. (AT/RT, atypical teratoid/rhabdoid tumor; CPP, choroid plexus papilloma; MPNST, malignant peripheral nerve sheath tumor; GBM, glioblastoma multiforme)
Gene expression signature recapitulates ACP histopathological characteristics
In a series of pair-wise comparisons (limma) with normal brain from a range of anatomic sites (including pituitary) and other brain (AT/RT, CPP, EPN, GBM, PA, MED, MEN, PNET), pituitary (PT) and peripheral solid tumors of childhood (MPNST, RMS) we identified genes that were overexpressed by ACP in all comparisons (FDR p < 0.05). These 384 ACP signature genes were then examined for enrichment (FDR < 0.005) of gene ontology terms curated by Gene Ontology (biological processes), Panther (biological processes) and KEGG (pathways) databases using DAVID. The majority of ontologic terms were comprised of ectodermal development-related genes (odontogenic, epidermal, epithelial development) (Table 3). Morphologic characteristics of odontogenesis in ACP range from deposits of calcium, which are evident on an x-ray, to development of whole teeth [38, 39]. Specific genes contributing to odontogenesis signature include DLX2, ODAM, AMBN, AMELX, ENAM, TP63, EDAR, SHH, FGF4. Epidermal morphology is also a defining histological feature of ACP, which is presumed to develop from nests of epithelium derived from Rathke’s cleft, with further development of non-viable wet keratin “ghost cells” that resemble polyhedral, anucleated corneocytes (final step of keratinocyte differentiation). The ACP genes that were highly expressed within these epidermal ontologies include numerous keratins (KRT5, KRT13, KRT14, KRT15, KRT16, KRT31, KRT34, KRT85) and laminins (LAMA3, LAMC2). TP63 expression is also extraordinarily high in ACP (Fig. 4a); as a regulator of odontogenic, epidermal and keratinocyte development, and in regulation of stemness, p63 may play a critical role in ACP development and morphogenesis [40]. We also found that a cluster of odontogenic, cytokine and EGF family proteins at Chromosome 4p5 was highly overexpressed (as much as 4,000 fold) in ACP (Fig. 4d). The genetic relevance of the high levels of expression at this locus is unclear.
Table 3.
Enriched ontology terms associated with ACP-exclusive genes. Limma identified 384 genes that were exclusively expressed in ACP compared to a normal brain tissues and a variety of CNS and peripheral malignancies. DAVID was used to identify 23 enriched ontologies (FDR < 0.005) that are shown ranked according to fold enrichment. Abbreviation: Ontology ID prefix GO, Gene ontology biological process; BP, panther biological process; HSA, KEGG pathway; FDR, Benjamini false discovery rate adjusted p-value
| Ontology term | ID | Fold enrichment | p-Value | FDR |
|---|---|---|---|---|
| Biomineral formation | GO:31214 | 13.38 | 2.53E-07 | 5.50E-05 |
| Odontogenesis of dentine-containing tooth | GO:42475 | 11.28 | 5.50E-06 | 8.69E-04 |
| Epidermis development | GO:8544 | 11.06 | 3.10E-27 | 2.70E-24 |
| Ectoderm development | GO:7398 | 10.78 | 2.62E-28 | 4.55E-25 |
| Epidermal cell differentiation | GO:9913 | 9.17 | 6.27E-08 | 1.56E-05 |
| Keratinocyte differentiation | GO:30216 | 9.17 | 2.74E-07 | 5.29E-05 |
| Odontogenesis | GO:42476 | 9.17 | 5.23E-06 | 9.09E-04 |
| Epithelial cell differentiation | GO:30855 | 8.43 | 5.25E-13 | 3.04E-10 |
| Skeletal development | BP:201 | 5.81 | 7.85E-07 | 1.96E-05 |
| Epithelium development | GO:60429 | 5.57 | 1.46E-10 | 6.33E-08 |
| Bone development | GO:60348 | 5.37 | 1.42E-05 | 0.00205 |
| Ossification | GO:1503 | 5.26 | 4.46E-05 | 0.00515 |
| Cell structure | BP:286 | 3.72 | 2.38E-14 | 2.98E-12 |
| Cell adhesion-mediated signaling | BP:120 | 3.66 | 4.34E-08 | 1.36E-06 |
| Cytokine-cytokine receptor interaction | HSA:4060 | 3.38 | 5.13E-05 | 0.00557 |
| Skeletal system development | GO:1501 | 3.28 | 2.06E-05 | 0.00274 |
| Cell structure and motility | BP:285 | 2.85 | 2.68E-13 | 1.68E-11 |
| Cell adhesion | GO:7155 | 2.83 | 4.39E-08 | 1.52E-05 |
| Biological adhesion | GO:22610 | 2.82 | 4.48E-08 | 1.30E-05 |
| Cell communication | BP:274 | 2.46 | 6.08E-10 | 2.53E-08 |
| Cell adhesion | BP:124 | 2.30 | 1.57E-04 | 0.00280 |
| Regulation of cell proliferation | GO:42127 | 2.24 | 3.69E-05 | 0.00457 |
| Signal transduction | BP:102 | 1.43 | 1.21E-04 | 0.00251 |
Fig. 4.
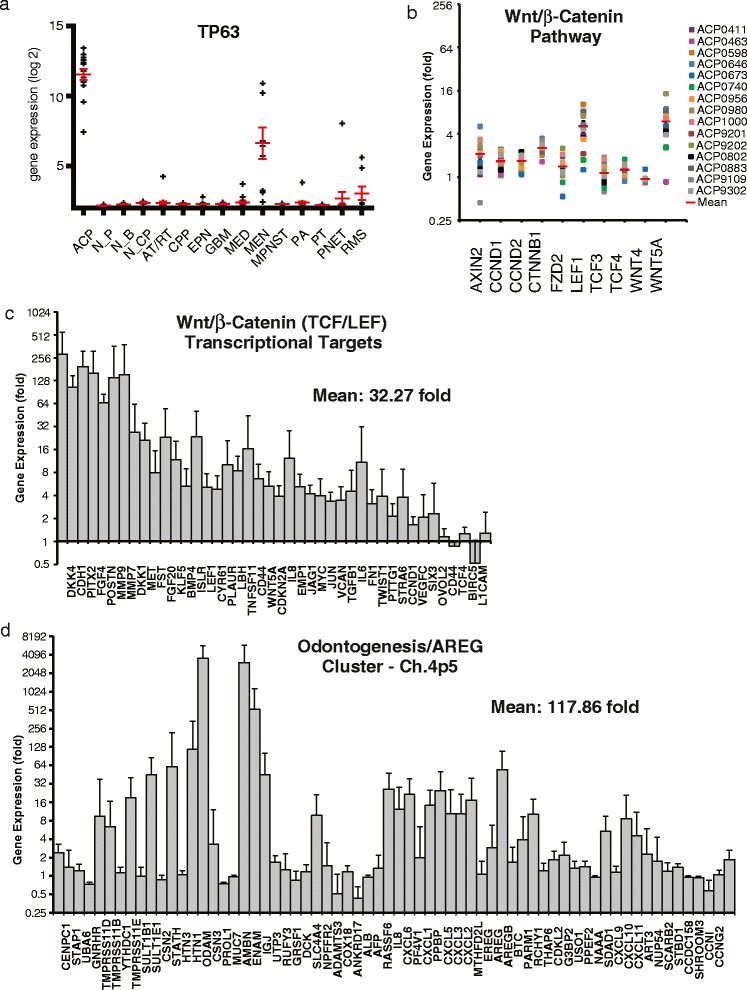
Expression of the indicated genes in pediatric ACP. p63 gene expression in the indicated tumor and normal tissue types (a). Wnt pathway (b) and β-catenin (TCF/LEF) target gene expression (c). Genes at chromosome 4p5 locus (d). Values are expressed as log2 (a) or as fold-difference of individual ACP samples relative to the average of all other tumor and normal samples (b-d)
Wnt pathway genes are expressed homogeneously and not at abnormally high levels in ACP (Fig. 4b), with the exception of the transcription factor (TCF)/lymphoid enhancer-binding factor (LEF) targets LEF1 and WNT5A. However, consistent with the hypothesis that aberrant Wnt signaling (via mutant β-catenin) is responsible for the pathogenesis of ACP, β-catenin/TCF/LEF target genes are overexpressed an average of 32 fold over the other samples (Fig. 4c).
The epigenetic profile of ACP may also give some insight into the developmental origins of this disease. Genes involved in pituitary development (Fig. 5a) are not highly expressed with the exception of PITX1 & 2 which are established TCF/LEF targets. Critical developmental and survival pathway gene expression patterns reveal a potential role for EGFR, Six family transcription factors, Shh and FGFs in ACP pathogenesis (Fig. 5b–f).
Fig. 5.
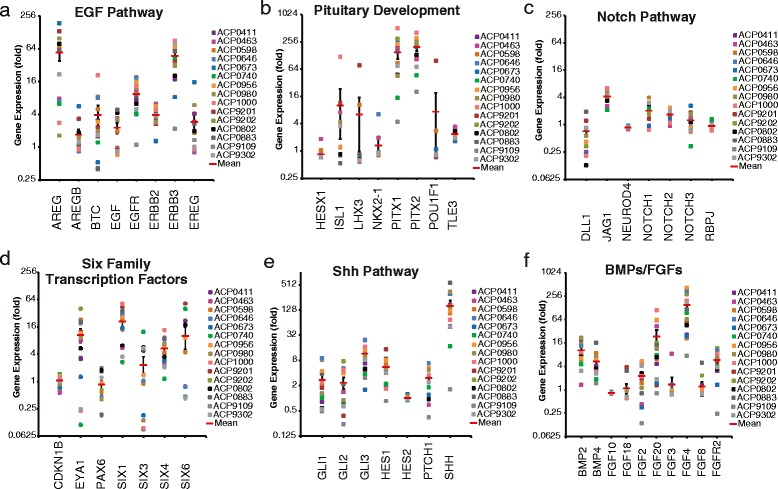
Expression of the indicated developmental and cancer-related genes in individual pediatric ACP samples. Epidermal growth factor (EGF) family genes (a). Genes involved in pituitary development (b). Developmental genes from the Notch (c), Six transcription factor (d), Sonic hedgehog (Shh) (e), Bone morphogenetic protein (BMP) and Fibroblast growth factor (FGF) (f) families. Values expressed are fold-difference of individual ACP samples relative to the average of all other tumor and normal samples
Discussion
Identification of potential therapeutic targets in our transcriptomic analysis confirmed findings of previous studies that had identified SHH and EGFR pathway activity in ACP and provide further evidence that therapies targeting these pathways could be used successfully in treating ACP. Transcriptomic analysis of ACP generated in a mouse model demonstrated high levels of SHH gene expression, suggesting a mitogenic autocrine/paracrine loop [25]. A subsequent study identified upregulation of members of the SHH signaling pathway in human specimens [41]. In vitro and xenotransplant model studies have demonstrated that EGFR activation is responsible for driving growth and migration in ACP [23, 24]. The proven clinical utility of EGFR inhibition in the treatment of cancer makes EGFR targeted drugs an attractive approach to ACP treatment. Our identification of high levels of EGFR ligand AREG provide a potential mechanism for EGFR activation in ACP that warrants further exploration. Furthermore, AREG has been implicated as a paracrine/juxtacrine regulator of cell survival in other cancers and epidermal cell types [42]. It has also recently been suggested that ACP may be paracrine in nature (i.e. the CTNNB1 mutant cells may promote the proliferation of another cell type that actually populates the tumor) [22]. This hypothesis could explain the extensive intratumoral heterogeneity in ACP and perhaps the difficulty we and others have found in obtaining “pure” tumor samples to accurately identify CTNNB1 mutations [15].
We also identified a number of additional novel potential drug targets. One group, including LCK, EPHA2 and SRC, is targeted by the kinase inhibitor dasatinib. Hundreds of trials are now underway using dasatinib to treat a wide variety of cancers beyond the few for which it currently has FDA approval. A second group of targets we identified are extracellular proteases, of which many were strongly represented in our analysis; two in particular, MMPs 9 and 12, are targeted by the drug AZD1236. MMP12, which we found expressed at very high levels, is a proteolytic factor that may contribute to the significant invasive phenotype that is a hallmark of ACP [43]. Previous studies have attempted to correlate proteinase activity with biological course in ACP [44] including a study that confirmed the presence of MMP9 by immunohistochemistry [45].
The results of this transcriptomic study of human pediatric ACP shed further light on the biology of this tumor, reflect the odontogenic and epithelial characteristics in the pathology of ACP and recapitulate the origins of ACP from oral ectoderm. The origins of ACP have long been the subject of speculation due to the oddity of their morphology and location. Many consider ACP to be a congenital midline developmental malformation, but the (albeit infrequent) occurrence of ACP in late adulthood and variable presentation are difficult to reconcile with this hypothesis. The recognition that β-catenin dysregulation is responsible for ACP have led to two conflicting mouse models for the formation of ACP – in mice an ACP-like tumor can develop from targeted CTNNB1 mutations in either pituitary oral ectoderm precursors in developing mice embryos or in pituitary stem cells in post-natal mice [46]. The data presented here are consistent with the theory that ACP is a congenital malformation that develops from the improper closure of the craniophyrangeal duct from the oral ectoderm-derived remnants of Rathke’s pouch [46–48] (Table 3). However, these data are not completely inconsistent with the hypothesis that ACP arises from an anterior pituitary stem cell that transdifferentiates toward an oral epithelial phenotype [25, 26]. Further analysis of more ACP samples, including adult ACP, in addition to transcriptomic studies of neuropathologically distinct cell subtypes present within ACP tumors will contribute to our understanding of how these tumors form and perhaps how to better treat them.
Conclusions
Current standard therapy for ACP is surgery and radiation, both of which lead to high morbidity in this sensitive region of the brain, especially in children, in whom these morbidities become a lifelong and life-altering disease. Intracystic delivery of therapeutic agents (interferon-alpha, bleomycin or Ytrrium90) has shown some efficacy in treating ACP [49], but this approach is limited by the requirement of a single cyst in the presenting tumor and requires stereotactic surgery to place a catheter and Ommaya reservoir for delivery. Systemic therapy could more safely and more effectively treat children with ACP. However, progress has been hindered by the absence of in vitro or in vivo models of this tumor that would enable the unbiased screening of drug libraries. This study forms the basis for further studies with rational therapies for ACP. The recent development of ACP xenotransplants in immune deficient mice [24] will enable pre-clinical testing of these rationally selected targeted therapies, providing further rationale for small studies on efficacy in augmenting surgery and radiation or in treating recurrent ACP. These efforts, combined with further collaborations between centers and consortiums will provide the foundation for a randomized clinical trial using targeted agents to treat ACP in the near future.
Acknowledgements
This work was supported by the Morgan Adams Foundation (J.M.G., T.C.H., N.F.), Colorado Clinical and Translational Sciences Institute/Children’s Hospital Colorado Research Institute KL2 Research Scholar Award NCATS/NIH UL1 TR001082 (T.C.H.), T32CA082086 (J.M.G.), American Cancer Society Postdoctoral Fellowship (J.M.G.) and the Molecular Pathology and Genomics and Microarray Shared Resources of the University of Colorado’s NIH/NCI Cancer Center support Grant P30CA046934.
We would like to thank Susan Staulcup, MS, for her extensive organizational and administrative support of this work. We also thank the laboratory of Dr. Margaret Wierman for the use of their pituitary microarray data.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JMG, AMD, BKK-D, LG, NF and TCH designed the study. AMD, VMA, AMG, BKK-D, MH, and TCH collected and processed the tumor samples. JMG, AMD, DKB, VMA, KKR, AMG conducted the experiments. JMG, AMD, DKB and TCH performed the transcriptomic data analysis. JMJ, RCEA and AR provided tumor samples. JMG, AMD and TCH drafted the manuscript with editorial input from all authors. All authors read and approved the final manuscript.
Contributor Information
Jacob M. Gump, Email: Jacob.Gump@ucdenver.edu
Todd C. Hankinson, Email: Todd.Hankinson@childrenscolorado.org
References
- 1.Hankinson TC, Fields EC, Torok MR, Beaty BL, Handler MH, Foreman NK, O’Neill BR, Liu AK. Limited utility despite accuracy of the national SEER dataset for the study of craniopharyngioma. J Neurooncol. 2012;110(2):271–278. doi: 10.1007/s11060-012-0966-5. [DOI] [PubMed] [Google Scholar]
- 2.Zacharia BE, Bruce SS, Goldstein H, Malone HR, Neugut AI, Bruce JN. Incidence, treatment and survival of patients with craniopharyngioma in the surveillance, epidemiology and end results program. Neuro Oncol. 2012;14(8):1070–1078. doi: 10.1093/neuonc/nos142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller HL. Childhood craniopharyngioma: treatment strategies and outcomes. Expert Rev Neurother. 2014;14(2):187–197. doi: 10.1586/14737175.2014.875470. [DOI] [PubMed] [Google Scholar]
- 4.Kawamata T, Kubo O, Hori T. Histological findings at the boundary of craniopharyngiomas. Brain Tumor Pathol. 2005;22(2):75–78. doi: 10.1007/s10014-005-0191-4. [DOI] [PubMed] [Google Scholar]
- 5.Muller HL, Gebhardt U, Schroder S, Pohl F, Kortmann RD, Faldum A, Zwiener I, Warmuth-Metz M, Pietsch T, Calaminus G, Kolb R, Wiegand C, Sorensen N, study committee of K Analyses of treatment variables for patients with childhood craniopharyngioma–results of the multicenter prospective trial KRANIOPHARYNGEOM 2000 after three years of follow-up. Horm Res Paediatr. 2010;73(3):175–180. doi: 10.1159/000284358. [DOI] [PubMed] [Google Scholar]
- 6.Ozyurt J, Thiel CM, Lorenzen A, Gebhardt U, Calaminus G, Warmuth-Metz M, Muller HL. Neuropsychological outcome in patients with childhood craniopharyngioma and hypothalamic involvement. J Pediatr. 2014;164(4):876–881. doi: 10.1016/j.jpeds.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Sughrue ME, Yang I, Kane AJ, Fang S, Clark AJ, Aranda D, Barani IJ, Parsa AT. Endocrinologic, neurologic, and visual morbidity after treatment for craniopharyngioma. J Neurooncol. 2011;101(3):463–476. doi: 10.1007/s11060-010-0265-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flitsch J, Muller HL, Burkhardt T. Surgical strategies in childhood craniopharyngioma. Front Endocrinol. 2011;2:96. doi: 10.3389/fendo.2011.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klimo P, Jr., Venable GT, Boop FA, Merchant TE (2015) Recurrent craniopharyngioma after conformal radiation in children and the burden of treatment. J Neurosurg Pediatr:1–7. doi:10.3171/2014.10.PEDS14384 [DOI] [PubMed]
- 10.Visser J, Hukin J, Sargent M, Steinbok P, Goddard K, Fryer C. Late mortality in pediatric patients with craniopharyngioma. J Neurooncol. 2010;100(1):105–111. doi: 10.1007/s11060-010-0145-5. [DOI] [PubMed] [Google Scholar]
- 11.Elliott RE, Sands SA, Strom RG, Wisoff JH. Craniopharyngioma Clinical Status Scale: a standardized metric of preoperative function and posttreatment outcome. Neurosurg Focus. 2010;28(4) doi: 10.3171/2010.2.FOCUS09304. [DOI] [PubMed] [Google Scholar]
- 12.Sterkenburg AS, Hoffmann A, Gebhardt U, Warmuth-Metz M, Daubenbuchel AM, Muller HL. Survival, hypothalamic obesity, and neuropsychological/psychosocial status after childhood-onset craniopharyngioma: newly reported long-term outcomes. Neuro Oncol. 2015 doi: 10.1093/neuonc/nov044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foreman NK, Faestel PM, Pearson J, Disabato J, Poole M, Wilkening G, Arenson EB, Greffe B, Thorne R. Health status in 52 long-term survivors of pediatric brain tumors. J Neurooncol. 1999;41(1):47–53. doi: 10.1023/A:1006145724500. [DOI] [PubMed] [Google Scholar]
- 14.Muller HL. Craniopharyngioma. Handb Clin Neurol. 2014;124:235–253. doi: 10.1016/B978-0-444-59602-4.00016-2. [DOI] [PubMed] [Google Scholar]
- 15.Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, Sunkavalli A, Shillingford N, Calicchio ML, Lidov HG, Taha H, Martinez-Lage M, Santi M, Storm PB, Lee JY, Palmer JN, Adappa ND, Scott RM, Dunn IF, Laws ER, Jr, Stewart C, Ligon KL, Hoang MP, Van Hummelen P, Hahn WC, Louis DN, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46(2):161–165. doi: 10.1038/ng.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y, Sakamoto M, Hirohashi S. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002;161(6):1997–2001. doi: 10.1016/S0002-9440(10)64477-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato K, Nakatani Y, Kanno H, Inayama Y, Ijiri R, Nagahara N, Miyake T, Tanaka M, Ito Y, Aida N, Tachibana K, Sekido K, Tanaka Y. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol. 2004;203(3):814–821. doi: 10.1002/path.1562. [DOI] [PubMed] [Google Scholar]
- 18.Buslei R, Nolde M, Hofmann B, Meissner S, Eyupoglu IY, Siebzehnrubl F, Hahnen E, Kreutzer J, Fahlbusch R. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 2005;109(6):589–597. doi: 10.1007/s00401-005-1004-x. [DOI] [PubMed] [Google Scholar]
- 19.Oikonomou E, Barreto DC, Soares B, De Marco L, Buchfelder M, Adams EF. Beta-catenin mutations in craniopharyngiomas and pituitary adenomas. J Neurooncol. 2005;73(3):205–209. doi: 10.1007/s11060-004-5232-z. [DOI] [PubMed] [Google Scholar]
- 20.Larkin SJ, Preda V, Karavitaki N, Grossman A, Ansorge O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol. 2014;127(6):927–929. doi: 10.1007/s00401-014-1270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holsken A, Kreutzer J, Hofmann BM, Hans V, Oppel F, Buchfelder M, Fahlbusch R, Blumcke I, Buslei R. Target gene activation of the Wnt signaling pathway in nuclear beta-catenin accumulating cells of adamantinomatous craniopharyngiomas. Brain Pathol. 2009;19(3):357–364. doi: 10.1111/j.1750-3639.2008.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Barbera JP (2015) Molecular and cellular pathogenesis of Adamantinomatous Craniopharyngioma. Neuropathol Appl Neurobiol. doi:10.1111/nan.12226. [DOI] [PMC free article] [PubMed]
- 23.Holsken A, Gebhardt M, Buchfelder M, Fahlbusch R, Blumcke I, Buslei R. EGFR signaling regulates tumor cell migration in craniopharyngiomas. Clin Cancer Res. 2011;17(13):4367–4377. doi: 10.1158/1078-0432.CCR-10-2811. [DOI] [PubMed] [Google Scholar]
- 24.Stache C, Holsken A, Schlaffer SM, Hess A, Metzler M, Frey B, Fahlbusch R, Flitsch J, Buchfelder M, Buslei R. Insights into the infiltrative behavior of adamantinomatous craniopharyngioma in a new xenotransplant mouse model. Brain Pathol. 2015;25(1):1–10. doi: 10.1111/bpa.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andoniadou CL, Gaston-Massuet C, Reddy R, Schneider RP, Blasco MA, Le Tissier P, Jacques TS, Pevny LH, Dattani MT, Martinez-Barbera JP. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012;124(2):259–271. doi: 10.1007/s00401-012-0957-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, Vernay B, Jacques TS, Taketo MM, Le Tissier P, Dattani MT, Martinez-Barbera JP. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A. 2011;108(28):11482–11487. doi: 10.1073/pnas.1101553108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voronkov A, Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des. 2013;19(4):634–664. doi: 10.2174/138161213804581837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geeleher P, Cox NJ, Huang RS. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014;15(3):R47. doi: 10.1186/gb-2014-15-3-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman LM, Donson AM, Nakachi I, Griesinger AM, Birks DK, Amani V, Hemenway MS, Liu AK, Wang M, Hankinson TC, Handler MH, Foreman NK. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol. 2014;127(5):731–745. doi: 10.1007/s00401-013-1212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, Stubbs H, McDermott U, Settleman J, Kwak EL, Clark JW, Isakoff SJ, Sequist LV, Engelman JA, Lynch TJ, Haber DA, Louis DN, Ellisen LW, Borger DR, Iafrate AJ. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;99:909–917. doi: 10.1198/016214504000000683. [DOI] [Google Scholar]
- 32.Michaelis KA, Knox AJ, Xu M, Kiseljak-Vassiliades K, Edwards MG, Geraci M, Kleinschmidt-DeMasters BK, Lillehei KO, Wierman ME. Identification of growth arrest and DNA-damage-inducible gene beta (GADD45beta) as a novel tumor suppressor in pituitary gonadotrope tumors. Endocrinology. 2011;152(10):3603–3613. doi: 10.1210/en.2011-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 35.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 36.Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA, Doremieux O. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31(1):334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 38.Beaty NB, Ahn E. Images in clinical medicine. Adamantinomatous craniopharyngioma containing teeth. N Engl J Med. 2014;370(9):860. doi: 10.1056/NEJMicm1308260. [DOI] [PubMed] [Google Scholar]
- 39.Muller C, Adroos N, Lockhat Z, Slavik T, Kruger H. Toothy craniopharyngioma: a literature review and case report of craniopharyngioma with extensive odontogenic differentiation and tooth formation. Childs Nerv Syst. 2011;27(2):323–326. doi: 10.1007/s00381-010-1296-6. [DOI] [PubMed] [Google Scholar]
- 40.Cao J, Lin JP, Yang LX, Chen K, Huang ZS. Expression of aberrant beta-catenin and impaired p63 in craniopharyngiomas. Br J Neurosurg. 2010;24(3):249–256. doi: 10.3109/02688690903576237. [DOI] [PubMed] [Google Scholar]
- 41.Gomes DC, Jamra SA, Leal LF, Colli LM, Campanini ML, Oliveira RS, Martinelli CE, Jr, Elias PC, Moreira AC, Machado HR, Saggioro F, Neder L, Castro M, Antonini SR. Sonic Hedgehog pathway is upregulated in adamantinomatous craniopharyngiomas. Eur J Endocrinol. 2015;172(5):603–608. doi: 10.1530/EJE-14-0934. [DOI] [PubMed] [Google Scholar]
- 42.Willmarth NE, Ethier SP. Autocrine and juxtacrine effects of amphiregulin on the proliferative, invasive, and migratory properties of normal and neoplastic human mammary epithelial cells. J Biol Chem. 2006;281(49):37728–37737. doi: 10.1074/jbc.M606532200. [DOI] [PubMed] [Google Scholar]
- 43.Adamson TE, Wiestler OD, Kleihues P, Yasargil MG. Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J Neurosurg. 1990;73(1):12–17. doi: 10.3171/jns.1990.73.1.0012. [DOI] [PubMed] [Google Scholar]
- 44.Lubansu A, Ruchoux MM, Brotchi J, Salmon I, Kiss R, Lefranc F. Cathepsin B, D and K expression in adamantinomatous craniopharyngiomas relates to their levels of differentiation as determined by the patterns of retinoic acid receptor expression. Histopathology. 2003;43(6):563–572. doi: 10.1111/j.1365-2559.2003.01751.x. [DOI] [PubMed] [Google Scholar]
- 45.Xia Z, Liu W, Li S, Jia G, Zhang Y, Li C, Ma Z, Tian J, Gong J. Expression of matrix metalloproteinase-9, type IV collagen and vascular endothelial growth factor in adamantinous craniopharyngioma. Neurochem Res. 2011;36(12):2346–2351. doi: 10.1007/s11064-011-0560-9. [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Barbera JP, Buslei R. Adamantinomatous craniopharyngioma: pathology, molecular genetics and mouse models. J Pediatr Endocrinol Metab. 2015;28(1–2):7–17. doi: 10.1515/jpem-2014-0442. [DOI] [PubMed] [Google Scholar]
- 47.Cushing H. Intracranial tumours. Baltimore, Md: C. C. Thomas, Springfield, Ill; 1932. [Google Scholar]
- 48.Bernstein ML, Buchino JJ. The histologic similarity between craniopharyngioma and odontogenic lesions: a reappraisal. Oral Surg Oral Med Oral Pathol. 1983;56(5):502–511. doi: 10.1016/0030-4220(83)90098-1. [DOI] [PubMed] [Google Scholar]
- 49.Steinbok P, Hukin J. Intracystic treatments for craniopharyngioma. Neurosurg Focus. 2010;28(4) doi: 10.3171/2010.1.FOCUS09315. [DOI] [PubMed] [Google Scholar]