

Abstract
C3 Glomerulonephritis (C3GN) is a recently described disorder that typically results from abnormalities in the alternative pathway of complement. Here, we describe the clinical features, kidney biopsy findings, alternative pathway abnormalities, glomerular proteomic profile, and follow-up in 12 cases of C3GN. This disorder equally affected all ages, both genders, and typically presented with hematuria and proteinuria. In both the short and long term, renal function remained stable in the majority of patients with native kidney disease. In two patients, C3GN recurred within one year of transplantation and resulted in a decline in allograft function. Kidney biopsy mainly showed a membranoproliferative pattern; although both mesangial proliferative and diffuse endocapillary proliferative glomerulonephritis were noted. Alternative pathway abnormalities were heterogeneous; both acquired and genetic. The most common acquired abnormality was the presence of C3 nephritic factors, while the most common genetic finding was the presence of H402 and V62 alleles of Factor H. In addition to these risk factors, other abnormalities included Factor H auto-antibodies and mutations in CFH, CFI and CFHR genes. Laser dissection and mass spectrometry of glomeruli from patients with C3GN showed accumulation of alternative pathway and terminal complement complex proteins. Thus, C3GN results from diverse abnormalities of the alternative complement pathway leading to subsequent glomerular injury.
Keywords: C3 glomerulonephritis, MPGN, C3GN, proteomics, mass spectrometry, alternative pathway of complement, terminal complement complex, C3 glomerulopathy
INTRODUCTION
C3 glomerulonephritis (C3GN) is a recently described entity that shows a glomerulonephritis on light microscopy (LM), bright C3 staining and the absence of C1q, C4 and immunoglobulins (Ig) on immunofluorescence microscopy (IF), and mesangial and/or subendothelial electron dense deposits on electron microscopy (EM). 1–3 Occasional intramembranous and subepithelial deposits are also frequently present.3 The term ‘C3 glomerulopathy’ is often used to include C3GN and Dense Deposit Disease (DDD), both of which result from dysregulation of the alternative pathway (AP) of complement.1, 3–5 C3GN and DDD may be difficult to distinguish from each other on LM and IF studies. However, EM shows mesangial and/or subendothelial, intramembranous and subepithelial deposits in C3GN, while dense osmiophilic deposits are present along the glomerular basement membranes (GBM) and in the mesangium in DDD. Both C3GN and DDD are distinguished from immune-complex mediated glomerulonephritis by the lack of immunoglobulin staining on IF.6
Very few papers have described the clinical and pathologic findings in C3GN.2, 3 The aim of this study was to present a comprehensive description of the clinical features, renal biopsy findings, complement abnormalities, treatment and follow-up in C3GN. We describe in detail 12 patients with C3GN, two of whom developed recurrent disease following allograft transplantation. We also performed laser dissection and mass spectrometry (LDMS) in eight cases to define the glomerular proteomic profile of this disease.
RESULTS
Clinical Features
Our study included 12 patients with C3GN (Table 1). All patients except one (case 12) were treated at the Mayo Clinic. Ten patients maintained native kidney function and in two patients, the disease was recurrent in allografts. Sex distribution was equal (six females and six males) and age ranged from 8 to 73 years (mean, 42.5 years). The serum creatinine at presentation ranged from 0.6 mg/dL to 3.1 mg/dL (mean, 1.5 mg/dL) with a glomerular filtration rate (GFR) of 31 to 161 ml/min (mean, 63.4 ml/min).
Table 1.
Laboratory evaluation of C3GN patients
| Patient | Age/Sex | Serum Cr at presentation mg/dL | Urinalysis RBC/HPF | Urinary protein (mg/24hours) | C3/C4 mg/dL | Serum Creatinine at follow-up |
|---|---|---|---|---|---|---|
| 1 | 71/F | 1.7 | 41–50, <25% dRBC | 614 | 46/24 | 0.78 (5 m) |
| 2 | 52/F | 1.44 | 50–100, > 25% dRBC | 6389 | 76/26 | 1.5 (3 m) |
| 3 | 14/M | 1.3 | 21–30, >25% dRBC | 15760 | 19/13 | 1.3 (2.5yrs) |
| 4 | 60/M | 1.1 | 50–100, >25% dRBC | 874 | 57/35 | 1.4 (4 yrs) |
| 5 | 47/F | 3.1 | 21–30, no dRBC | 204 while on dialysis | 56/47 | on dialysis 4 m after presentation |
| 6 | 22/M | 2.02 | 50–100 | 10390 | 12/normal | 1.6 (4 m) |
| 7 | 73/M | 2.1 | 31–40, >25% dRBC | 1700 | 39/33 | 1.7 (8 m) |
| 8 | 8/F | 0.5 | NA* | 631 | 42/14 | 0.7 (23 yrs) |
| 9 | 36/M | 1.1 | 3–10, >25% dRBC | 9740 | 85 /14 | 0.9 (2 yr) |
| 10 | 42/F | 0.6 | 4–10, >25% dRBC | 614 | 17/18 | 0.69 (4 yrs) |
| 11 | 31/M | 1.7 (baseline) | 41–50, <25% dRBC | 2360 (1.7 gms 4 years later) | 72/6 | 2.9 (66 m) Recurrence within 1 yr |
| 12 | 21/F | 1.6 (baseline) | 51–100, >25% dRBC | 5730 | 40/27 | 2.1 (17 m) Recurrence within 1 yr |
normal C3/C4 as per notes, dRBC= dysmorphic RBC, C3 normal range (75–175 mg/dL). C4 normal range (14–40 mg/dL), m=month, yr= year,
UA at presentation at not available, latest at follow up 23 years alter shows on 1-3RBC/HPF
Of the 10 patients with native kidney function, nine were hypertensive. All had hematuria and proteinuria (six patients had >20 RBCs/high power field (HPF) and >25% dysmorphic RBC). Twenty-four hour urinary protein ranged from 615 to 15000 mg/24 hours (mean, 5762 mg/24 hours). C3 levels were low in nine patients, varying from 12 to 76 mg/dL (mean, 40.4 mg/dL; normal range 75–175 mg/dL). C4 levels were borderline low in only one patient (13 mg/dL; normal range 14–40 mg/dL). Evaluation for hepatitis B and C, antinuclear antigen (ANA), double stranded DNA (dsDNA), and cryoglobulins were negative uniformly negative. Two patients showed a small monoclonal gammopathy spike – one patient had IgG kappa and the other had IgM kappa on serum immunofixation studies. Bone marrow studies in these two patients showed less than 10% plasma cells. Two patients also had strong family history of renal disease. None of patients had ocular drusen.
Of the two patients with recurrent C3GN (patients 11 and 12), one was a 31-year male patient and the other patient was a 21-year old female patient. Recurrence of C3GN was noted in both of these patients within 1–1.5 years of renal allograft transplantation and was heralded by a rise in serum creatinine, the development of non-nephrotic range proteinuria and low serum C3 levels.
Kidney Biopsy Findings
In the 10 patients with native kidney function, biopsies uniformly showed a proliferative glomerulonephritis on LM. In eight cases, the pattern was membranoproliferative glomerulonephritis (MPGN) with mesangial and endocapillary proliferation with mononuclear cells, glomerular capillary wall remodeling with double contour formation and lobular accentuation of the capillary tufts. In the remaining two cases, the biopsy showed a diffuse endocapillary proliferative glomerulonephritis with endocapillary proliferation and influx of both mononuclear cells and neutrophils. One case showed small crescents, and one case showed a focal area of fibrinoid necrosis. Tubular atrophy and interstitial fibrosis ranged from 0 to 25% of the cortex, with a mean of 15% (Table 2).
Table 2.
Kidney biopsy findings of C3GN patients
| Patient | Pattern of Injury, GS/total glomeruli | Tubulo- interstitial Scarring | Immunofluorescence Microscopy (CW and mesangial) | Electron Microscopy |
|---|---|---|---|---|
| 1 | MPGN, 2/10 | 25 | C3 (2+) | SE, SU, MES |
| 2 | DPGN, 4/23 | 25 | C3+, trace to 1+ lambda and C1q | SE, SU, IN, MES |
| 3 | MPGN,2/29 | 10 | C3 (3+) | SE, IN, MES, TBM |
| 4 | MPGN, 0/18 | 10 | C3 (3+) | SE, SU, IN, MES,TBM |
| 5 | DPGN, 3/10 | 20 | C3 (3+) | SE, SU, MES |
| 6 | MPGN, with crescents, 2/9 | 5 | C3 (3+) | SE, SU, IN, MES |
| 7 | MPGN, 0/3 | 25 | C3 (3+) | SE, SU, IN, MES |
| 8 | MPGN, 0/12 | 0 | C3 (intensity not documented) | SE, IN, MES |
| 9 | MPGN, 1/23, with secondary FSGS | 20 | C3 (3+) | SE, SU, IN, MES |
| 10 | MPGN, 1/12 | 5 | C3 (3+) | SE, SU, IN, MES |
| 11 (tx) | Mesangial Proliferative GN | 10 | C3 (3+) | SE, MES |
| 12 (tx) | MPGN, 3/23 | 25 | C3 (3+) | SE, SU, IN, MES |
SE- subendothelial, SU- subepithelial, IN- intramembranous, MES- mesangial, MPGN- membranoproliferative glomerulonephritis, DPGN- diffuse proliferative glomerulonephritis, CW-capillary wall, GS-globally sclerosed.
IF microscopy for C3 is the hallmark of C3GN. All cases showed bright staining for C3 in the mesangium and glomerular capillary walls. In two cases C3 staining was also present along the tubular basement membranes. No case showed significant staining for IgG, IgM, C1q, kappa and lambda light chains. Trace staining for IgG and light chains was present in two cases.
EM showed subendothelial and mesangial electron dense deposits in all cases. Eight cases also showed subepithelial deposits and eight cases showed a few intramembranous deposits. The deposits were amorphous and appeared lighter and less sharply demarcated than immune-type electron-dense deposits. The subepithelial deposits raised the possibility of post-infectious glomerulonephritis (see discussion). Double contour formation was present in eight cases with subendothelial expansion by cellular elements, electron dense deposits, and new basement membrane formation. Representative LM, IF and EM are shown in Figure 1.
Figure 1.

Representative light, immunofluorescence, and electron microscopy in C3GN. A, B, C. Light microscopy showing different pattern of injury of the 3 different cases of C3GN. (A) shows a predominantly mesangial proliferative glomerulonephritis (PAS 20x), (B) shows a membranoproliferative glomerulonephritis (PAS 40x), and (C) shows a diffuse endocapillary proliferative glomerulonephritis with numerous infiltrating neutrophils within the glomerular capillaries (PAS 40x). D, E, F. Three different cases of C3GN showing bright C3 in the mesangium and/or along capillary walls (40x). G, H, I. Three different cases of C3GN showing large mesangial (black arrow), subendothelial deposits (thick black arrow), and subepithelial deposits (white arrow) on electron microscopy.
Renal biopsy of the two transplant cases showed recurrent C3GN. LM showed a membranoproliferative glomerulonephritis, IF showed bright C3 staining in the mesangium and capillary walls, and EM showed mesangial and capillary wall deposits. In patient 12 (transplant case 2) subepithelial deposits and intramembranous deposits were present. Recurrent C3GN first appeared as a mesangial proliferative glomerulonephritis and subsequently evolved and progressed to a MPGN. This course is in keeping with the previously described biopsy findings in cases of recurrent MPGN.7
Evaluation of the Alternative Pathway of Complement
Functional and genetic studies of the AP identified autoantibodies or mutations in complement genes in eight of 12 patients. The remaining patients (patients 3, 4, 5 and 11) carried CFH variants that are associated with an increase in baseline AP activity. (1) (Table 3)
Table 3.
Complement abnormalities of C3GN patients
| Patient | CFH | CFI | MCP | CFB | CFHR5 | C3 | MLPA | FH AA | Hemolytic assay |
APFA | C3Nef | sMAC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.2171delC, p.Thr724fsS STOP725; V62 -1 copy H402-0 copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Negative | ND | ND | Negative | 0.24 |
| 2 | No mutations | No mutations | No mutations | No mutations | c.646-647 AA>TT, p.Asn216Pro | No mutations | No mutations | Negative | Normal | 63.40% Slightly low | Positive | 0.21 |
| 3 | No mutations V62-2 copies H402-0 copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Negative | Normal | 63% Slightly low | Negative | ND |
| 4 | No mutations H402 -2 copies | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Negative | Normal | 1.0 % Very low | ND | 1.23 |
| 5 | No mutations V62-1 copy H402 -1copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | ND | ND | ND | Negative | 0.48 |
| 6 | No mutations V62-1 copy H402-0 copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Negative | Normal | 14.1% Very low | Positive | ND |
| 7 | No mutations V62-1 copy H402 -1copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Positive 1:200 | Normal | 6.6% Very low | Negative | 0.46 |
| 8 | ND | No mutations | ND | ND | ND | ND | ND | ND | ND | 12.7 % Very low | Positive | ND |
| 9 | No mutations V62-2 copies H402 -1copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Negative | Positive | 109% | Positive | 0.41 |
| 10 | No mutations H402-1 copy | Exon 6 c. 782G>A, p.Gly 261Asp) | No mutations | No mutations | No mutations | ND | ND | ND | ND | ND | ND | ND |
| 11 | No mutations V62-2 copies H402-0 copy | No mutations | No mutations | No mutations | No mutations | Risk allele c.463A>C, p.Lys155Gln | CFHR3-1 deletion | Negative | Normal | 60.5% Slightly low | Negative | 0.28 |
| 12 | No mutations V62-2 copies H402-0 copy | No mutations | No mutations | No mutations | No mutations | No mutations | No deletions or duplications | Negative | Normal | 28.2% low | Positive | 0.23 |
sMAC= serum membrane attack complex (normal 0.3 mg/L), ND= not done, FHAA= Factor H autoantibody (normal – titer <1:50), MLPA= Multiplex Ligation-dependent Probe Amplification, APFA=alternate pathway functional assay (normal 65% – 130%)
Five patients, including one patient with recurrent C3GN, were positive for C3Nefs and one patient was positive for FHAAs. Mutations were found in three patients and included a frameshift in CFH (c.2171delC, p.Thr724fsSTOP725) (patient 1) that has not been described in C3GN or DDD and missense variants in exon 6 of CFI (c.782G>A, p.Gly261Asp)(patient 10) and CFHR5 (c.646-647AA>TT, p.Asn216Phe)(patient 2). Risk alleles that were identified included Factor H risk polymorphisms H402 (c.1204C, p.His402) in 4 patients and V62 (c.184G, p.Val62) in 8 patients. One patient carried the C3 risk allele G102, L314 (c.304G, p.Gly102; c.941T, p.Leu314) (1).
Laser Dissection and Mass-Spectrometry
We performed laser dissection and mass spectrometry (LDMS) to determine the glomerular proteomic profile in eight cases (Figure 2). All patients showed accumulation of AP and TCC proteins. The deposition of C3 and C9 was extensive in all cases, with an average of 51.3 and 13.6 spectra, respectively. C5, C6, C7 and C8 were also present in all cases, with an average of 8.5, 3.5, 4.8, and 9.3 spectra, respectively. The “spectra” value indicates the total number of mass spectra collected on the mass spectrometry that matched the protein in question utilizing the proteomics software. Complement regulating proteins vitronectin, clusterin and apolipoprotein E were present in abundance. Vitronectin and clusterin are fluid phase regulators of TCC. CFHR-1 was present in all cases with an average of 15.3 spectra, and CFHR-5 was present in seven of eight cases with an average of 6 spectra.
Figure 2.



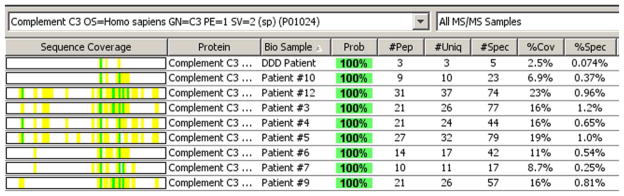
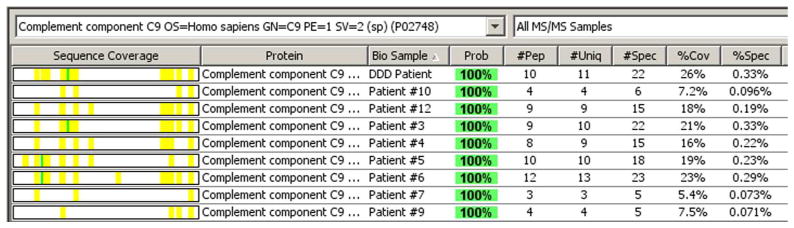
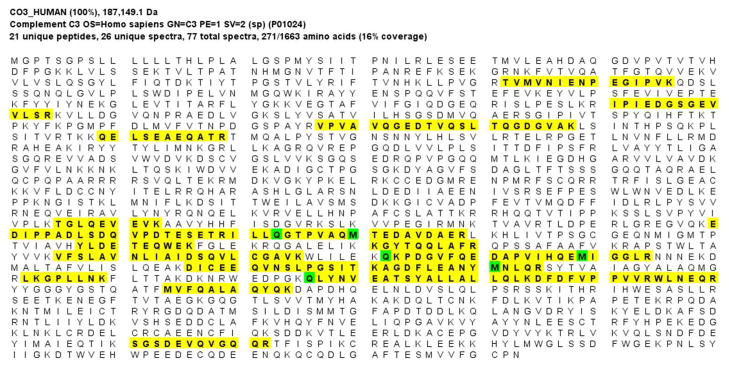

Laser microdissection and mass spectrometry analysis of glomerular proteins in 8 patients of C3GN and 1 patent of Dense Deposit Disease (DDD). (A) Glomeruli marked prior to dissection in patient 5, and (B) empty space following microdissection. (C) Representative scaffold readout of proteins of interest for 8 patients of C3GN and 1 patient of DDD (last column). The proteomic data show extensive accumulation of proteins of AP including C3, C9, C8, C5, C7 and C6 in order of abundance, with >95% probability. CFHR-1, CFHR-5, Vitronectin, Apolipoprotein E, clusterin are also present in relative abundance, with >95% probability. Yellow stars indicate proteins of interest, while red stars indicate protein ambiguity when two proteins share conserved regions. (D, E) Sequence coverage for C3 and C9 in all patients showing the number of peptides, number of unique peptides, number of spectra and percentage of coverage of peptide sequence for C3 (Figure 2 D) and C9 (Figure 2E). (F) Analysis of C3 in one sample showing 21 unique peptides, 26 unique spectra and 77 total spectra, all of which result in 16% peptide coverage with 100% probability for C3. (G) Analysis of C9 in one sample showing 9 unique peptides, 10 unique spectra and 22 total spectra, all of which result in 21% peptide coverage with 100% probability for C9. The yellow highlighted areas in F and G shows the actual peptides detected by the mass spectrometry, and the green highlight shows oxidized or methylated amino acids.
There was little or no significant accumulation of complement factors of the classical complement pathway, such as C1, C2 or C4. In addition, there was little or no Ig present. The minor spectra of various Ig, kappa and lambda light chain noted in a few cases likely represent protein reabsorption granules present in the podocytes of dissected glomeruli. In aggregate, these findings are similar to those noted in patients with DDD. 8 The last lane in figure 2 shows the glomerular proteomic profile in a recent case of DDD for comparison.
Treatment and Follow-up
Eight of the 10 patients with native renal function were treated with ACE inhibitors and angiotensin II blockers (renin-angiotensin system (RAS) blockade) and seven received prednisone for 4 weeks to 1 year. In addition, three patients (patients 1, 2, 3) were treated with mycophenolate mofetil (Cellcept) following prednisone taper. One patient (patient 6) received prednisone followed by Cyclophosphamide (Cytoxan) with stabilization of kidney function. Two patients (patients 9 and 10) were treated conservatively with RAS blockade only and did not receive steroids or other forms of immunosuppressive therapy.
Follow-up ranged from 4 months to 23 years after renal biopsy with a mean follow-up was 26.4 months (calculated using a maximum of 48 months in patients followed for longer times to avoid skewing results) (Figure 3). In general, the patients did well with no significant decline in renal function, both in the short and long term. The mean serum creatinine (except patient 5) on presentation was 1.5 mg/dL while the mean serum creatinine on follow-up decreased to 1.22mg/dL. Patient 5 presented with a serum creatinine of 3.1 mg/dL and was on dialysis within 3 months of presentation. In one case (patient 8), kidney biopsy done at the age of 8 years was interpreted as MPGN type I at the time. On review of the biopsy, the findings fulfilled all the criteria of C3GN. A repeat kidney biopsy done 17 years later continued to show C3GN. In this case, follow-up is available for 23 years after the first biopsy and 6 years after the 2nd biopsy.
Figure 3.

Serum creatinine at presentation and follow-up (in months) of all patients. Patient 5 was on dialysis soon after presentation. * Patient 8 follow-up is for 23 years, with stable kidney function.
Both patients with recurrent C3GN showed evidence of recurrent disease within 1 to 1.5 years. Review of their native kidney biopsies revealed MPGN with bright C3 and no Ig or light chain staining on IF, and mesangial and subendothelial deposits on EM, fulfilling the criteria for C3GN. The native disease was present for greater than 10 years in both patients. The first patient received a living donor kidney transplant and was treated with prednisone, tacrolimus and mycophenolate mofetil. Within one year, the patient developed recurrent C3GN and skin (purpuric) vasculitis. For the past five years, he has been maintained on plasmapheresis during which time his serum creatinine has risen from a baseline of 1.7 mg/dL to 2.9 mg/dL. The second patient received a living-related one-haplotype match kidney transplant. The patient is on prednisone, tacrolimus and mycophenolate mofetil. The serum creatinine has gradually increased from 1.6 mg/dL to 2.1 mg/dL during 17 months of follow up.
Discussion
In this study, we have described the clinical features, kidney biopsy findings, complement abnormalities, glomerular protein profile, treatment and follow-up in 12 patients with C3GN. We found that C3GN occurred at all ages, did not have a sex predilection and presented with a nephritic-nephrotic picture. Most patients had hypertension, proteinuria and hematuria.
Kidney biopsy showed a predominantly MPGN pattern of injury, although mesangial proliferative, diffuse proliferative (exudative), and crescentic glomerulonephritis were also present, a finding consistent with other recent studies on AP-mediated glomerulonephritis.2, 3, 9, 10 In addition, although not included in our series, we have seen cases of AP-mediated glomerulonephritis that showed only mesangial expansion with minimal hypercellularity. It is pertinent to note that in the patients in our series, the initial diagnosis was MPGN type I in four, MPGN type III in one, and post-infectious glomerulonephritis in four, but in all cases review of the biopsy clearly showed C3GN. The absence of significant Ig, including kappa and lambda light chains on IF, should exclude the diagnoses immune-complex MPGN types I and III, while the misdiagnosis of post-infectious glomerulonephritis was likely due to the presence of subepithelial deposits.
From the pathology standpoint, the finding of bright mesangial and capillary wall C3 in the absence of significant Ig should prompt an evaluation of AP. Low C3 and normal C4 serum levels also point towards AP dysfunction. By history, there was no antecedent infection to justify a diagnosis of post-infectious glomerulonephritis, with concurrent infections often exacerbating clinical symptoms like hematuria. There did not appear to be a disease-triggering event in these patients, and in none was there evidence of persistent infection. Evaluation for an autoimmune disease was also negative. Monoclonal gammopathy was present in two patients and has been described in the setting of dysregulation of the AP.11
Three patients had multiple kidney biopsies and in this group it is pertinent that there was no observed progression of glomerulosclerosis and tubulointerstitial scarring, even though the glomeruli continue to show proliferative features. These histologic findings are consistent with the stable kidney function and indeed long-term follow-up in these patients has shown stabilization of the renal function. These data suggest that C3GN may be less aggressive than DDD, which progresses to end-stage renal failure in ~25% of patients after 5 years and ~50% of patients after 10 years. 4, 12, 13
Complement evaluation in all patients showed findings consistent with AP dysfunction.2, 5, 14 Based on our studies, we find that serum complement levels and an assay of AP function (alternate pathway function assay, APFA) serve as good screening tests to assess AP dysfunction. We found that C3 was low and C4 was normal in 11 of 12 patients and that APFA was abnormal in seven of eight tested patients. The most common AP abnormality was the presence of C3Nef, an autoantibody directed against C3 convertase. By stabilizing C3 convertase and preventing its factor H-mediated degradation, C3Nefs cause a dysregulation of AP control.4 Their detection in 50% of C3GN patients in this series is lower than their prevalence of ~80% in DDD, where they have been strongly implicated in disease pathogenesis.15
Another common abnormality, identified in 10 of 11 patients, was the presence of specific polymorphisms in several complement genes. Although the small sample size precludes definitively implicating these variants in C3GN, they have been causally associated with DDD and age-related macular degeneration. 16–19 These polymorphisms lead to increased baseline AP activity even in normal controls, and studies comparing the H402 and Y402 variants of Factor H have shown that the former is associated with poorer AP control secondary to decreased binding to both endothelial cells and lipid peroxidation products like malondialdehyde, which accumulates in many pathophysiological processes.17, 18, 20, 21 Larger studies are required to definitely implicate these polymorphisms in C3GN. In addition to these specific polymorphisms, we identified a reported disease-causing variant in CFI in one patient and novel variants in CFH and CFHR5 in two other patients. 2
There are little data on recurrent C3GN, with recurrent C3 glomerulopathy being reported in only one patient with CFHR5 glomerulopathy.22 In the two patients we present, both had low serum C3 and abnormal APFAs, consistent with ongoing AP dysregulation. In one patient, C3Nefs were identified and while the genetic analysis found risk alleles for AP dysregulation, there was nothing unique to either transplant recipient to suggest that their disease might be more aggressive. Recurrent C3GN was noted within 18 months of the transplant, a surprising outcome given the fact that C3GN was present in both patients for more than 10 years prior to development of end-stage kidney disease. This phenomenon of early recurrence with rapid loss of allograft function compared to slow progression of disease in the native kidney occurs with many renal diseases including membranous nephropathy, MPGN and focal segmental glomerulosclerosis. 7 This outcome may reflect advanced disease at the time of transplantation and/or intra-renal hypertension and hyperfiltration in the solitary kidney resulting in deposition of the complement factors in the allograft in the face of persistent disease.
The LDMS findings revealed a pattern of glomerular complement and regulatory proteins remarkable for its similarity to DDD.8 The two most abundant complement proteins were C3 and C9, but there were also significant amounts of C5, C6, C7 and C8, in aggregate consistent with fluid-phase dysregulation of C3 and C5 convertases resulting in accumulation of AP and TCC complement factors in the mesangium and capillary wall with ensuing glomerular inflammation. Both CFHR1 and CFHR5 were also present in abundance. These findings are in stark contrast to immune-complex mediated MPGN, which show large spectra of Ig in all cases, although the type of Ig (IgG, IgA, IgM, Ig kappa light chains, Ig lambda light chains, Ig heavy chains) may vary depending on the etiology of immune-complex mediated MPGN. In addition, C3 and C4 are abundant complement factors in immune-complex mediated MPGN while TCC complement factors and complement regulatory proteins such vitronectin and apolipoprotein E are rarely present.8
Because C3GN is a recently described entity, it is difficult to make treatment recommendations. Early reports on the treatment of ‘idiopathic’ MPGN cannot be applied to our patients since those studies were performed in an era when the use of RAS blockade was rare or non-existent and at a time where investigators were unaware that complement abnormalities lead to MPGN. The majority of early studies on treatment also included patients with all types of MPGN.23 While some of the patients in this study were treated with corticosteroids or other immunossuppressive agents, in the absence of a properly conducted study it is difficult to acertain their benefit. It is perhaps relevant that a few patients who were not treated with immunossupressive therapy also did relatively well over time. Our impression that C3GN is less aggressive than DDD suggests that detailed comparisons between these two AP-mediated renal diseases are warranted to define their commonalities and differences.
To summarize, we have presented the most comprehensive description to date of the clinical features, pathologic findings, complement abnormalities, glomerular proteomic profile, treatment and follow-up in C3GN and recurrent C3GN. Our data show that C3GN: (1) occurs at all ages with no sex predilection; (2) presents with hematuria and proteinuria; (3) shows a predominantly MPGN pattern on injury on kidney biopsy; (4) is associated with fairly stable kidney function both in the short and long term; (5) occurs as a result of AP dysregulation that leads to glomerular accumulation of AP and TCC proteins; and (6) results from diverse abnormalities of the AP.
Material and Methods
Renal biopsies from 12 Mayo Clinic patients were evaluated. In all cases, routine work up including LM, IF and EM was performed. Clinical information was obtained from the charts. Although three patients have been previously described, longer follow-up and detailed evaluation are now available. 3 Evaluation of the AP was performed as previously described.3 The Institutional Review Boards at the Mayo Clinic and University of Iowa approved the study.
C3 Nephritic Factors
The presence of C3Nefs was analyzed by enzyme-linked immuno-sorbent assay (ELISA). Purified human C3b (Complement Technology Inc., Tyler, TX) was coated on a 96-well micro-titer plate (pre-treated with 0.2% glutaraldehyde) overnight in 4°C at a concentration of 10μg/ml. After washing three times with 1X PBS, free reactive sites were blocked with 1% BSA in 1X PBS for 1 hour at 37°C. To form C3 convertase, complement factor B (5μg/ml) and complement factor D (0.25μg/ml) in GVB buffer supplemented with 2mM NiCl2 were added to the washed wells followed by a 30 minute incubation at 37°C (a non-convertase well was also included for each sample by omitting this step). Patient serum (1:100 dilution) was added for 25 minutes incubation at 37°C. Plates were washed with PBST (1X PBS with 0.5% Triton-X) and incubated for 30 minutes at room temperature with horseradish peroxidase-labeled goat anti-human IgG antibody specific for the γ chain. Enzymatic activity was measured using TMB (3, 3′, 5, 5′-tetramethylbenzidine) and the optical density (OD) was read at λ450. An OD of +2 SD above normal based on 50 healthy controls was considered positive.
Alternative Pathway Functional Assay (APFA)
AP complement activity was evaluated using the Wieslab complement AP assay kit. The method combines principles of the hemolytic assay for complement activation with the use of labeled antibodies specific for neoantigen produced as a result of complement activation. The amount of neoantigen generated is proportional to the functional activity of the AP. In brief, patient serum was diluted in diluents containing specific blockers to ensure only AP activity. Activation was initiated during incubation of diluted patient serum in microtiter wells coated with specific activators of AP. The wells were washed and C5b- 9 (membrane attack complex, MAC) was detected with a specific phosphatase-labeled antibody to the neoantigen expressed during MAC formation. After further washing, specific antibodies were detected by incubation with an alkaline phosphatase substrate solution. The amount of AP activation was correlated with color intensity and was measured as absorbance (optical density). The value reported was expressed as a percentage of normal control samples. Control values were based on 50 normal sera samples with a normal reference range of 65–130%.
Hemolytic Assay
The sheep erythrocyte lysis assay measures complement-mediated lysis of sheep erythrocytes secondary to AP activation. Sheep erythrocytes generally act as non-activators of complement-mediated lysis in human serum. A small number of C3b molecules spontaneously generated through AP tick-over are deposited on the surface of sheep erythrocytes. In normal human serum, factor H binds to C3b molecules through its N-terminal domains and to sheep erythrocytes through its C-terminal domains. These interactions result in efficient protection of sheep erythrocytes against complement and no lysis is observed. The test was performed under conditions specific to activation of the AP. In brief, 20 μl of patient serum was diluted in 30ul of GVB/Mg2+/EGTA buffer (AP activation possible). A duplicate dilution was prepared in GVB/EDTA as a blank (AP activation not possible). 50 μl of sheep erythrocytes (1 × 108 cells/ml in GVB/Mg2+/EGTA buffer) were added to the mixture and incubated at 37°C for 30 min, after which the reaction was stopped by adding 150 μl of ice-cold GVB/EDTA buffer and the sample was centrifuged. A415 was measured and the amount of lysis was calculated by subtracting A415 of the corresponding blank and dividing by A415 associated with total lysis (50 μl of sheep erythrocytes and 200 μl of water). Reference ranges were: normal (<3%); 1+ (3%–20%); 2+ (20%–40%); 3+ (40%–60%); 4+ (60%–80%); 5+ (80%–100%, complete hemolysis).
Factor H Autoantibodies
The presence of FHAA was analyzed by ELISA. Purified human FH (Complement Technology Inc, Tyler, TX) was coated on a 96-well micro-titer plate overnight in 4°C at a concentration of 5μg/ml. After washing three times with 1X PBST (1XPBS containing 0.5% Triton-X), free reactive sites were blocked with 1% BSA in 1X PBST for 1 hour at room temperature. Patient serum (1:50 dilution) was added for 1 hour incubation at room temperature. Plates were then washed and incubated for 1 hour at room temperature with a horseradish peroxidase-labeled goat anti-human IgG antibody specific for the γ chain. After a final washing, enzymatic activity was measured using TMB (3, 3′, 5, 5′-tetramethylbenzidine) and the optical density (OD) was read at λ450. An OD of + 2 SD above normal based on 50 healthy controls was considered positive (normal reference range: <300 units).
Mutational Analysis
Coding regions and intron-exon boundary junctions of CFH (MIM#134370; NM_000186), CFHR5 (MIM#608593; NM_030787.3), CFI (MIM#217030; NM_000204.3), CD46 (MIM#120920; NM_002389.3), CFB (MIM#138470; NM_001710.5), C3 (MIM#120700; NM_000064.2) and THBD (MIM#188040; NM_000361.2) were amplified and screened for mutations and polymorphisms using bi-directional sequencing. Polymerase chain reaction (PCR) amplification of genomic DNA was completed using 20ng genomic DNA, 2x NH4 buffer, 3mM MgCl2, 400μM each dNTP, 50U/ml TAQ polymerase and 10% DMSO. Reaction conditions consisted of initial denaturation at 95°C for 5 minutes, 35 cycles of denaturation at 95°C for 30 seconds, annealing at 60°C for 30 seconds and extension at 72°C for 30 seconds. These steps were followed by a final extension at 72°C for 10 minutes.
Specimen Preparation, Laser Microdissection and Mass Spectrometry Proteomic Analysis
The methods have been published previously. 8, 24 Briefly, for each case, slides were made using 10μm-thick sections of formalin-fixed paraffin-embedded tissues stained with hematoxylin and eosin. Laser microdissection (LD) was performed by selecting two glomeruli per slide and studying two slides per case. The LD material was collected in 0.5ml micro-centrifuge tube caps containing 35 μL Tris/EDTA/0.002% Zwittergent buffer. Microdissected fragments were digested into tryptic peptides overnight and analyzed by liquid chromatography electrospray tandem mass spectrometry (MS). MS raw data files were queried using three different algorithms (Sequest, Mascot and X!Tandem), the results were combined and assigned peptide and protein probability scores in Scaffold (Proteome Software Inc., Portland, OR). For each case, a list of proteins based on peptides identified by MS was generated. Peptide identifications were accepted if they could be established at >90.0% probability as specified by the Peptide Prophet algorithm. 24–26 Protein identifications <90% confidence level and those with single peptide identification were not considered. The ‘spectra’ value indicates the total number of mass spectra collected on the MS and matched to the protein utilizing the proteomics software. A higher number of mass spectra is indicative of greater abundance and will typically yield greater amino acid sequence coverage. A higher mass spectra value also indicates a higher confidence in the protein identification. Testing required a minimum number of four spectra in all samples before the protein identification was deemed clinically valid. The proteins were ranked according to abundance based on the number of assigned spectra.
Acknowledgments
This research was supported in part by NIH grant DK074409 to SS and RJHS, and Fulk Family Foundation award (Mayo Clinic) to SS
Footnotes
Disclosure- None
References
- 1.Fakhouri F, Fremeaux-Bacchi V, Noel L-H, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol. 2010;6(8):494–9. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 2.Servais A, Fremeaux-Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. Journal of Medical Genetics. 2007;44(3):193–9. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sethi S, Fervenza FC, Zhang Y, et al. Proliferative Glomerulonephritis Secondary to Dysfunction of the Alternative Pathway of Complement. Clinical Journal of the American Society of Nephrology. 2011;6(5):1009–17. doi: 10.2215/CJN.07110810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith RJH, Alexander J, Barlow PN, et al. New Approaches to the Treatment of Dense Deposit Disease. Journal of the American Society of Nephrology. 2007;18(9):2447–56. doi: 10.1681/ASN.2007030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. The Lancet. 2010;376(9743):794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sethi S, Fervenza FC. Membranoproliferative Glomerulonephritis: Pathogenetic Heterogeneity and Proposal for a New Classification. Seminars in Nephrology. 2011;31(4):341–8. doi: 10.1016/j.semnephrol.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz EC, Sethi S, Leung N, Dispenzieri A, Fervenza FC, Cosio FG. Recurrent membranoproliferative glomerulonephritis after kidney transplantation. Kidney Int. 2010;77(8):721–8. doi: 10.1038/ki.2010.1. [DOI] [PubMed] [Google Scholar]
- 8.Sethi S, Gamez JD, Vrana JA, et al. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney Int. 2009;75(9):952–60. doi: 10.1038/ki.2008.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker PD, Ferrario F, Joh K, Bonsib SM. Dense deposit disease is not a membranoproliferative glomerulonephritis. Mod Pathol. 2007;20(6):605–16. doi: 10.1038/modpathol.3800773. [DOI] [PubMed] [Google Scholar]
- 10.Servais A, Noël L-H, Dragon-Durey M-A, et al. Heterogeneous pattern of renal disease associated with homozygous Factor H deficiency. Human Pathology. 2011 doi: 10.1016/j.humpath.2010.11.023. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 11.Sethi S, Sukov WR, Zhang Y, et al. Dense Deposit Disease Associated With Monoclonal Gammopathy of Undetermined Significance. American Journal of Kidney Diseases. 2010;56(5):977–82. doi: 10.1053/j.ajkd.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nasr SH, Valeri AM, Appel GB, et al. Dense Deposit Disease: Clinicopathologic Study of 32 Pediatric and Adult Patients. Clinical Journal of the American Society of Nephrology. 2009;4(1):22–32. doi: 10.2215/CJN.03480708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu D-F, Moon M, Lanning L, McCarthy A, Smith R. Clinical features and outcomes of 98 children and adults with dense deposit disease. Pediatric Nephrology. 2011:1–9. doi: 10.1007/s00467-011-2059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Habbig S, Mihatsch MJ, Heinen S, et al. C3 deposition glomerulopathy due to a functional Factor H defect. Kidney Int. 2009;75(11):1230–4. doi: 10.1038/ki.2008.354. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Meyer NC, Wang K, et al. Causes of Alternative Pathway Dysregulation in Dense Deposit Disease. Clinical Journal of the American Society of Nephrology. 2012 doi: 10.2215/CJN.07900811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abrera-Abeleda MA, Nishimura C, Smith JLH, et al. Variations in the Complement Regulatory Genes Factor H (CFH) and Factor H Related 5 (CFHR5) are Associated with Membranoproliferative Glomerulonephritis Type II (Dense Deposit Disease) J Med Genet. 2005;43(7):582–9. doi: 10.1136/jmg.2005.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heurich M, Martinez-Barricarte R, Francis NJ, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proceedings of the National Academy of Sciences. 2011;108(21):8761–6. doi: 10.1073/pnas.1019338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abrera-Abeleda MA, Nishimura C, Frees K, et al. Allelic Variants of Complement Genes Associated with Dense Deposit Disease. Journal of the American Society of Nephrology. 2011 doi: 10.1681/ASN.2010080795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hageman GS, Hancox LS, Hardisty LI, et al. Common haplotype in the complement regulatory gene, factor H (HF1/CFH), predisposes individuals to age-related macular degeneration. Proceedings of the National Academy of Sciences. 2005;102(20):7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skerka C, Lauer N, Weinberger A, et al. Defective complement control of Factor H (Y402H) and FHL-1 in age-related macular degeneration. Molecular Immunology. 2007;44(13):3398–406. doi: 10.1016/j.molimm.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478(7367):76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vernon KA, Gale DP, de Jorge EG, et al. Recurrence of Complement Factor H-Related Protein 5 Nephropathy in a Renal Transplant. American Journal of Transplantation. 2011;11(1):152–5. doi: 10.1111/j.1600-6143.2010.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatric Nephrology. 2010;25(8):1409–18. doi: 10.1007/s00467-009-1322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, III, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957–9. doi: 10.1182/blood-2009-07-230722. [DOI] [PubMed] [Google Scholar]
- 25.Choi NH, Nakano Y, Tobe T, Mazda T, Tomita M. Incorporation of SP-40,40 into the soluble membrane attack complex (SMAC, SC5b-9) of complement. Int Immunol. 1990;(12):413–7. doi: 10.1093/intimm/2.5.413. [DOI] [PubMed] [Google Scholar]
- 26.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]