

Abstract
We investigate the influence of uniaxial strain on the site occupancy of hydrogen in vanadium, using density functional theory. The site occupancy is found to be strongly influenced by the strain state of the lattice. The results provide the conceptual framework for the atomistic description of the observed hysteresis in the  to
to 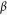 phase transition in bulk, as well as the preferred octahedral occupancy of hydrogen in strained V layers.
phase transition in bulk, as well as the preferred octahedral occupancy of hydrogen in strained V layers.
Vanadium is a transition metal with electronic configuration 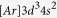 that forms a body-centered cubic structure (bcc). The bcc structure contains tetrahedral and octahedral interstitial sites that can accommodate hydrogen1,2. In bulk vanadium, hydrogen is found to reside in tetrahedral sites at low concentrations (
that forms a body-centered cubic structure (bcc). The bcc structure contains tetrahedral and octahedral interstitial sites that can accommodate hydrogen1,2. In bulk vanadium, hydrogen is found to reside in tetrahedral sites at low concentrations ( -phase), while in the
-phase), while in the 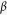 -phase H occupies octahedral sites1,2. The symmetry of the hydrogen induced local strain field is strongly depending on the site occupancy, which is reflected in the hydrogen induced expansion of the lattice1,2. When hydrogen resides in a tetrahedral site, the local strain field is close to spherical, while it is almost uniaxial when hydrogen resides in the octahedral sites.
-phase H occupies octahedral sites1,2. The symmetry of the hydrogen induced local strain field is strongly depending on the site occupancy, which is reflected in the hydrogen induced expansion of the lattice1,2. When hydrogen resides in a tetrahedral site, the local strain field is close to spherical, while it is almost uniaxial when hydrogen resides in the octahedral sites.
In a body-centered tetragonal structure there are three types of tetrahedral sites and three types of octahedral sites, and there are in total three octahedral and six tetrahedral sites per metal atom (Fig. 1). The six tetrahedral sites comprise four 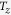 and two
and two 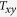 sites (
sites (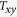 refers to either one of the equivalent
refers to either one of the equivalent 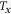 or
or 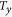 sites). The three octahedral sites comprise one
sites). The three octahedral sites comprise one 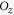 and two
and two 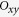 sites. The local elastic response of the lattice arising from the presence of hydrogen in these interstitial sites3,4,5,6,7,8 gives rise to a local strain field, which is the cause of the hydrogen induced expansion4. The expansion can be viewed as the sum of the hydrogen induced local strain fields and can therefore, in principle, be used to determine the preferred site occupancy of hydrogen.
sites. The local elastic response of the lattice arising from the presence of hydrogen in these interstitial sites3,4,5,6,7,8 gives rise to a local strain field, which is the cause of the hydrogen induced expansion4. The expansion can be viewed as the sum of the hydrogen induced local strain fields and can therefore, in principle, be used to determine the preferred site occupancy of hydrogen.
Figure 1.
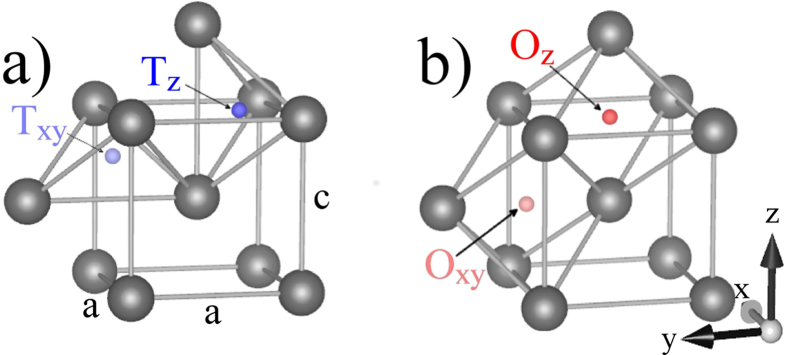
The different types of (a) tetrahedral and (b) octahedral interstitial sites in bcc vanadium are illustrated here. Large dark spheres represent vanadium atoms and small red, blue, light red, and light blue spheres represent (according to their respective labels) different interstitial positions that hydrogen can occupy. The  -axis is aligned along the vertical direction, while the
-axis is aligned along the vertical direction, while the  - and
- and  -axes lie in the horizontal plane.
-axes lie in the horizontal plane.
The volume changes depend strongly on the boundary conditions and site occupancy, enabling the polarization of the local strain fields. This can, for example, be experimentally accomplished by the use of clamping 9,4 as the preferred site occupancy of hydrogen in vanadium is linked to the strain state of the structure10,11,12,13. The hydrogen induced change in volume of a clamped epitaxial film is restricted to the direction perpendicular to the surface. For example, a single crystal vanadium (001) film on a MgO(001) substrate will exhibit a lattice expansion (or contraction) in the (001) direction, independent of site occupancy10,9. Clamping of V can therefore be used to change the volume expansion, as only 1/3 of the strain field will propagate to the surface when hydrogen is residing in tetrahedral sites. By the same token, the uniaxial component of the local strain field arising from an 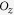 site occupancy will reach the free boundaries and therefore not restrict the expansion. Furthermore, when hydrogen resides in
site occupancy will reach the free boundaries and therefore not restrict the expansion. Furthermore, when hydrogen resides in 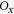 or
or 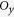 sites, the local strain field cannot give rise to any expansion due to the constraint implemented through the elastic boundary conditions (i.e., clamping) imposed by the substrate. Clamping and straining a V layer will therefore strongly affect the site occupancy, and thereby the observed volume changes9,10,13,4.
sites, the local strain field cannot give rise to any expansion due to the constraint implemented through the elastic boundary conditions (i.e., clamping) imposed by the substrate. Clamping and straining a V layer will therefore strongly affect the site occupancy, and thereby the observed volume changes9,10,13,4.
In the present work we use an atomistic framework and first-principles methodology to investigate the polarization of the local strain fields generated by hydrogen in clamped vanadium and the implications for both site occupancy and lattice expansion. The results allow us to provide a plausible atomistic understanding of the observed hysteresis2 in the hydrogen absorption and desorption in bulk V as well as to explain the 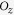 site occupancy at low concentrations in strained V layers9. Since in the underlying experiments, the vanadium layers are composed of 21 monolayers9, we neglect here any direct interface effects and effectively treat the vanadium host as a bulk material.
site occupancy at low concentrations in strained V layers9. Since in the underlying experiments, the vanadium layers are composed of 21 monolayers9, we neglect here any direct interface effects and effectively treat the vanadium host as a bulk material.
Methods
The calculations were performed using the Vienna Ab initio Simulation Package (VASP)14,15,16,17. We have used a modified version of VASP in which the National Supercomputer Centre (NSC) at Linköping University had implemented the possibility to perform constrained cell relaxations with one or more lattice vectors fixed, as described in further detail below.
The interactions between the electrons and the nuclei was obtained using the projector-augmented-wave method18,19. The generalized gradient approximation (GGA) in the parametrization of the Perdew-Burke-Ernzerhof (PBE)20,21 approach was employed to approximate the exchange and correlation terms in the density functional theory (DFT)22,23 method. The GGA-PBE method has earlier been shown to be reliable when calculating electronic properties of transition metal hydrides8,9. A conjugate gradient algorithm was used to relax the atomic nuclei positions to a local minimum in the total energy landscape.
In order to reduce H-H interactions resulting from the imposed periodic boundary conditions while still studying a system that is small enough to be computationally manageable for a large number of calculations, a supercell consisting of 128 vanadium atoms (4 × 4 × 4 bcc unit cells) was constructed to mimic bulk vanadium in which the lowest possible ratio of hydrogen to vanadium [H/V] is 1/128 (corresponding to 0.775 at.% of hydrogen). Due to the rather large dimensions (11.9 Å × 11.9 Å × 11.9 Å) of the supercell, only the  point was used in sampling the Brillouin zone. Comparisons of the total energy, using a 3 × 3 × 3 k-point mesh further established that
point was used in sampling the Brillouin zone. Comparisons of the total energy, using a 3 × 3 × 3 k-point mesh further established that  point sampling of the Brillouin zone is sufficient for a quantitative investigation.
point sampling of the Brillouin zone is sufficient for a quantitative investigation.
Zero-point energy corrections to the total energy are included for the hydrogen atoms and have been calculated from an harmonic approximation of the potential energy change as a function of atomic displacement.
Calculations for higher hydrogen concentrations than [H/V] = 1/128 were carried out by randomly distributing hydrogen atoms into the vanadium supercell and calculating the resulting average volume and energy of 50 structures, with different hydrogen distributions. Hydrogen concentrations [H/V] of 8/128 (5.88 at.%), 16/128 (11.11 at.%), 32/128 (20.00 at.%) and 64/128 (33.33 at.%) were investigated. This corresponds to a disordered state, mimicking the conditions above the phase boundaries of a 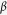 -phase.
-phase.
The  and
and  lattice vectors were fixed for all calculations, only allowing lattice relaxation in the
lattice vectors were fixed for all calculations, only allowing lattice relaxation in the  -direction. The motivation for this approach is to mimic the conditions of hydrogen uptake in a superlattice where the bottom layer of a thin film of vanadium is held in place through strong bonds to a substrate9. This constraint results in a one-dimensional lattice expansion, perpendicular to the plane of the substrate.
-direction. The motivation for this approach is to mimic the conditions of hydrogen uptake in a superlattice where the bottom layer of a thin film of vanadium is held in place through strong bonds to a substrate9. This constraint results in a one-dimensional lattice expansion, perpendicular to the plane of the substrate.
It is sufficient to consider the strain in one direction, for example the  -direction, to capture the effect of strain on site occupancy. This implies also that we can treat the
-direction, to capture the effect of strain on site occupancy. This implies also that we can treat the 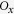 and
and 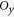 sites to be equivalent when hydrogen is occupying
sites to be equivalent when hydrogen is occupying 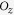 sites in the
sites in the 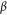 -phase. These are identical in the sense that a rotation by 90°around the
-phase. These are identical in the sense that a rotation by 90°around the  -axis will map the
-axis will map the 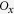 site onto the
site onto the 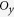 site and vice versa.
site and vice versa.
Results & discussion
Strain and site occupancy
Before presenting the results from our ab initio calculations we will provide a conceptual framework for the effect of strain on site occupancy, using a simple hard-spheres model. We will use this approach to estimate the relative energetics of hydrogen occupation in octahedral and tetrahedral sites, solely based on the available interstitial space in a V single crystal. The maximum sphere radius that can be accommodated in the interstitial space formed by metal atom spheres arranged in a bcc lattice is 0.155 for octahedral and 0.291 for tetrahedral sites, in units of the metal atom sphere radius. In the atomistic model used here the vanadium has a Wigner-Seitz radius of 1.217 Å and the corresponding value for hydrogen is 0.370 Å. An octahedral site in V has therefore  Å = 0.189Å of spherical radius available, which is much smaller than the hydrogen radius. A tetrahedral site provides
Å = 0.189Å of spherical radius available, which is much smaller than the hydrogen radius. A tetrahedral site provides  Å = 0.35 Å, which is close to the hydrogen radius. From this consideration, one can see directly that it is not energetically favourable for hydrogen to occupy octahedral sites in an unstrained lattice, because of a large overlap between H and V electrons, raising the total energy through a Born-Mayer repulsion24. In the tetrahedral sites, the corresponding density overlap is much smaller, favouring occupation of tetrahedral sites. When the lattice is under uniaxial tensile strain (i.e.,
Å = 0.35 Å, which is close to the hydrogen radius. From this consideration, one can see directly that it is not energetically favourable for hydrogen to occupy octahedral sites in an unstrained lattice, because of a large overlap between H and V electrons, raising the total energy through a Born-Mayer repulsion24. In the tetrahedral sites, the corresponding density overlap is much smaller, favouring occupation of tetrahedral sites. When the lattice is under uniaxial tensile strain (i.e., 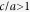 ) the maximum sphere radius that can be accommodated in the
) the maximum sphere radius that can be accommodated in the 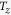 and
and 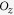 sites is more and more shifted in favour of the
sites is more and more shifted in favour of the 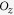 sites. The maximum sphere radius that can be accommodated in
sites. The maximum sphere radius that can be accommodated in 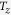 and
and 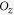 sites becomes equal for c/a = 1.118
sites becomes equal for c/a = 1.118
When the lattice is expanded in the  -direction the
-direction the 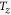 and
and 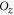 sites are energetically favoured in comparison to their
sites are energetically favoured in comparison to their  - and
- and  -oriented counterparts. This is rather obvious for the octahedral sites but not immediately clear for the tetrahedral sites. For the
-oriented counterparts. This is rather obvious for the octahedral sites but not immediately clear for the tetrahedral sites. For the 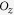 sites, a tensile strain in the
sites, a tensile strain in the  -direction will increase the spacing between the vanadium atoms that sit closest to hydrogen, while for
-direction will increase the spacing between the vanadium atoms that sit closest to hydrogen, while for 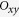 sites, the closest vanadium atoms lie in the
sites, the closest vanadium atoms lie in the 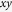 -plane, which are geometrically unaffected by the uniaxial strain in the
-plane, which are geometrically unaffected by the uniaxial strain in the  -direction.
-direction.
Figure 2 shows the ab initio calculated local strain fields in vanadium caused by hydrogen occupying either a 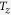 or an
or an 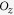 site in a 128 vanadium atoms supercell. The arrows indicate the direction and the magnitude of the displacement of the vanadium atoms. Only the strain on the vanadium atoms in the
site in a 128 vanadium atoms supercell. The arrows indicate the direction and the magnitude of the displacement of the vanadium atoms. Only the strain on the vanadium atoms in the 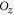 and
and 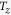 sites are shown (i.e., 4 atoms for a tetrahedral site and 6 atoms for an octahedral site). The isotropic strain field from hydrogen occupying a tetrahedral site and the strongly anisotropic strain field from occupying an octahedral site can be clearly seen in Fig. 2. The “top” and “bottom” vanadium atoms in the octahedron (i.e., the two vanadium atoms that possess the same
sites are shown (i.e., 4 atoms for a tetrahedral site and 6 atoms for an octahedral site). The isotropic strain field from hydrogen occupying a tetrahedral site and the strongly anisotropic strain field from occupying an octahedral site can be clearly seen in Fig. 2. The “top” and “bottom” vanadium atoms in the octahedron (i.e., the two vanadium atoms that possess the same  and
and  coordinates) are much closer to the hydrogen than the vanadium atoms in the tetrahedron; hence, the former are pushed farther away. In the absence of hydrogen the calculated lattice parameter is 2.99 Å . When hydrogen is placed in the
coordinates) are much closer to the hydrogen than the vanadium atoms in the tetrahedron; hence, the former are pushed farther away. In the absence of hydrogen the calculated lattice parameter is 2.99 Å . When hydrogen is placed in the 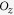 site, the “top” and “bottom” V-atoms are displaced, increasing their mutual distance to 3.35 Å. This local strain corresponds to an increase of 12.1% in spacing between the V atoms which is in excellent agreement with the experimental results of 12.7% for
site, the “top” and “bottom” V-atoms are displaced, increasing their mutual distance to 3.35 Å. This local strain corresponds to an increase of 12.1% in spacing between the V atoms which is in excellent agreement with the experimental results of 12.7% for 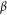 -phase VH0.5 obtained by EXAFS10.
-phase VH0.5 obtained by EXAFS10.
Figure 2.
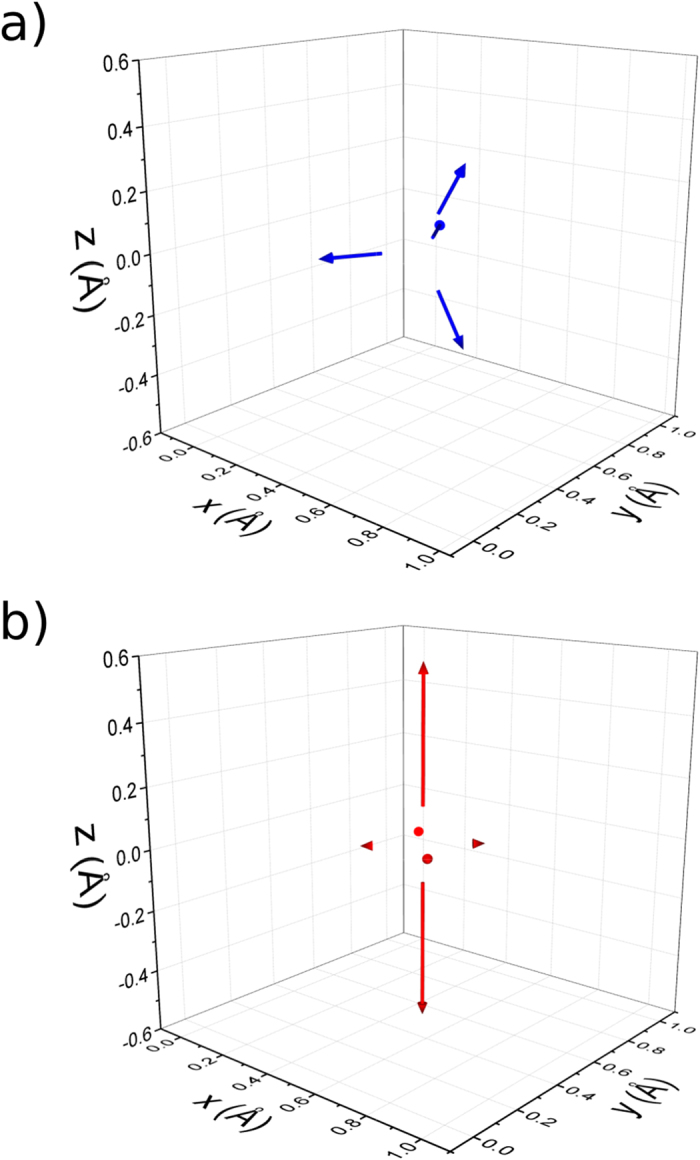
The strain on surrounding vanadium atoms by hydrogen occupying a (a) 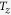 site or (b)
site or (b) 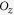 site. The arrows represent the displacement vectors, i.e., by what distance the V atoms have been repelled by the H atom. For clearer visibility, the length of the arrows has been scaled up by a factor of 30.
site. The arrows represent the displacement vectors, i.e., by what distance the V atoms have been repelled by the H atom. For clearer visibility, the length of the arrows has been scaled up by a factor of 30.
To quantify the qualitative ideas obtained from the hard-sphere model, we used ab initio total energy calculations to determine the preferred hydrogen occupancy. Fig. 3 shows a plot of the energy for a single hydrogen in a supercell (i.e., a concentration of [H/V] = 1/128, corresponding to 0.775 at.% of hydrogen) occupying a 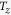 ,
, 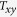 ,
, 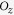 or an
or an 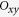 site as a function of the uniaxial strain given in the form of the
site as a function of the uniaxial strain given in the form of the 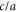 ratio. The vertical axis shows
ratio. The vertical axis shows 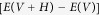 where
where 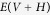 is the total energy of the metal-hydrogen system and
is the total energy of the metal-hydrogen system and 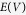 is the total energy of the hydrogen-free vanadium supercell, both calculated at the same
is the total energy of the hydrogen-free vanadium supercell, both calculated at the same 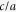 ratio. When the lattice is uniaxially strained (
ratio. When the lattice is uniaxially strained (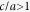 ), the
), the 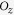 sites will “open up” as described above and become more energetically favoured. This is easily inferred from the results in Fig. 3 since the slope of the
sites will “open up” as described above and become more energetically favoured. This is easily inferred from the results in Fig. 3 since the slope of the 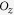 line is larger than that of
line is larger than that of 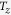 , thus at some
, thus at some 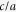 ratio the site occupancy of
ratio the site occupancy of 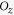 will become lower in energy compared to the
will become lower in energy compared to the 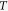 sites. As seen in Fig. 2, the strain is very large in the
sites. As seen in Fig. 2, the strain is very large in the  -direction for hydrogen occupying an
-direction for hydrogen occupying an 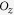 site. A comparison of the strain fields from hydrogen occupation of
site. A comparison of the strain fields from hydrogen occupation of 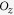 and
and 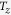 sites shows a larger increase of available spherical radius for the hydrogen occupying an
sites shows a larger increase of available spherical radius for the hydrogen occupying an 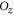 site. This, together with the favourable effect of the uniaxial tensile strain for the occupation of the
site. This, together with the favourable effect of the uniaxial tensile strain for the occupation of the 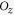 sites makes the
sites makes the 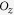 sites energetically favorable already when c/a = 1.043. The hard sphere model yielded a transition at c/a = 1.118 which can be considered as satisfying agreement when considering the simplicity of the model.
sites energetically favorable already when c/a = 1.043. The hard sphere model yielded a transition at c/a = 1.118 which can be considered as satisfying agreement when considering the simplicity of the model.
Figure 3.
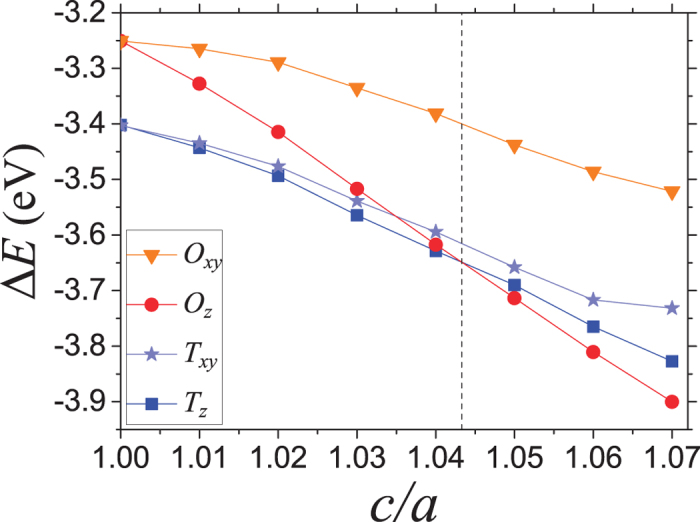
Energy difference as a function of an externally applied global uniaxial lattice strain 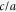 where the difference in total energy
where the difference in total energy 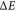 is defined as
is defined as 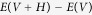 . The dashed vertical line at c/a = 1.043 marks the critical uniaxial lattice strain for which hydrogen occupancy of
. The dashed vertical line at c/a = 1.043 marks the critical uniaxial lattice strain for which hydrogen occupancy of 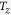 and
and 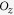 sites becomes energetically equivalent.
sites becomes energetically equivalent.
Concentration dependence of site occupancy
It is not only the initial strain state which is the source of tetragonal distortion. The hydrogen induced volume changes will also influence the 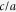 ratio in clamped samples and thereby alter the energy balance between the
ratio in clamped samples and thereby alter the energy balance between the 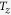 and
and 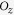 sites. Fig. 4 compares the energies of
sites. Fig. 4 compares the energies of 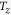 and
and 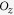 site occupancy at optimal
site occupancy at optimal 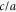 ratios to identify the critical hydrogen concentration where change in site occupancy occurs. The average energy of 50 structures with random hydrogen distributions for four different hydrogen concentrations is calculated (in a disordered phase). Change in site occupancy is approximated to occur between [H/V] of 0.28
ratios to identify the critical hydrogen concentration where change in site occupancy occurs. The average energy of 50 structures with random hydrogen distributions for four different hydrogen concentrations is calculated (in a disordered phase). Change in site occupancy is approximated to occur between [H/V] of 0.28 0.07 as this is where the total energy of
0.07 as this is where the total energy of 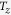 and
and 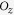 site occupancy becomes equal.
site occupancy becomes equal.
Figure 4.
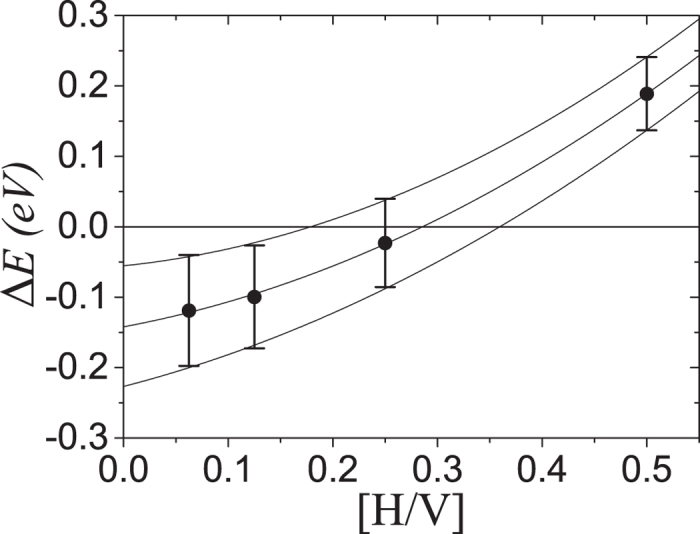
Energy difference between 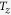 and
and 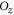 occupancy at optimal
occupancy at optimal 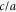 ratios as a function of hydrogen concentration. Here, the energy difference
ratios as a function of hydrogen concentration. Here, the energy difference 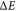 is defined as
is defined as 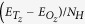 where
where 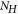 is the number of hydrogen atoms included in the simulation. Data points are for average values and the bars indicate
is the number of hydrogen atoms included in the simulation. Data points are for average values and the bars indicate  one standard deviation (calculated as the square root of the variance). Connecting lines are second-order polynomial fitting functions.
one standard deviation (calculated as the square root of the variance). Connecting lines are second-order polynomial fitting functions.
The shift in the site occupancy is found to be driven by energetics rather than entropy. The configurational entropy was determined using Boltzmann’s entropy formula and the internal energy was approximated as the number of hydrogen atoms in  -sites times an energy penalty of 0.2 eV (i.e., we approximate that moving a hydrogen atom from a
-sites times an energy penalty of 0.2 eV (i.e., we approximate that moving a hydrogen atom from a 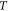 site to an
site to an  site will raise the energy by 0.2 eV, in accordance with the difference in energy between
site will raise the energy by 0.2 eV, in accordance with the difference in energy between 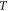 and
and  site occupancy, cf. Fig. 3). For all tested hydrogen concentrations and for a broad temperature range, the internal energy is always found to dominate the entropy part, so that coexistence of
site occupancy, cf. Fig. 3). For all tested hydrogen concentrations and for a broad temperature range, the internal energy is always found to dominate the entropy part, so that coexistence of 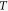 and
and  sites is concluded unlikely to occur in the low concentration region. The hydrogen induced lattice expansion can however give rise to change in site occupancy. This can take place in both ordered and disordered phases, thus a
sites is concluded unlikely to occur in the low concentration region. The hydrogen induced lattice expansion can however give rise to change in site occupancy. This can take place in both ordered and disordered phases, thus a 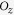 occupancy does not need to imply an ordered
occupancy does not need to imply an ordered 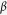 -phase and a change of site does therefore not by necessity imply a disorder-order phase transition.
-phase and a change of site does therefore not by necessity imply a disorder-order phase transition.
Volume expansion and hysteresis effects
Figure 5 shows the resulting uniaxial lattice expansion (quantified by the 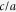 ratio) as a function of hydrogen concentration [H/V]. The relationship between calculated
ratio) as a function of hydrogen concentration [H/V]. The relationship between calculated 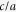 ratio and hydrogen concentration [H/V] is to a very good approximation linear with a slope of 0.120 for
ratio and hydrogen concentration [H/V] is to a very good approximation linear with a slope of 0.120 for 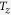 sites and 0.236 for
sites and 0.236 for 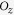 sites, see Fig. 5. These results are in good agreement with the experimental results by Pálsson et al.99 which determined the expansion to be 0.1189(7) for hydrogen occupation in tetrahedral sites and 0.19(1) for octahedral sites. The calculated change in total volume due to hydrogen occupation of
sites, see Fig. 5. These results are in good agreement with the experimental results by Pálsson et al.99 which determined the expansion to be 0.1189(7) for hydrogen occupation in tetrahedral sites and 0.19(1) for octahedral sites. The calculated change in total volume due to hydrogen occupation of 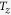 and
and 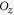 sites are 1.61 Å3 and 3.14 Å3, respectively, per added hydrogen atom when the expansion is restricted to the
sites are 1.61 Å3 and 3.14 Å3, respectively, per added hydrogen atom when the expansion is restricted to the  -direction (due to clamping).
-direction (due to clamping). 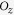 occupancy gives rise to a larger increase in volume, as compared to
occupancy gives rise to a larger increase in volume, as compared to 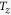 occupancy, due to the anisotropy of the local strain field as seen in Fig. 2. The strain component in the
occupancy, due to the anisotropy of the local strain field as seen in Fig. 2. The strain component in the  -direction is larger for
-direction is larger for 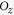 than for
than for 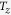 sites, implying that a shift from
sites, implying that a shift from 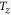 to
to 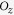 occupancy is accompanied by an increased
occupancy is accompanied by an increased 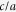 ratio, which favours
ratio, which favours 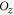 occupancy. The shift in site occupancy from
occupancy. The shift in site occupancy from 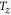 to
to 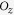 can thus be viewed as a self-amplified process and resembles therefore in many ways a first-order phase transition.
can thus be viewed as a self-amplified process and resembles therefore in many ways a first-order phase transition.
Figure 5.
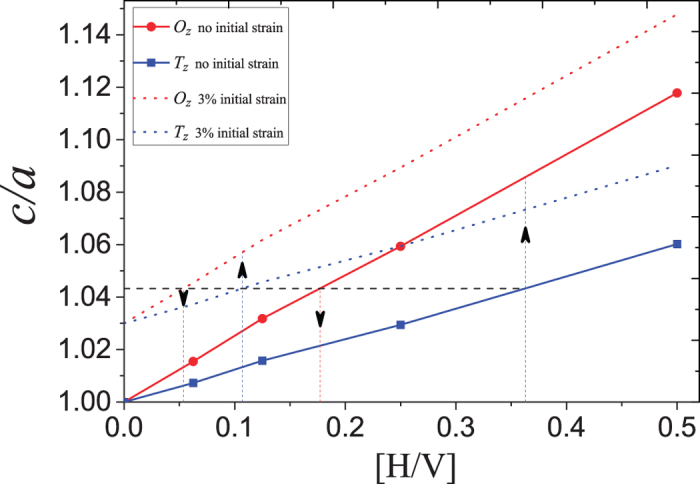
The uniaxial lattice strain 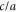 resulting from varying the concentration of hydrogen occupying exclusively either
resulting from varying the concentration of hydrogen occupying exclusively either 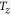 sites (blue data points and lines) or
sites (blue data points and lines) or 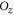 sites (red data points and lines) in clamped vanadium. The horizontal black dashed line at c/a = 1.043 marks the critical uniaxial lattice strain for which the hydrogen occupancy of
sites (red data points and lines) in clamped vanadium. The horizontal black dashed line at c/a = 1.043 marks the critical uniaxial lattice strain for which the hydrogen occupancy of 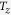 and
and 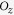 site becomes equal in energy, as seen in Fig. 3. The vertical coloured lines indicate at which hydrogen concentration the critical
site becomes equal in energy, as seen in Fig. 3. The vertical coloured lines indicate at which hydrogen concentration the critical 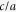 ratio of 1.043 is reached for occupancy of
ratio of 1.043 is reached for occupancy of 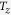 ([H/V] = 0.363) and
([H/V] = 0.363) and 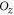 ([H/V] = 0.177) sites, respectively, when there is no initial strain (i.e. c/a = 1.00). The dotted lines represent the case of an initial strain of c/a = 1.03 before any hydrogen has entered the system. The critical
([H/V] = 0.177) sites, respectively, when there is no initial strain (i.e. c/a = 1.00). The dotted lines represent the case of an initial strain of c/a = 1.03 before any hydrogen has entered the system. The critical 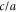 ratio is then reached at [H/V] = 0.107 for
ratio is then reached at [H/V] = 0.107 for 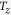 occupancy and 0.054 for
occupancy and 0.054 for 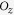 occupancy.
occupancy.
Now we will discuss the difference in the lattice response when the hydrogen concentration is increased or decreased. In an unstrained or nearly unstrained lattice hydrogen is exclusively found in 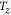 sites. When increasing the hydrogen concentration from low concentrations in clamped V layers, the expansion will open up the
sites. When increasing the hydrogen concentration from low concentrations in clamped V layers, the expansion will open up the 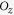 sites, which become energetically favoured above the critical
sites, which become energetically favoured above the critical 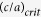 value of 1.043. The uniaxial lattice expansion will therefore result in a shift in site occupancy from
value of 1.043. The uniaxial lattice expansion will therefore result in a shift in site occupancy from 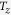 to
to 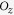 at that concentration.
at that concentration. 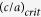 for the change from
for the change from 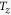 to
to 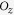 occupancy is marked by a vertical line in Fig. 3 and a horizontal dashed line in Fig. 5.
occupancy is marked by a vertical line in Fig. 3 and a horizontal dashed line in Fig. 5.
When starting at a high concentration, all the hydrogen will reside in 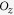 sites. When decreasing the hydrogen concentration,
sites. When decreasing the hydrogen concentration, 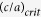 = 1.043 will be reached at [H/V] = 0.177, resulting in a shift in site occupancy from
= 1.043 will be reached at [H/V] = 0.177, resulting in a shift in site occupancy from 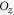 to
to 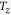 . Thus, when increasing the concentration the shift from
. Thus, when increasing the concentration the shift from 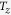 to
to 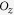 is reached at a different concentration as compared to the change of sites from
is reached at a different concentration as compared to the change of sites from 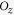 to
to 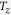 sites when decreasing the hydrogen concentration. Therefore, a hysteresis with respect to lattice expansion is expected when loading and unloading H under the specified conditions and when the thermal excitations are smaller than the energy difference between the two states. These effects do resemble the
sites when decreasing the hydrogen concentration. Therefore, a hysteresis with respect to lattice expansion is expected when loading and unloading H under the specified conditions and when the thermal excitations are smaller than the energy difference between the two states. These effects do resemble the  to
to 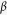 phase transition in bulk V, with respect to both change of sites as well as observed hysteresis2. Furthermore, these results clearly illustrate the effect of clamping on the site occupancy, which can be changed without entering the
phase transition in bulk V, with respect to both change of sites as well as observed hysteresis2. Furthermore, these results clearly illustrate the effect of clamping on the site occupancy, which can be changed without entering the 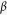 -phase in V. When the initial strain of the sample is changed, these boundaries will move as illustrated in Fig. 5: With a biaxial compressive strain in the
-phase in V. When the initial strain of the sample is changed, these boundaries will move as illustrated in Fig. 5: With a biaxial compressive strain in the 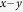 plane, the boundaries will move to lower concentrations and the hysteresis gap will decrease. When
plane, the boundaries will move to lower concentrations and the hysteresis gap will decrease. When 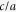 will be larger than a threshold value, hydrogen will solely reside in
will be larger than a threshold value, hydrogen will solely reside in 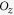 sites, as inferred from experiments9
sites, as inferred from experiments9
Summary
The preferred interstitial site occupancy in vanadium with constrained boundaries has been studied using calculations based on density functional theory. The energetics of hydrogen atoms in a bcc-bct supercell were investigated to provide a conceptual understanding of the experimentally observed shifts in site occupancy9. In the investigated range of 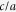 from 1.00 up to 1.07, the tetrahedral (
from 1.00 up to 1.07, the tetrahedral (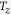 ) sites are energetically favoured for hydrogen occupation in comparison to the octahedral (
) sites are energetically favoured for hydrogen occupation in comparison to the octahedral (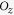 ) sites in the
) sites in the 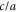 range from 1.00 to 1.043. The octahedral sites are energetically favoured for hydrogen occupation when c/a >1.043. The forces exerted on the vanadium lattice by hydrogen atoms occupying interstitial sites will alter the global strain state which in turn triggers a shift in site occupancy above the critical value of c/a = 1.043. This self-amplified process can be understood by the obtained strain field from octahedral (
range from 1.00 to 1.043. The octahedral sites are energetically favoured for hydrogen occupation when c/a >1.043. The forces exerted on the vanadium lattice by hydrogen atoms occupying interstitial sites will alter the global strain state which in turn triggers a shift in site occupancy above the critical value of c/a = 1.043. This self-amplified process can be understood by the obtained strain field from octahedral (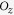 ) site occupancy which has a larger
) site occupancy which has a larger  -component than that obtained for a tetrahedral (
-component than that obtained for a tetrahedral (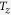 ) site occupancy. The increase in
) site occupancy. The increase in 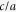 as a function of hydrogen concentration [H/V] is linear and in good agreement with previously obtained experimental results.
as a function of hydrogen concentration [H/V] is linear and in good agreement with previously obtained experimental results.
The different rate at which the 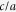 ratio changes as a function of [H/V] for tetrahedral (
ratio changes as a function of [H/V] for tetrahedral (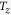 ) and octahedral (
) and octahedral (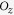 ) site occupancy has the consequence that the condition for shift in site occupancy is met at different hydrogen concentrations [H/V] when starting from high (
) site occupancy has the consequence that the condition for shift in site occupancy is met at different hydrogen concentrations [H/V] when starting from high (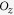 ) or low concentrations (
) or low concentrations (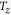 ). This leads to the theoretical prediction of a hysteresis in the hydrogen loading-unloading process, in which the switch from
). This leads to the theoretical prediction of a hysteresis in the hydrogen loading-unloading process, in which the switch from 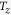 to
to 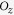 site occupancy and the reverse switch from
site occupancy and the reverse switch from 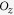 to
to 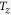 occur at different hydrogen concentrations. The results therefore provide an insight into the interplay between site occupancy and ordering in both bulk and thin films of bcc lattices. The experimentally observed9 coexistence of tetrahedral and octahedral hydrogen occupation in the [H/V] concentration range of 0.065–0.068 is an indication that such a hysteresis behavior could indeed be found in vanadium.
occur at different hydrogen concentrations. The results therefore provide an insight into the interplay between site occupancy and ordering in both bulk and thin films of bcc lattices. The experimentally observed9 coexistence of tetrahedral and octahedral hydrogen occupation in the [H/V] concentration range of 0.065–0.068 is an indication that such a hysteresis behavior could indeed be found in vanadium.
Author Contributions
All authors designed the research, analyzed the data, and reviewed the manuscript; R.J. performed research; R.J., R.H.S. and B.H. wrote the paper.
Additional Information
How to cite this article: Johansson, R. et al. Effect of uniaxial strain on the site occupancy of hydrogen in vanadium from density-functional calculations. Sci. Rep. 5, 10301; doi: 10.1038/srep10301 (2015).
Acknowledgments
Financial support from the Swedish Research Council is gratefully acknowledged. The project is part of the COST Action MP1103. O.E. also acknowledges the KAW foundation, eSSENCE, STandUP for Energy, and the ERC (project 247062 - ASD). The calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC).
References
- Alefeld G. & Völkl J. Hydrogen in Metals I, vol. 28 ( Springer, 1978). [Google Scholar]
- Alefeld G. & Völkl J. Hydrogen in Metals II, vol. 29 ( Springer, 1978). [Google Scholar]
- Buck H. & Alefeld G. Hydrogen in palladiumsilver in the neighbourhood of the critical point. Phys. Stat. Sol. 49, 317–327 (1972). [Google Scholar]
- Alefeld G. Ber. Bunsenges. Phys. Chem. 76, 746 (1972). [Google Scholar]
- Laudahn U., Fähler S., Krebs H. U. & Pundt A. Appl. Phys. Lett. 74, 647 (1999). [Google Scholar]
- Dornheim M. et al. Stress development in thin yttrium films on hard substrates during hydrogen loading. Journal of Applied Physics 93, 8958–8965 (2003). [Google Scholar]
- Laudahn U. et al. Hydrogen-induced stress in nb single layers. Journal of Alloys and Compounds 293–295, 490–494 (1999). [Google Scholar]
- Blomqvist A., Pálsson G. K., Araújo C. M., Ahuja R. & Hjörvarsson B. Significance of self-trapping on hydrogen diffusion. Phys. Rev. Lett. 105, 185901 (2010). [DOI] [PubMed] [Google Scholar]
- Pálsson G. K. et al. Hydrogen site occupancy and strength of forces in nanosized metal hydrides. Phys. Rev. B 85, 195407 (2012). [Google Scholar]
- Burkert T., Miniotas A. & Hjörvarsson B. Hydrogen-induced changes of the local structure in Fe/V (001) superlattices. Physical Review B 63, 125424 (2001). [Google Scholar]
- Hjörvarsson B., Andersson G. & Karlsson E. Metallic superlattices: Quasi two-dimensional playground for hydrogen. J. Alloys Compd. 253, 51–57 (1997). [Google Scholar]
- Andersson G., Andersson P. H. & Hjörvarsson B. Effects of varying compressive biaxial strain on the hydrogen uptake of thin vanadium (001) layers. Journal of Physics 11, 6669–6677 (1999). [Google Scholar]
- Olsson S. & Hjörvarsson B. Effect of biaxial elastic constraints on H–H interactions in ultrathin vanadium. Phys. Rev. B 71, 035414 (2005). [Google Scholar]
- Kresse G. & Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Hafner J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- Kresse G., D. J. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- Blöchl P. E. Projector augmented-wave method . Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- Perdew J. P., Burke K. & Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- Perdew J. P., Burke K. & Ernzerhof M. Erratum: Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396 (1997). [DOI] [PubMed] [Google Scholar]
- Hohenberg P. & Kohn W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964). [Google Scholar]
- Kohn W. & Sham L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). [Google Scholar]
- Abrahamson A. A. Born-mayer-type interatomic potential for neutral ground-state atoms with z = 2 to z = 105. Phys. Rev. 178, 76–79 (1969). [Google Scholar]