

Abstract
Medication-related osteonecrosis of the jaw (MRONJ) is a severe devastating complication for which the exact pathogenesis is not completely understood. Multiple systemic and local factors may contribute to the development of MRONJ. A growing body of evidence supports diabetes mellitus (DM) as an important risk factor for this complication; however, the exact mechanism by which DM may promote MRONJ has yet to be determined. The current review elucidates the role of DM in the pathogenesis of MRONJ and the mechanisms by which DM may increase the risk for MRONJ. Factors related to DM pathogenesis and treatment may contribute to poor bone quality through multiple damaged pathways, including microvascular ischemia, endothelial cell dysfunction, reduced remodeling of bone, and increased apoptosis of osteoblasts and osteocytes. In addition, DM induces changes in immune cell function and promotes inflammation. This increases the risk for chronic infection in the settings of cancer and its treatment, as well as antiresorptive medication exposure, thus raising the risk of developing MRONJ. A genetic predisposition for MRONJ, coupled with CYP 450 gene alterations, has been suggested to affect the degradation of medications for DM such as thiazolidinediones and may further increase the risk for MRONJ.
Keywords: diabetes complications, microvascular disease, bone remodeling, inflammation, angigenesis, immune responses
Introduction
In the early 2000s, teams led by Marx and Ruggiero first reported nonhealing exposed bone in the maxillofacial region of patients treated with intravenous (IV) bisphosphonates (BPs) (Marx 2003; Ruggiero et al. 2004). Numerous studies have since been published on medication-related osteonecrosis of the jaw (MRONJ), mainly BP related. The cumulative incidence of this severe bone disease among antiresorptive users is estimated to range from 0.7% to 18% (Aragon-Ching et al. 2009; Migliorati et al. 2010). More recent antiresorptive therapies, including the denosumab- receptor activator of nuclear factor (NF)–κB ligand-RANKL monoclonal antibody inhibitor and antiangiogenic therapies, are associated with an increasing incidence of MRONJ (AAOMS 2014). Among the characteristics of BP and other antiresorptive therapies that have been suggested as risk factors for MRONJ are high drug potency, the IV route of administration, and extended duration of exposure (Bamias et al. 2005; Saad et al. 2012). A number of patient-related systemic factors have also been suggested, including cancer diagnosis, older age, chemotherapy treatment (antiangiogenic treatments), and factors relating to comorbidities (malnutrition, obesity, anemia, dialysis), lifestyle (alcohol, tobacco), and pharmacotherapy, such as the use of glucocorticoids (Bamias et al. 2005; Barasch et al. 2011; AAOMS 2014). Patient-related local risk factors include concomitant oral disease, poor oral hygiene, and major dental procedures (e.g., dental extraction) (Bamias et al. 2005; AAOMS 2014).
Potential mechanisms for the development of MRONJ involve impaired bone repair, suppression of osteoclast activity, infection and inflammation, and impaired angiogenesis or vascular repair (Allen and Burr 2009) (Table 1). This review describes the role of diabetes mellitus (DM) in the pathogenesis of MRONJ and the mechanisms by which DM may increase the risk for osteonecrosis (Figure, Table 2). The main focus is microvascular ischemia of the bone, endothelial cell dysfunction and apoptosis, decreased bone turnover and remodeling, and increased apoptosis of osteoblasts and osteocytes. Factors that may contribute to the development of MRONJ, such as local factors, drug-related factors, immune suppression, inflammation, and genetic factors, are discussed.
Table 1.
Potential Mechanisms of Medication-Related Osteonecrosis of the Jaw (MRONJ).
| Mechanism | |
|---|---|
| Altered bone turnover and remodeling |
|
| Increased osteoblast and osteocyte apoptosis |
|
| Altered immune responses and increased inflammation |
|
| Angiogenesis, vascularization, and endothelial damage |
|
| Gene regulation |
|
OPG, osteoprotegerin; BP, bisphosphonate.
Figure.
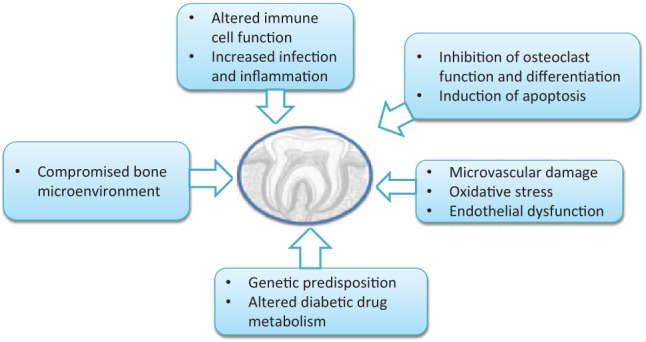
Role of diabetes in the pathogenesis of medication-related osteonecrosis of the jaw (MRONJ).
Table 2.
Potential Diabetes-Related Mechanisms Involved in the Development of Medication-Related Osteonecrosis of the Jaw (MRONJ).
| Mechanism | |
|---|---|
| Altered bone turnover and remodeling |
|
| Increased osteoblast and osteocyte apoptosis |
|
| Altered immune responses and increased inflammation |
|
| Angiogenesis, vascularization, and endothelial damage |
|
| Gene regulation |
|
DM, diabetes mellitus; AGE, advanced glycation end products; TNFα, tumor necrosis factor α; MCP-1, monocyte chemoattractant protein-1; VEGF, vascular endothelial growth factor.
Diabetes as a Risk Factor for MRONJ
Most studies investigating the relationship between DM and MRONJ reported a positive association (Khamaisi et al. 2007; Bocanegra-Pérez et al. 2012; Molcho et al. 2013; Watters et al. 2013; Berti-Couto et al. 2014); however, one study reported no association (Wilkinson et al. 2007) (Table 3). In a case-control study, DM was detected in 17% (33/191) of those with osteonecrosis of the jaw, compared with 11% (63/573) of the controls (Barasch et al. 2011). In an observational study, DM was detected in 35% (15/44) of patients with MRONJ (Bocanegra-Pérez et al. 2012). From a long-term follow-up of 109 individuals with MRONJ, a marginally significant correlation was found between poor prognosis and a diagnosis of DM (Watters et al. 2013). Among 68 patients with a progressive or unchanged clinical course of MRONJ, 18 (26.5%) had diabetes, compared to only 4 (9.4%) of the 29 with partial resolution or resolved MRONJ. Our team recently reported a prevalence of 58% (18/31) of DM (mainly type 2 DM) or impaired fasting glucose (IFG) among patients with MRONJ, compared to 12% among those treated with BP without MRONJ; diabetic nephropathy conferred an increased risk of MRONJ by up to 3.9-fold (Molcho et al. 2013) (Table 3).
Table 3.
A Summary of the Studies That Reported an Association Between Diabetes and Medication-Related Osteonecrosis of the Jaw (MRONJ) in Humans.
| Study | Study Design | Patients, n | Proportion of DM or IFG With MRONJ (%) | Strength of Correlation Between DM and MRONJ | Comments and Limitations |
|---|---|---|---|---|---|
| Khamaisi et al. (2007) | Case control | 31 | 58 | P < 0.01 | Both DM and IFG were analyzed. |
| Data on glucose levels and HbA1c are lacking. | |||||
| Wilkinson et al. (2007) | Population-based cohort | 865 (total DM patients) | 19 (DM with complications) | HR = 1.60 (0.48 to 5.31) | Multiple inflammatory/surgical oral complications accounted for the analysis. |
| Barasch et al. (2011) | Case control | 191 | 17 | OR = 1.7 (1.1 to 2.8) | Data on glucose levels and HbA1c are lacking. |
| Bocanegra-Pérez et al. (2012) | Case control | 44 | 35 | NA | Limited data on the DM group |
| Molcho et al. (2013) | Case control | 46 | 67 | P = 0.009 | Both DM and IFG were analyzed. |
| No correlation with HbA1c | |||||
| Watters et al. (2013) | Case control | 109 | 22 | P = 0.05 | Data on glucose levels and HbA1c are lacking. |
DM, diabetes mellitus; IFG, impaired fasting glucose; HbA1c, hemoglobin A1c; HR, hazard ratio; OR, odds ratio; NA, not applicable.
The current level of evidence is still premature and does not indicate a cause-and-effect relationship between DM and MRONJ. In a recently documented study of rats that were treated with alendronate and then underwent tooth extractions, osteonecrosis presented in 7 of 9 (78%) of those with streptozotocin-induced DM, compared to 0 of 11 of those without induced DM (Berti-Couto et al. 2014). The exact mechanism by which diabetes increases the risk of osteonecrosis of the jaw has yet to be determined.
Decreased Bone Turnover and Remodeling
Diabetic patients treated with antiresorptive medications are challenged by both the metabolic disorder and by the potential for bone remodeling complications conferred by the treatment. DM specifically has been found to have a profound effect on bone metabolism via reductions in bone turnover, in remodeling, and in alveolar bone formation. Several clinical studies showed that while low bone mineral density (BMD) is consistently observed in type 1 diabetes (T1DM) at the radius and femur, BMD in type 2 diabetes (T2DM) is similar to or higher than that in nondiabetic persons (Isidro and Ruano 2010). Altered BMD in diabetic patients, together with bone quality deterioration, may be linked to an increased risk for fractures.
Multiple pathways can affect bone metabolism in DM and may explain the deterioration in bone quality, among them changes in insulin levels, elevated levels of advanced glycation end products (AGEs) in collagen, increased urinary excretion coupled with lower intestinal absorption of calcium, inappropriate homeostatic response of parathyroid hormone secretion, complex alterations of vitamin D regulation, reduced renal function, lower insulin-like growth factor-1 (IGF-1), microangiopathy, and inflammation (Isidro and Ruano 2010). Osteoblasts, which were found to express insulin receptors, mediate an anabolic effect of insulin on the bone. By promoting bone formation, bone density and strength are maintained or increased (Thrailkill et al. 2005). Poor metabolic control can exert deleterious effects on the bone through a number of mechanisms. Chronic hyperglycemia leads to glycosylation of proteins, producing AGEs in various tissues, including bone. AGEs contribute to the inhibition of osteoblastic adhesion to the bone matrix, which can exacerbate diabetic osteopenia by inhibiting growth, differentiation, and mineralization in the bone matrix (McCarthy et al. 2004). These effects are enhanced by impaired calcium homeostasis and abnormalities in vitamin D metabolism, which are associated with DM. Reduced hemostasis was suggested to induce negative feedback on the parathyroid gland, resulting in hypoparathyroidism and leading to reduced parathyroid hormone (PTH) secretion and further negative calcium balance (McNair et al. 1981). Glucosuric-induced osmotic diuresis causes renal calcium leak, which also contributes to negative calcium balance (Okazaki et al. 1997). In addition, hyperosmolarity induced by hyperglycemia suppresses the expression of osteocalcin, MMP-13, and vascular endothelial growth factor (VEGF), genes associated with osteoblast maturation (Botolin and McCabe 2006).
Accumulating evidence indicates that the glucose-lowering medications, thiazolidinediones (TZDs), may negatively affect bone metabolism. TZDs induce bone loss by affecting the bone remodeling process, suppressing new bone formation by osteoblasts, and increasing bone resorption by osteoclasts. Furthermore, TZDs decrease BMD and bone volume, as well as induce changes in bone microarchitecture (Rzonca et al. 2004). In marrow cells of mesenchymal lineage, TZDs suppressed bone anabolic signaling by decreasing activity of Wnt and by transforming growth factor–β (TGF-β)/bone morphogenic protein and IGF-1 pathways, while inducing production of RANKL (Shockley et al. 2009). In patients with T2DM, TZDs decreased serum osteocalcin and femoral and radial BMD (Kanazawa et al. 2010). In addition, a high risk for fractures was described in T2DM women older than 65 years who were treated with TZDs (Habib et al. 2010).
Increased Apoptosis of Osteoblasts and Osteocytes
Osteocyte apoptosis was found to be more prevalent in histological specimens of patients diagnosed with MRONJ than in specimens of healthy bone (Lesclous et al. 2009). Associations between changes in bone metabolism and elevated blood glucose levels support a role of both T1DM and T2DM in MRONJ. These changes include inhibition of osteoclast differentiation and function, as well as induction of osteoblast and osteocyte apoptosis. AGEs are increased in individuals with DM and are believed to contribute to the development and progression of chronic complications that include a decrease in bone quality. In an in vitro system, Gangoiti et al. (2013) reported that AGEs induce disruption in the osteoblastic actin cytoskeleton and alterations in cell morphology. The subsequent decrease in cell-substratum interactions was shown to increase apoptosis of osteoblasts and decrease osteoblastic proliferation; the addition of alendronate enhanced such activity (Gangoiti et al. 2013). Many of the effects of AGEs are receptor dependent and involve a multiligand member of the immunoglobulin superfamily of cell surface molecules. The best characterized of these is the receptor for advanced glycation end products (RAGE). Based on studies of RAGE-deficient mice, RAGE appears to play a central role in oral infection, exaggerated inflammatory host responses, and the destruction of alveolar bone in DM (Lalla et al. 2001).
Sorbitol, a by-product of high-glucose flux through the polyol pathway, has been implicated in the development of DM complications. Inaba (2006) suggested that intracellular sorbitol accumulation in individuals with DM might be involved in the development of osteoblast dysfunction and osteoclast formation. This was further supported by the protective effect of aldose reductase inhibitor against the development of galactose-induced bone diseases.
Increased Inflammation
Periodontal disease is the most prevalent oral complication of T2DM. T2DM confers a 2.81 increased risk of destructive periodontitis (Papapanou 1996). Both rat models and clinical settings demonstrated that decreased metabolic control in T2DM negatively affects all clinical measures of periodontal health and disease severity (Cutler et al. 1999). A bidirectional relationship between DM and periodontitis seems very likely; periodontal diseases can aggravate insulin resistance and adversely affect glycemic control, and periodontal treatment improves glycemic control in T2DM (Stanko and Izakovicova Holla 2014).
Activated T and B cells in the gingival tissues are the primary sources of RANKL, which induces osteoclastogenesis, osteoclast activation, and bone loss in periodontitis (Kawai et al. 2006). Inhibiting the function of RANKL produced by activated T cells can prevent alveolar bone loss (Lin et al. 2011). These studies provide strong evidence that T-cell activation mediates bone loss via recruitment and activation of osteoclasts. Elevated levels of high-sensitivity C-reactive protein in gingival crevicular fluid were recently reported in T2DM patients with chronic periodontitis (Kalra et al. 2013). In addition, interleukin (IL)–6 and the RANK/osteoprotegerin ratio were higher in the oral mucosa of patients with jaw necrosis than in those without (Mozzati et al. 2013).
Altered Immune Responses
BPs share structural homologies with previously identified γδ T-cell ligands (Kunzmann et al. 2000). Long-term or high-dose use of BPs has been associated with loss of peripheral blood Vγ9Vδ2 T cells; this is apparently due to drug-induced immune dysfunction. Kalyan et al. (2014) recently reported that neutrophils present in human peripheral blood readily take up zoledronate and that this phenomenon is associated with the potent immune suppression of human peripheral blood Vγ9Vδ2 T cells. Furthermore, BPs were shown to inhibit T-lymphocyte activation and proliferation, to suppress monocyte production of various proinflammatory cytokines (Sansoni et al. 1995), and to increase production of proinflammatory cytokines by lymphocytes (Coxon et al. 2006). Neutrophil chemotaxis and survival were found to be compromised following exposure to BPs (Kuiper et al. 2012). The result was altered activity and chronic tissue damage that can lead to MRONJ. Similarly, reduced neutrophil activity and impaired chemotaxis were demonstrated in individuals diagnosed with MRONJ, independent of BP treatment (Favot et al. 2013). Furthermore, compromised macrophage function at MRONJ sites was also recently demonstrated (Hoefert et al. 2014). In patients with metastatic prostate cancer treated with docetaxel and zoledronic acid, leukopenia induced by chemotherapy was suggested as a risk factor for MRONJ, perhaps by aggravating oral cavity infection mediated by mucositis (Miyazaki et al. 2012). Furthermore, Kikuiri et al. (2010) showed that systemic infusion of mesenchymal stem cells (MSCs) in MRONJ-like mice prevents and cures MRONJ-like disease, possibly via induction of peripheral tolerance—specifically, by inhibition of Th17 and enhancement of Treg cells. This demonstrates an immunity-based mechanism of MRONJ disease and supports the rationale for in vivo immune-modulatory therapy for MRONJ, using Tregs or MSCs.
Diabetes induces changes in neutrophil, monocyte, and macrophage function. Neutrophil adherence, chemotaxis, and phagocytosis are often impaired. Conversely, the monocyte-macrophage cell line may be hyperresponsive to bacterial antigens, resulting in significantly increased production of proinflammatory cytokines (Nassar et al. 2007). Elevated levels of monocyte chemoattractant protein-1 (MCP-1) in gingival tissues, increased gingival inflammatory cell infiltration, and alveolar bone loss were reported in rats with either DM or periodontitis (Sakallioğlu et al. 2008).
Factors contributing to an altered immune response may increase the risk for chronic infection in the setting of DM, cancer and its treatment, and BP exposure, thereby increasing the risk of developing MRONJ in this population.
Angiogenesis, Vascularization, and Endothelial Damage
Antiresorptive medications may inhibit angiogenesis by impairing endothelial cell (EC) functions. BPs were shown to inhibit EC proliferation, leading to altered EC adhesion and migration (Wood et al. 2002). Zoledronic acid was found to inhibit integrin-mediated adhesion and migration in human EC (Bezzi et al. 2003). Furthermore, reduced proliferation, increased apoptosis, and decreased capillary-like tube formation in ECs treated with BPs was described (Fournier et al. 2002). Among patients treated with BPs, proangiogenic factors—specifically, hydroxymethylglutaryl coenzyme A reductase and VEGF—were reduced (Mozzati et al. 2013). Histological evaluation of MRONJ tissue revealed decreased p63 gene expression, indicating a reduction in basal cell progenitors and leading to impaired healing of the oral mucosa (Scheller et al. 2011). In patients with multiple myeloma (MM) and MRONJ, EC apoptosis was reported to increase following administration of BPs. This supports the hypothesis that BPs inhibit angiogenesis by interfering with EC proliferation and survival, leading to loss of blood vessels and avascular necrosis (Allegra et al. 2010).
The above findings demonstrate antiangiogenic properties of potent BPs, which may contribute to the treatment of malignant bone disease, as well as to other diseases with an angiogenic component. Several studies assessed the relationship between MRONJ and other antiangiogenic drugs. A number of anecdotal publications documented MRONJ in patients treated with bevacizumab (Christodoulou et al. 2009), a humanized monoclonal antibody against VEGF; however, investigation of the databases of 3 large prospective trials of advanced breast cancer, comprising 3,560 patients, did not reveal a significant effect of bevacizumab on the risk of MRONJ, beyond that of BPs alone (Guarneri et al. 2010). Antiangiogenic agents, including sunitinib and sorafenib, in combination with bisphosphonates, have been suggested to induce osteonecrosis of the jaw more frequently than do BPs alone (Christodoulou et al. 2009). A particularly high rate of 18% MRONJ was noted in patients with prostate cancer on zoledronic acid treated with a drug regimen combining bevacizumab, docetaxel, thalidomide, and prednisone (Aragon-Ching et al. 2009).
Microvascular ischemia of the bone is recognized in DM. This is especially relevant since oxidative damage plays a fundamental pathogenic role in MRONJ, as established by detection of mitochondrial DNA damage in the gingival tissue of patients with periodontitis (Hill 1998) and increased systemic markers of oxidative stress (Bagan et al. 2014). Okamoto et al. (2002) showed that BPs inhibit AGE-induced angiogenesis processes via DNA synthesis, as well as through the formation of microvascular endothelial cells. This is accelerated in individuals with diabetes, most likely leading to poorly functioning blood vessels. That study also demonstrated the inhibition of transcriptional activation of RANK and activator protein-1, as well as the subsequent upregulation of VEGF messenger RNA levels in AGE-exposed endothelial cells. Decreased endothelial VEGF expression and microvessel density were detected in gingival samples from patients with generalized, severe, and chronic periodontitis with T2DM (Aspriello et al. 2009). These findings may result from insulin resistance and endothelial dysfunction, which often present in patients with T2DM. More recently, using a technique for in vivo detection of free radical reactions and laser Doppler flowmetry to evaluate gingival reactive hyperemia, Sugiyama et al. (2012) found that vascular endothelial function was decreased in DM and in periodontal disease due to increasing oxidative stress in gingival circulation.
Our recent findings (Molcho et al. 2013) of higher rates of microvascular complications among MRONJ patients with DM or IFG than among those without DM or IFG suggest an increased probability of developing MRONJ among individuals with microvascular complications of DM.
Genetic Factors
In individuals with MM treated with antiresorptive therapy, 54,600 genes were analyzed in an exploratory transcriptional profiling of mononuclear cells in peripheral blood (Raje et al. 2008). Among the MM patients with MRONJ, downregulation of genes involved in osteoclast or osteoblast signaling, activation, or differentiation was demonstrated, as well as upregulation of osteoclast-inhibiting factors. A single-nucleotide polymorphism (SNP) analysis, comparing 24 MM patients with MRONJ to 651 treated with BP and without MRONJ, revealed 4 SNPs that were associated with MRONJ: rs1934951, rs1934980, rs1341162, and rs17110453 (Sarasquete et al. 2008). The SNP rs1934951 showed a particularly high risk for developing MRONJ, with an odds ratio (OR) of 12.75. All SNPs mapped within the cytochrome P450 (CYP 450), by subfamily 2C polypeptide 8 genes (CYP2C8). CYP2C8 is responsible for the metabolism of several drugs, including TZDs. Thus, variability in genes encoding CYP2C8 may affect drug pharmacokinetics, including glucose-lowering drugs (Ingelman-Sundberg et al. 2007). BP taken up by osteoclasts inhibits the activity of the enzyme farnesyl pyrophosphate synthase, which is involved in cholesterol synthesis. This enzyme also prevents lipid modification of signaling proteins, which ultimately induces osteoclast apoptosis. These enzymatic factors may contribute to the high bone turnover compromise in susceptible bones, such as the maxilla and mandible, and to the development of MRONJ. Additional genetic polymorphisms to be considered are the metabolic enzymes involved in oral health and inflammation: glutathione transferases M1 and T1, N-acetyl transferase 2, and CYP 1A1. The genes that encode these metalloproteases (involved in periodontal tissue remodeling and degradation), cytokines (involved in inflammation), prothrombin, and DNA repair activities, may be associated with susceptibility to dental diseases that have a genetic base (Baldi et al. 2009).
Genetic pathways involved in drug degradations in prominent members of the CYP 450 isoenzyme family were found to be altered in T2DM, thus further challenging the diabetic patient treated with BPs. A significant increase in the prevalence of CYP2C8 mutations in T2DM patients was reported. For example, the CYP2C8 *4 allele was found to be mutated in 15% and 2% of those with and without DM, respectively (Weise et al. 2010). These differences may result in altered drug degradation and efficacy. In vitro analysis showed different alleles to result in altered metabolism of the TZD, pioglitazone (Muschler et al. 2009).
Future Directions
The identification of numerous cofactors associated with MRONJ has prompted the initiation of hypothesis-driven research aimed to elucidate the pathogenesis of this debilitating disease (Table 1). Accumulating evidence supports an association between DM and MRONJ (Table 3); however, prospective clinical trials are needed to define this relationship, specifically with modern antiresorptive medications. Angiogenesis and endothelial dysfunction in individuals with DM, as well as the relationship to MRONJ, need to be investigated in the setting of tooth extraction. Alterations of vasculature reconstitution should be assessed in the setting of suppression of angiogenesis and hypoxia-related gene expression. More accurate identification of patients who are at particularly high risk for developing MRONJ is needed (Figure, Table 2). This may be accomplished by defining a molecular risk panel that will enable a more profound consideration of the antiresorptive treatment in such patients, including those with DM. Improved therapy of MRONJ has a promising venue in the field of stem cell therapy. Further exploration of MSC therapy in human clinical trials is needed, since stem cells can induce ectopic bone formation as well as angiogenesis. Stem cells might become a promising tool in the treatment of MRONJ.
Author Contributions
A. Peer, contributed to design and data interpretation, drafted the manuscript; M. Khamaisi, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
The authors received no financial support and declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- AAOMS. (2014). Position paper on medication-related osteonecrosis of the jaw [accessed 2014 Oct 30]. http://www.aaoms.org/docs/position_papers/mronj_position_paper.pdf?pdf=MRONJ-Position-Paper
- Allegra A, Alonci A, Penna G, Granata A, Nastro Siniscalchi E, Oteri G, Loddo S, Teti D, Cicciù D, De Ponte FS, et al. 2010. Bisphosphonates induce apoptosis of circulating endothelial cells in multiple myeloma patients and in subjects with bisphosphonate-induced osteonecrosis of the jaws. Acta Haematol. 124(2):79–85. [DOI] [PubMed] [Google Scholar]
- Allen MR, Burr DB. 2009. The pathogenesis of bisphosphonate-related osteonecrosis of the jaw: so many hypotheses, so few data. J Oral Maxillofac Surg. 67(5 Suppl):61–70. [DOI] [PubMed] [Google Scholar]
- Aragon-Ching JB, Ning YM, Chen CC, Latham L, Guadagnini JP, Gulley JL, Arlen PM, Wright JJ, Parnes H, Figg WD, et al. 2009. Higher incidence of osteonecrosis of the jaw (ONJ) in patients with metastatic castration resistant prostate cancer treated with anti-angiogenic agents. Cancer Invest. 27(2):221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspriello SD, Zizzi A, Lucarini G, Rubini C, Faloia E, Boscaro M, Tirabassi G, Piemontese M. 2009. Vascular endothelial growth factor and microvessel density in periodontitis patients with and without diabetes. J Periodontol. 80(11):1783–1789. [DOI] [PubMed] [Google Scholar]
- Bagan J, Sáez GT, Tormos MC, Gavalda-Esteve C, Bagan L, Leopoldo-Rodado M, Calvo J, Camps C. 2014. Oxidative stress in bisphosphonate-related osteonecrosis of the jaws. J Oral Pathol Med. 43(5):371–377. [DOI] [PubMed] [Google Scholar]
- Baldi D, Izzotti A, Bonica P, Pera P, Pulliero A. 2009. Degenerative periodontal-diseases and oral osteonecrosis: the role of gene-environment interactions. Mutat Res. 667(1–2):118–131. [DOI] [PubMed] [Google Scholar]
- Bamias A, Kastritis E, Bamia C, Moulopoulos LA, Melakopoulos I, Bozas G, Koutsoukou V, Gika D, Anagnostopoulos A, Papadimitriou C, et al. 2005. Osteonecrosis of the jaw in cancer after treatment with bisphosphonates: incidence and risk factors. J Clin Oncol. 23(34):8580–8587. [DOI] [PubMed] [Google Scholar]
- Barasch A, Cunha-Cruz J, Curro FA, Hujoel P, Sung AH, Vena D, Voinea-Griffin AE; CONDOR Collaborative Group; Beadnell S, Craig RG, DeRouen T, Desaranayake A, Gilbert A, Gilbert GH, Goldberg K, Hauley R, Hashimoto M, Holmes J, et al. 2011. Risk factors for osteonecrosis of the jaws: a case-control study from the CONDOR dental PBRN. J Dent Res. 90(4):439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti-Couto SA, Vasconcelos AC, Iglesias JE, Figueiredo MA, Salum FG, Cherubini K. 2014. Diabetes mellitus and corticotherapy as risk factors for alendronate-related osteonecrosis of the jaws: a study in Wistar rats. Head Neck. 36(1):84–93. [DOI] [PubMed] [Google Scholar]
- Bezzi M, Hasmim M, Bieler G, Dormond O, Rüegg C. 2003. Zoledronate sensitizes endothelial cells to tumor necrosis factor–induced programmed cell death: evidence for the suppression of sustained activation of focal adhesion kinase and protein kinase B/Akt. J Biol Chem. 278(44):43603–43614. [DOI] [PubMed] [Google Scholar]
- Bocanegra-Pérez MS, Vicente-Barrero M, Sosa-Henríquez M, Rodríguez-Bocanegra E, Limiñana-Cañal JM, López-Márquez A, Pérez-Plasencia D, Ramos-Macías A. 2012. Bone metabolism and clinical study of 44 patients with bisphosphonate-related osteonecrosis of the jaws. Med Oral Patol Oral Cir Bucal. 17(6):e948–e955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. 2006. Chronic hyperglycemia modulates osteoblast gene expression through osmotic and non-osmotic pathways. J Cell Biochem. 99(2):411–424. [DOI] [PubMed] [Google Scholar]
- Christodoulou C, Pervena A, Klouvas G, Galani E, Falagas ME, Tsakalos G, Visvikis A, Nikolakopoulou A, Acholos V, Karapanagiotidis G, et al. 2009. Combination of bisphosphonates and antiangiogenic factors induces osteonecrosis of the jaw more frequently than bisphosphonates alone. Oncology. 76(3):209–211. [DOI] [PubMed] [Google Scholar]
- Coxon FP, Thompson K, Rogers MJ. 2006. Recent advances in understanding the mechanism of action of bisphosphonates. Curr Opin Pharmacol. 6(3):307–312. [DOI] [PubMed] [Google Scholar]
- Cutler CW, Machen RL, Jotwani R, Iacopino AM. 1999. Heightened gingival inflammation and attachment loss in type 2 diabetics with hyperlipidemia. J Periodontol. 70(11):1313–1321. [DOI] [PubMed] [Google Scholar]
- Favot CL, Forster C, Glogauer M. 2013. The effect of bisphosphonate therapy on neutrophil function: a potential biomarker. Int J Oral Maxillofac Surg. 42(5):619–626. [DOI] [PubMed] [Google Scholar]
- Fournier P, Boissier S, Filleur S, Guglielmi J, Cabon F, Colombel M, Clézardin P. 2002. Bisphosphonates inhibit angiogenesis in vitro and testosterone-stimulated vascular regrowth in the ventral prostate in castrated rats. Cancer Res. 62(22):6538–6544. [PubMed] [Google Scholar]
- Gangoiti MV, Anbinder PS, Cortizo AM, McCarthy AD. 2013. Morphological changes induced by advanced glycation endproducts in osteoblastic cells: effects of co-incubation with alendronate. Acta Histochem. 115(7):649–657. [DOI] [PubMed] [Google Scholar]
- Guarneri V, Miles D, Robert N, Diéras V, Glaspy J, Smith I, Thomssen C, Biganzoli L, Taran T, Conte P. 2010. Bevacizumab and osteonecrosis of the jaw: incidence and association with bisphosphonate therapy in three large prospective trials in advanced breast cancer. Breast Cancer Res Treat. 122(1):181–188. [DOI] [PubMed] [Google Scholar]
- Habib ZA, Havstad SL, Wells K, Divine G, Pladevall M, Williams LK. 2010. Thiazolidinedione use and the longitudinal risk of fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 95(2):592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill PA. 1998. Bone remodelling. Br J Orthod. 25(2):101–107. [DOI] [PubMed] [Google Scholar]
- Hoefert S, Schmitz I, Weichert F, Gaspar M, Eufinger H. 2014. Macrophages and bisphosphonate-related osteonecrosis of the jaw (BRONJ): evidence of local immunosuppression of macrophages in contrast to other infectious jaw diseases. Clin Oral Investig [epub ahead of print 24 June 2014] in press. [DOI] [PubMed] [Google Scholar]
- Inaba M. 2006. Effect of aldose reductase on the abnormality of calcium metabolism in diabetic patients [in Japanese]. Clin Calcium. 16(8):1360–1365. [PubMed] [Google Scholar]
- Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. 2007. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 116(3):496–526. [DOI] [PubMed] [Google Scholar]
- Isidro ML, Ruano B. 2010. Bone disease in diabetes. Curr Diabetes Rev. 6(3):144–155. [DOI] [PubMed] [Google Scholar]
- Kalra N, Pradeep AR, Priyanka N, Kumari M. 2013. Association of stem cell factor and high-sensitivity C reactive protein concentrations in crevicular fluid and serum in patients with chronic periodontitis with and without type 2 diabetes. J Oral Sci. 55(1):57–62. [DOI] [PubMed] [Google Scholar]
- Kalyan S, Chandrasekaran V, Quabius ES, Lindhorst TK, Kabelitz D. 2014. Neutrophil uptake of nitrogen-bisphosphonates leads to the suppression of human peripheral blood γδ T cells. Cell Mol Life Sci. 71(12):2335–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa I, Yamaguchi T, Yano S, Yamamoto M, Yamauchi M, Kurioka S, Sugimoto T. 2010. Baseline atherosclerosis parameter could assess the risk of bone loss during pioglitazone treatment in type 2 diabetes mellitus. Osteoporos Int. 21(12):2013–2018. [DOI] [PubMed] [Google Scholar]
- Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, Goncalves RB, Valverde P, Dibart S, Li YP, et al. 2006. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 169(3):987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamaisi M, Regev E, Yarom N, Avni B, Leitersdorf E, Raz I, Elad S. 2007. Possible association between diabetes and bisphosphonate-related jaw osteonecrosis. J Clin Endocrinol Metab. 92(3):1172-1175. [DOI] [PubMed] [Google Scholar]
- Kikuiri T, Kim I, Yamaza T, Akiyama K, Zhang Q, Li Y, Chen C, Chen W, Wang S, Le AD, et al. 2010. Cell-based immunotherapy with mesenchymal stem cells cures bisphosphonate-related osteonecrosis of the jaw-like disease in mice. J Bone Miner Res. 25(7):1668–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper JW, Forster C, Sun C, Peel S, Glogauer M. 2012. Zoledronate and pamidronate depress neutrophil functions and survival in mice. Br J Pharmacol. 165(2):532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. 2000. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. 96(2):384–392. [PubMed] [Google Scholar]
- Lalla E, Lamster IB, Stern DM, Schmidt AM. 2001. Receptor for advanced glycation end products, inflammation, and accelerated periodontal disease in diabetes: mechanisms and insights into therapeutic modalities. Ann Periodontol. 6(1):113–118. [DOI] [PubMed] [Google Scholar]
- Lesclous P, Abi Najm S, Carrel JP, Baroukh B, Lombardi T, Willi JP, Rizzoli R, Saffar JL, Samson J. 2009. Bisphosphonate-associated osteonecrosis of the jaw: a key role of inflammation? Bone. 45(5):843–852. [DOI] [PubMed] [Google Scholar]
- Lin X, Han X, Kawai T, Taubman MA. 2011. Antibody to receptor activator of NF-κB ligand ameliorates T cell-mediated periodontal bone resorption. Infect Immun. 79(2):911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx RE. 2003. Pamidronate (Aredia) and zoledronate (Zometa) induced avascular necrosis of the jaws: a growing epidemic. J Oral Maxillofac Surg. 61(9):1115–1117. [DOI] [PubMed] [Google Scholar]
- McCarthy AD, Uemura T, Etcheverry SB, Cortizo AM. 2004. Advanced glycation endproducts interefere with integrin-mediated osteoblastic attachment to a type-I collagen matrix. Int J Biochem Cell Biol. 36(5):840–848. [DOI] [PubMed] [Google Scholar]
- McNair P, Christensen MS, Madsbad S, Christiansen C, Transbøl I. 1981. Hypoparathyroidism in diabetes mellitus. Acta Endocrinol (Copenh). 96(1):81–86. [DOI] [PubMed] [Google Scholar]
- Migliorati CA, Woo SB, Hewson I, Barasch A, Elting LS, Spijkervet FK, Brennan MT; Bisphosphonate Osteonecrosis Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). 2010. A systematic review of bisphosphonate osteonecrosis (BON) in cancer. Support Care Cancer. 18(8):1099–1106. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Nishimatsu H, Kume H, Suzuki M, Fujimura T, Fukuhara H, Enomoto Y, Ishikawa A, Igawa Y, Hirano Y, et al. 2012. Leukopenia as a risk factor for osteonecrosis of the jaw in metastatic prostate cancer treated using zoledronic acid and docetaxel. BJU Int. 110(11 Pt B):E520–E525. [DOI] [PubMed] [Google Scholar]
- Molcho S, Peer A, Berg T, Futerman B, Khamaisi M. 2013. Diabetes microvascular disease and the risk for bisphosphonate-related osteonecrosis of the jaw: a single center study. J Clin Endocrinol Metab. 98(11):E1807–E1812. [DOI] [PubMed] [Google Scholar]
- Mozzati M, Martinasso G, Maggiora M, Scoletta M, Zambelli M, Carossa S, Oraldi M, Muzio G, Canuto RA. 2013. Oral mucosa produces cytokines and factors influencing osteoclast activity and endothelial cell proliferation, in patients with osteonecrosis of jaw after treatment with zoledronic acid. Clin Oral Investig. 17(4):1259–1266. [DOI] [PubMed] [Google Scholar]
- Muschler E, Lal J, Jetter A, Rattay A, Zanger U, Zadoyan G, Fuhr U, Kirchheiner J. 2009. The role of human CYP2C8 and CYP2C9 variants in pioglitazone metabolism in vitro. Basic Clin Pharmacol Toxicol. 105(6):374–379. [DOI] [PubMed] [Google Scholar]
- Nassar H, Kantarci A, van Dyke TE. 2007. Diabetic periodontitis: a model for activated innate immunity and impaired resolution of inflammation. Periodontology. 2000;43:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Yamagishi S, Inagaki Y, Amano S, Takeuchi M, Kikuchi S, Ohno S, Yoshimura A. 2002. Incadronate disodium inhibits advanced glycation end products-induced angiogenesis in vitro. Biochem Biophys Res Commun. 297(2):419–424. [DOI] [PubMed] [Google Scholar]
- Okazaki R, Totsuka Y, Hamano K, Ajima M, Miura M, Hirota Y, Hata K, Fukumoto S, Matsumoto T. 1997. Metabolic improvement of poorly controlled noninsulin-dependent diabetes mellitus decreases bone turnover. J Clin Endocrinol Metab. 82(9):2915–2920. [DOI] [PubMed] [Google Scholar]
- Papapanou PN. 1996. Periodontal diseases: epidemiology. Ann Periodontol. 1(1):1–36. [DOI] [PubMed] [Google Scholar]
- Raje N, Woo SB, Hande K, Yap JT, Richardson PG, Vallet S, Treister N, Hideshima T, Sheehy N, Chhetri S, et al. 2008. Clinical, radiographic, and biochemical characterization of multiple myeloma patients with osteonecrosis of the jaw. Clin Cancer Res. 14(8):2387–2395. [DOI] [PubMed] [Google Scholar]
- Ruggiero SL, Mehrotra B, Rosenberg TJ, Engroff SL. 2004. Osteonecrosis of the jaws associated with the use of bisphosphonates: a review of 63 cases. J Oral Maxillofac Surg. 62(5):527–534. [DOI] [PubMed] [Google Scholar]
- Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. 2004. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 145(1):401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad F, Brown JE, Van Poznak C, Ibrahim T, Stemmer SM, Stopeck AT, Diel IJ, Takahashi S, Shore N, Henry DH, et al. 2012. Incidence, risk factors, and outcomes of osteonecrosis of the jaw: integrated analysis from three blinded active-controlled phase III trials in cancer patients with bone metastases. Ann Oncol. 23(5):1341–1347. [DOI] [PubMed] [Google Scholar]
- Sakallioğlu EE, Ayas B, Lütfioğlu M, Keleş GC, Açikgöz G, Firatli E. 2008. Gingival levels of monocyte chemoattractant protein-1 (MCP-1) in diabetes mellitus and periodontitis: an experimental study in rats. Clin Oral Investig. 12(1):83–89. [DOI] [PubMed] [Google Scholar]
- Sansoni P, Passeri G, Fagnoni F, Mohagheghpour N, Snelli G, Brianti V, Engleman EG. 1995. Inhibition of antigen-presenting cell function by alendronate in vitro. J Bone Miner Res. 10(11):1719–1725. [DOI] [PubMed] [Google Scholar]
- Sarasquete ME, García-Sanz R, Marín L, Alcoceba M, Chillón MC, Balanzategui A, Santamaria C, Rosiñol L, de la Rubia J, Hernandez MT, et al. 2008. Bisphosphonate-related osteonecrosis of the jaw is associated with polymorphisms of the cytochrome P450 CYP2C8 in multiple myeloma: a genome-wide single nucleotide polymorphism analysis. Blood. 112(7):2709–2712. [DOI] [PubMed] [Google Scholar]
- Scheller EL, Baldwin CM, Kuo S, D’Silva NJ, Feinberg SE, Krebsbach PH, Edwards PC. 2011. Bisphosphonates inhibit expression of p63 by oral keratinocytes. J Dent Res. 90(7):894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. 2009. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 106(2):232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanko P, Izakovicova Holla L. 2014. Bi directional association between diabetes mellitus and inflammatory periodontal disease: a review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 158(1):35–38. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Takahashi SS, Tokutomi FA, Yoshida A, Kobayashi K, Yoshino F, Wada-Takahashi S, Toyama T, Watanabe K, Hamada N, Todoki K, et al. 2012. Gingival vascular functions are altered in type 2 diabetes mellitus model and/or periodontitis model. J Clin Biochem Nutr. 51(2):108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrailkill KM, Lumpkin CK, Bunn RC, Kemp SF, Fowlkes JL. 2005. Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am J Physiol Endocrinol Metab. 289(5):E735–E745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters AL, Hansen HJ, Williams T, Chou JF, Riedel E, Halpern J, Tunick S, Bohle G, Huryn JM, Estilo CL. 2013. Intravenous bisphosphonate-related osteonecrosis of the jaw: long-term follow-up of 109 patients. Oral Surg Oral Med Oral Pathol Oral Radiol. 115(2):192–200. [DOI] [PubMed] [Google Scholar]
- Weise A, Prause S, Eidens M, Weber MM, Kann PH, Forst T, Pfützner A. 2010. Prevalence of CYP450 gene variations in patients with type 2 diabetes. Clin Lab. 56(7–8):311–318. [PubMed] [Google Scholar]
- Wilkinson GS, Kuo Y-F, Freeman JL, Goodwin JS. 2007. Intravenous bisphosphonate therapy and inflammatory conditions or surgery of the jaw: a population-based analysis. J Natl Cancer Inst. 99(13):1016–1024. [DOI] [PubMed] [Google Scholar]
- Wood J, Bonjean K, Ruetz S, Bellahcène A, Devy L, Foidart JM, Castronovo V, Green JR. 2002. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 302(3):1055–1061. [DOI] [PubMed] [Google Scholar]