

SUMMARY
Antiangiogenic therapy is commonly used in the clinic but its beneficial effects are short-lived leading to tumor relapse within months. Here, we demonstrate that the efficacy of angiogenic inhibitors targeting the VEGF/VEGFR pathway is dependent on the induction of the angiostatic and immunestimulatory chemokine CXCL14 in mouse models of pancreatic neuroendocrine and mammary tumors. In response, tumors reinitiated angiogenesis and immune-suppression by activating PI3K-signaling in all CD11b+ cells rendering tumors non-responsive to VEGF/VEGFR inhibition. Adaptive resistance was also associated with an increase in Gr1+CD11b+ cells, but targeting Gr1+ cells was not sufficient to further sensitize angiogenic blockade as TAM would compensate for their lack and vice versa leading to an oscillating pattern of distinct immune cell populations. However, PI3K inhibition in CD11b+ myeloid cells generated an enduring angiostatic and immune-stimulatory environment in which anti-angiogenic therapy remained efficient.
Graphical Abstract
INTRODUCTION
Antiangiogenic therapy represents one of the most widely used anti-cancer strategies today, with most approved therapies targeting the vascular endothelial growth factor (VEGF) signaling pathway. However, the beneficial effects observed across the multitude of cancers that respond are typically short-lived; therefore much effort has focused on uncovering the various mechanisms whereby tumors bypass the tumor-inhibitory effects of therapy (Bergers and Hanahan, 2008; Kerbel, 2008). One such resistance mechanism involves reinstatement of angiogenesis by tumor-infiltrating innate immune cells (Dierickx et al., 1963; Fischer et al., 2007; Shojaei et al., 2007a; Shojaei et al., 2007b).
Tumors can contain a significant percentage of different infiltrating myeloid cells with bivalent functions but predominantly are thought to support tumor progression by promoting angiogenesis and suppressing anti-tumor immunity. Tumor-associated macrophages (TAM) are typically characterized as either “classically” activated tumoricidal macrophages (M1) or “alternatively” activated protumorigenic macrophages (M2) (Mantovani et al., 2008). Extending upon this nomenclature, neutrophils (TAN) have also been categorized as N1 or N2 based on their anti-or pro-tumor activity in tumors (Fridlender et al., 2009). In addition, immature Gr1+ cells with either a mononuclear or granular morphology have been identified in tumors that convey immune-suppressive functions and are therefore also termed myeloid-derived suppressor cells (M-MDSC and G-MDSC respectively) (Talmadge and Gabrilovich, 2013). Typically, surface marker profiling based on expression of CD11b, F4/80, Gr1, Ly6C, and Ly6G is used to categorize these subsets of tumor-infiltrating myeloid cells (Fridlender et al., 2009; Talmadge and Gabrilovich, 2013; Wynn et al., 2013).
There is mounting evidence that tumors recruit these distinct populations where they become an additional source of angiogenic chemokines and cytokines to promote angiogenesis (Coussens et al., 2000; Du et al., 2008; Giraudo et al., 2004; Lin et al., 2006; Shojaei et al., 2007b). As hypoxia is a major driver of myeloid cell recruitment (Du et al., 2008; Mazzieri et al., 2011) it is conceivable that therapy-induced hypoxia via an angiogenic blockade can induce factors that mobilize cells from the bone marrow and attract them to the tumor site. Indeed, tumor-associated myeloid cells have been shown to sustain angiogenesis in the face of antiangiogenic therapy, in part by stimulating VEGF-independent pathways. For example, macrophages induced expression of several angiogenic molecules, including FGF-1,2, MMP9, and Ang2 in response to antiangiogenic therapy (Casanovas et al., 2005; Fischer et al., 2007; Rigamonti et al., 2014), while Gr1+ myeloid cells were found to convey resistance to anti-VEGF treatment via secretion of the angiogenic PKR-1/2 ligand Bv8 (Shojaei et al., 2007a; Shojaei et al., 2007b). As much as inhibitors of macrophages or Gr1+ cells enhanced the effects of antiangiogenic therapy, in many of these models tumor growth was still apparent at a slower pace throughout the duration of treatment. Here, we investigated the overall contributions of the different tumor-associated myeloid populations to evasion of antiangiogenic therapy. We analyzed the composition and function of TAM, TAN, and two Gr1+ immature monocyte populations in two distinct tumor models that responded differently to angiogenic inhibition. In the Rip1Tag2 model of pancreatic neuroendocrine tumors (PNET), angiogenic blockade was able to transiently reduce vessel density and block tumor growth (response) followed by reinstatement of neovascularization and robust tumor growth (relapse) thereby enabling us to evaluate true “response” and “relapse” phases in a single model. In the PyMT mammary carcinoma model, angiogenic blockade was only able to slow down tumor growth with some reduction in vessel density, a feature that is commonly observed in various tumor models. Analysis of myeloid cell content within tumors revealed that the angiogenic relapse was associated with an increase in tumor-specific subsets of Gr1+ myeloid cells. By investigating the role of these cells during relapse, we were able to uncover a compensatory nature of myeloid cell-mediated resistance to antiangiogenic therapy. In the present study, we inquired about the nature and mechanisms by which distinct innate immune cells compensate for each other to maintain resistance and identify means that modulate inflammation to sustain the effects of antiangiogenic therapy.
RESULTS
Targeting distinct myeloid subtypes leads to a compensatory oscillation between innate immune cells enabling reneovascularization during antiangiogenic therapy
We established a model of evasive resistance to antiangiogenic therapy in the Rip1Tag2 (RT2) model of pancreatic neuroendocrine tumors (PNET) whereby treatment of 13-week old tumor-bearing mice with the broadspectrum angiokinase inhibitor sorafenib induced a two-to-three week period of response characterized by tumor stasis, followed by robust tumor growth and subsequent death (Figure 1A). The onset of sorafenib response and relapse is similar to that observed with DC101, a VEGFR2-neutralizing antibody, supporting the notion that sorafenib is predominantly blocking the VEGF/VEGFR2 pathway in PNET ((Casanovas et al., 2005; Rigamonti et al., 2014) and Bergers unpublished data). Tumor analysis during the response phase (two week treatment; 15 weeks of age) with sorafenib revealed substantial reduction in vessel density and hemorrhage formation while relapsing tumors (four week treatment; 17 weeks of age) exhibited a vascular pattern similar to untreated tumors with high vessel density and hemorrhage recurrence (Figure 1B). Thus, we defined “responding” tumors as those treated from 13 to 15 weeks of age (two weeks), while “relapse” tumors as those treated from 13 to 17 weeks of age (four weeks). Therefore, RT2-PNETs overcome vascular growth restrictions by gaining the capability to reinitiate neovascularization. Investigating the intratumoral composition of innate immune cells, FACS analysis of PNET during the course of treatment revealed no substantial difference in the number or composition of myeloid cells during the response phase (Figures 1C, S1A, S1B, and S2A). However, a substantial increase in intratumoral Gr1+Ly6CHi monocytes (SSCLowCD11b+Gr1LowLy6CHighLy6GLow) accompanied the angiogenic relapse (Figures 1C, S1A, S2A). As elevated levels of infiltrating Gr1+ myeloid cells had been found to promote tumor resistance to anti-VEGF therapy (Shojaei et al., 2007a), we targeted relapse-associated Gr1+ cells two weeks after the start of antiangiogenic therapy, when tumors were still responding to sorafenib (Figure 1D). Despite blocking the increased accumulation of Gr1+ cells within the tumor (Figures 1F, S1A, S1B, and S2B), this approach did not prolong responsiveness to sorafenib and led to a proangiogenic relapse similar to sorafenib alone (Figure 1E). Furthermore, reducing Gr1+ cell infiltration induced a compensatory increase in TAM to potentially confer therapeutic resistance. (Figures 1F, S1A, S1B, and S2B). Hence we investigated whether TAM-depletion with a CSF1-neutralizing antibody would be more beneficial in sustaining sorafenib response; however, this approach was also unable to prolong response to antiangiogenic therapy (Figures 1G and 1H), as it induced an increase in intratumoral Gr1+Ly6GHi and Gr1+Ly6CHi monocytes and TAN (Figures 1I, S1A, and S2C). Thus, Gr1+ cells compensate for TAM depletion and vice versa, a phenomenon reminiscent of an oscillating pattern of innate immune cells to convey therapeutic resistance to antiangiogenic therapy. These results imply that several myeloid cell populations regulate reneovascularization and promote it in a compensatory manner.
Figure 1. Targeting distinct myeloid populations induces compensatory oscillation.

(A) Tumor burden of RT2 mice. Response indicates tumor stasis (two week sorafenib treatment) and relapse indicates tumor regrowth (four week sorafenib treatment). (B) Microvessel density of immunofluorescent anti-CD31 stained tumors. (C) Myeloid cell composition in RT2 PNET by FACS. P=0.0248 for relapse versus response phase Gr1+Ly6CHi cells. (D–F) Tumor burden (D), microvessel density (E), and myeloid cell composition (F) in mice treated with sorafenib plus anti-Gr1. P=0.01 for TAM versus sorafenib response phase TAM; p=0.007 for Gr1+Ly6CHi cells and TAM versus cognate populations in sorafenib relapse tumors. (G–I) Tumor burden (G), microvessel density (H) and myeloid cell composition (I) in mice treated with sorafenib plus anti-CSF1. P<0.05 for TAM, Gr1+Ly6CHi and Gr1+Ly6GHi cells versus cognate populations in both sorafenib response and relapse phase tumors. Scale bars,100μm. Mean±SEM is presented for all quantitation. For Figures 1D–1I, data is presented with untreated and sorafenib response tumor data from Figures 1A–1C. See also Figures S1 and S2, and Experimental Procedures for details.
Upregulation of angiostatic chemokines is associated with antiangiogenic effects of sorafenib in PNET
To elicit how myeloid cell oscillation promotes a proangiogenic relapse during sorafenib therapy, we compared gene expression of several prominent angiogenic factors between naïve, responding, and relapsing PNET tumors of RT2 mice treated with sorafenib alone or with either anti-Gr1 or anti-CSF1. Interestingly, response phase tumors compared to naïve tumors already displayed enhanced levels of several prominent proangiogenic factors, despite exhibiting substantial vessel reduction (Figures 2A, S3A, and S3B). They remained at similar levels in the relapse phase independent of mice receiving sorafenib alone or in combination with either anti-Gr1 or anti-CSF1 treatment (Figure 2A, S3A, and S3B). In contrast, expression analysis of various angiostatic chemokines in tumors exposed CXCL14 to be dominantly upregulated in responding tumors, followed to a lesser extent by CXCL4 (Figures 2A, S3C, and S3D). Both chemokines returned to base level upon tumor relapse with either sorafenib alone or in combination with anti-Gr1 or anti-CSF1 treatment. Expression of the angiogenic factor BV8 and antiangiogenic molecules thrombospondin-1 and 2 did not change throughout the duration of treatment (data not shown). These results suggest that in PNET tumors, the angiogenic response and relapse during sorafenib treatment is not determined by upregulation of proangiogenic factors, but rather by the levels of angiostatic chemokines, specifically CXCL14.
Figure 2. Antiangiogenic therapy regulates both angiogenic and angiostatic gene expression in myeloid cells.

(A) QPCR-based expression analyses of proangiogenic and angiostatic genes in RT2 tumors. (B–E) QPCR expression analysis of FACS-sorted TAMs (B), Gr1+Ly6CHi monocytes (C), Gr1+Ly6GHi monocytes (D), and TAN (E) isolated from tumors treated as indicated. Dotted lines indicate baseline gene expression in untreated samples. Mean±SEM is presented for quantitation and p values calculated comparing each treatment group to untreated (UT). See also Figure S3.
Myeloid cells regulate angiogenic response and relapse in PNET
We then asked to which extent intratumoral myeloid cells are the source of intratumoral angiogenic factors. We isolated TAM, Gr1+Ly6CHi and Gr1+Ly6GHi monocytes, and TAN from RT2 PNET of untreated, two-week sorafenib, and four-week sorafenib treated mice (alone or with anti-Gr1 or anti-CSF1), and compared their angiogenic gene expression profile to that of the entire tumor (Figures 2A–2E). Surprisingly, proangiogenic and angiostatic factor expression among all four myeloid populations was very similar and reflected those found in whole tumor extracts. Furthermore, proangiogenic factor gene expression was increased in all four myeloid cell types isolated from responding tumors and remained unchanged when tumors became refractory. In contrast, angiostatic CXCL chemokine expression was low in myeloid cells of untreated and relapsing tumors but was upregulated in cells from responding tumors, with CXCL14 being the most upregulated factor followed by CXCL4. (Figures 2A–2E). As expression of all CXCL chemokines was undetectable in CD45+ cell-depleted tumor samples, innate immune cells appear to be the primary source of these chemokines (data not shown). These results have several implications. First, as the angiogenic expression profile of all tumor-associated innate immune cells reflects that of the whole tumor, myeloid cells appear to be the dominant regulators of the angiostatic and proangiogenic status of PNET during antiangiogenic therapy. Second, each myeloid population expressed similar angiogenic factors at comparable levels arguing that they compensate for each other by redundant expression of these genes. Third, all four innate immune cell types induced antiangiogenic activities in responding tumors concomitant with CXCL14 upregulation and became proangiogenic in relapsing tumors when they lost their ability to enhance CXCL14 expression.
CXCL14 thwarts angiogenic activity of intratumoral myeloid cells
Therefore, we next inquired whether blocking CXCL14 would be sufficient to abrogate the anti-angiogenic effects of sorafenib in PNET. We first addressed this question using aortic slices cultured ex vivo in the presence of protein extracts made from CD11b+ cells isolated from untreated, two-week sorafenib treated (response phase) or four-week sorafenib treated (relapse phase) tumors (Figure 3A). Only CD11b+ cells from refractory tumors induced endothelial cell outgrowth and migration from aortic slices (Figure 3A). The addition of a CXCL14-neutralizing antibody rendered CD11b extracts from responding tumors angiogenic but had no impact on the activities of CD11b extracts from untreated or refractory tumors (Figure 3A). Furthermore, recombinant CXCL14 blocked the activity of CD11b extracts from refractory tumors (Figure 3A). These results argue that CXCL14 levels determine the angiogenic activity of myeloid cells in PNET tumors. We then treated RT2 mice with sorafenib in the presence of a neutralizing anti-CXCL14 antibody or control IgG beginning at 13-weeks of age. Mice receiving the control combination exhibited a typical therapeutic response characterized by reduced tumor burden at 15-weeks and a reduction in microvessel density (Figures 3B and 3C). Neutralization of CXCL14, however, abrogated the therapeutic effects of sorafenib because both tumor burden and microvessel density were comparable to that of untreated mice (Figures 3B and 3C). Thus, myeloid cell-produced CXCL14 is necessary to endorse an antiangiogenic response and therefore needs to be suppressed to enable a proangiogenic tumor relapse during antiangiogenic therapy.
Figure 3. Myeloid CXCL14 induction is necessary for sorafenib to elicit an antiangiogenic response.

(A) Aortic slice assay using CD11b+ cell extracts from untreated and treated RT2 tumors as indicated. Endothelial cell migration from the aortic slice into the surrounding collagen matrix was assessed by immunofluorescent staining of CD31. Quantitation of migrating cells per slice is presented in the right panel. *p<0.01. (B) Tumor burden of RT2 mice treated with either anti-CXCL14 or control IgG antibody plus sorafenib. (C) CD31 staining and quantitation of microvessel density of RT2 tumors. Data from untreated and four week sorafenib-treated mice is presented from Figures 1A and 1B in Figures 3B and 3C for comparison. Mean±SEM is presented for all quantitation.
Antiangiogenic therapy promotes an immune-stimulating phenotype in myeloid cells during response that becomes immune-suppressive concomitant with tumor relapse
CXCL14 not only is an angiostatic factor but has also been described to induce dendritic cell maturation, thus stimulating immune response (Schaerli et al., 2005). As angiogenesis has been found to correlate with immune-suppression in different tumors (Motz and Coukos, 2011), we hypothesized that anti-angiogenic therapy changes the immune status of tumor-associated myeloid cells thereby inducing angiostatic activities in these populations. Congruently, tumors responding to antiangiogenic therapy exhibited an induction in proinflammatory gene expression and a reduction in immunosuppressive gene expression. In refractory tumors, proinflammatory gene expression levels went to baseline while the expression of immunosuppressive genes was enhanced (Figures 4A, S4E, and S4F).
Figure 4. Antiangiogenic therapy induces myeloid cell polarization.

(A–E) QPCR RNA analyses of immune-modulating genes from RT2 tumors (A), RT2 tumor-isolated TAM (B), Gr1+Ly6CHi (C) and Gr1+Ly6GHi monocytes (D), or TAN (E) untreated and treated as indicated. P values are calculated by compariing total untreated gene expression. (F) FACS-analysis of intratumoral CD8+ CTLs. (G) QPCR analysis of Perforin in CD8+ CTLs. (H) CFSE-based proliferation assay of CD8+ T-cells cocultured with the splenic myeloid populations from RT2 mice treated as indicated. (I–K) Tumor burden (I), apoptosis (J), and microvessel density from mice treated concurrently with sorafenib plus anti-CD8a compared to untreated and sorafenib relapse data from Figures 1A and 1B. Mean±SEM is presented for all quantitations. UT=untreated, Resp=response (two week sorafenib), Rel=relapse (four week sorafenib), mito=Mitomycin C. See also Figure S4 and Supplementary Experimental Procedures for details.
To confirm that the changes in inflammatory gene expression in whole tumors were a reflection of those in innate immune cells, we examined the immune expression profile in the four myeloid cell populations isolated from responding and relapsing tumors. Surprisingly, not only TAM and TAN but also both Gr1+ monocyte populations displayed an immune-stimulating phenotype in responding tumors that reverted upon tumor relapse (Figure 4B–4E). TAM exhibited increased expression of the proinflammatory cytokines IL-12, IL-23 and the proinflammatory response gene TNFα in responding tumors that remained elevated albeit at a lower level in relapsing tumors. This was counteracted by the upregulation of the immunosuppressive factors MMR, CCL22, and Arg1 in relapsing tumors (Figure 4B). Similar to TAM, all three Gr1+ populations increased expression of proinflammatory genes (TNFα, CCL11, IL-12, and IL-23) in responding tumors that became reduced to or below baseline levels in relapsing tumors. In contrast, expression of immunosuppressive factors including IL-10 and Arg1 was reduced in all three Gr1+ populations from responding tumors and returned to baseline levels upon tumor relapse (Figures 4C–4E).
To validate that the different myeloid cells were immune-stimulating in responding and immune-suppressive in relapsing tumors, we analyzed T-cell numbers and activity. Infiltrating CD8+ cytotoxic T-cells (CTLs) increased in tumors responding to therapy and then dropped to levels observed in untreated tumors as tumors relapsed (Figure 4F). Concomitantly, CD8+ cells isolated from responding but not from untreated or relapsing tumors expressed elevated Perforin mRNA, and Granzyme B protein levels, indicative of activation (Figure 4G; Figure S4A). CD4+ T-cells did not change in number during sorafenib treatment but exhibited lower IL-10 expression during response suggestive of a reduction in CD4+ TRegs (Figures S4B and S4C).
We next tested whether the changes in myeloid cell phenotype were responsible for the differing T-cell behaviors in responding and relapsing tumors by assessing their ability to stimulate CD8+ T-cell proliferation. Using a co-culture assay in which myeloid cells from RT2 spleens were mixed with naive CD8+ T-cells, we found that macrophages and Gr1+ monocytes isolated from untreated and relapse phase RT2 tumors suppressed CD8+ T-cell proliferation, while response phase myeloid cells permitted CD8+ T-cell proliferation (Figure 4H). We obtained similar results with CD11b+ cells isolated from untreated, responding, or refractory tumors (Figure S4D). These results suggest that CTLs contribute to the therapeutic effects of antiangiogenic therapy. Indeed, CD8+ T-cell depletion in RT2 mice resulted in a lower response to sorafenib because tumor burden was increased and tumor cell apoptosis decreased compared to tumors treated with sorafenib plus IgG control, albeit at statistically non-significant levels (Figures 4I and 4J). This intermediate response stemmed from sorafenib’s ability to reduce overall vessel density and induce apoptosis in the absence of CTL cells, due to CXCL14 upregulation (Figure 4K). These results demonstrate that antiangiogenic therapy drives the polarization of TAM, TAN and both Gr1+ monocytes to an immune-stimulating type promoting CTL proliferation and activation that coincides with their angiostatic activity, leading to vessel reduction, enhanced tumor cell apoptosis and subsequent tumor growth blockade. However, this effect is transient as all four myeloid cell populations become skewed towards an immunosuppressive phenotype in refractory PNET leading to immune suppression, reneovascularization and tumor regrowth.
Activation of PI3K-signaling in myeloid cells promotes immune suppression and reneovascularization during antiangiogenic therapy
Our results revealed that myeloid cells in untreated tumors differ from those in relapsing tumors undergoing sorafenib treatment. Not only were myeloid cells increased in relapsing tumors, they also displayed elevated levels of immune-suppressive and proangiogenic factors and lower levels of immune-stimulating molecules compared to myeloid cells in untreated tumors. Thus, we hypothesized that PNET tumors activate myeloid cells to further promote inflammation and subsequent tumor relapse. Indeed, we identified enhanced levels of tumor-derived growth factors and chemokines, including SDF1α and IL-6 (Figures S5A–S5C) in relapsing tumors. These have previously been shown to mediate myeloid infiltration and subsequent tumor progression through activation of PI3Kγ in myeloid cells (Schmid et al., 2011). Congruent with the finding that both γ and δ isoforms of PI3K are immune cell specific, we found TAM, TAN and GR1+ monocytes expressed both while PNET tumor cells did not (Figure S5D) (Mencke et al., 2009; Rommel et al., 2007). To determine PI3K-activation in intratumoral myeloid cells we assessed phosphorylation of ribosomal protein S6, a downstream target of PI3K, in CD11b+ cells of untreated and sorafenib-responding and relapsing PNET tumors (Figure 5A and 5B). While only about 18–20% of tumor-associated CD11b+ cells exhibited PI3K activity in untreated or responding tumors, up to 80% of myeloid cells were activated in relapsing tumors (Figure 5B). Importantly, analysis of human PNET biopsies of naïve patients, or patients that were treated with the chemotherapeutic 5-FU or bevacizumab until relapse, revealed that only patients that had received bevacizumab displayed an increase of activated intratumoral myeloid cells as visualized by phosphorylated S6 (pS6) staining in CD45+ immune cells, confirming the results in the RT2 PNET model (Figure 5C). These data suggest that PNET tumors become refractory to anti-angiogenic therapy by activating PI3K-signaling in myeloid cells.
Figure 5. The PI3K γ/δ inhibitor IPI145 enhances efficacy of antiangiogenic therapy.

(A) Key downstream effectors of the PI3K signaling pathway. (B) Phospho-S6 (pS6) and CD11b immunofluorescent staining in RT2 PNET. Quantitation of CD11b+ phospho-S6+ cells is shown to the right; **p<0.005 versus sorafenib 2 weeks; ###p<0.0005 versus sorafenib four weeks alone and plus antibodies. (C) CD45 and phospho-S6 staining and quantitation from PNET tumor samples of patients treated as indicated. Each dot represents one patient.. Scale bars, 7.5 um. (D and E) Tumor microvessel density (D) and burden (E) from mice treated with sorafenib plus IPI145 versus untreated and sorafenib-response phase tumors from Figures 1B and 1A. (F) Survival of RT2 mice treated with vehicle (n=4; median survival=102 days), sorafenib (n=4; median survival=118.5 days), sorafenib+IPI145 (n=10; median survival=144 days), or IPI145 alone (n=4; median survival=102 days). ns=no significance versus vehicle, *p=0.0266 versus vehicle, #p=0.00068 versus sorafenib. Vertical dashed line indicates the start of sorafenib treatment (91 days); red dotted line indicates 50% survival. (G) Myeloid cell composition of tumors treated with sorafenib plus IPI145 compared to untreated and sorafenib-treated tumors from Figure 1C. P≤0.05 for Gr1+Ly6CHi monocytes and TAM from four week sorafenib alone versus four week sorafenib plus IPI145. (H and I) QPCR- analyses of immune- (H) or angiogenesis- (I) modulating genes from RT2 tumors. PNET treated with sorafenib plus IPI145 are compared to untreated and sorafenib-treated tumors from Figure 3A. P values were calculated by comparing each treatment to untreated (UT). Dotted lines indicate baseline gene expression in untreated samples. Mean±SEM is presented for all quantitations. See also Figure S5.
Inhibiting PI3K in myeloid cells enhances and endures efficacy of anti-angiogenic therapy
If induced PI3K-signaling mediates an immunosuppressive and proangiogenic switch in myeloid cells that blocks tumor-inhibitory effects of sorafenib, then inhibition of PI3K activity should suffice to prolong tumor response. To test this proposition, we pharmacologically blocked PI3K in myeloid cells using the small molecule inhibitor IPI145, which targets both PI3K γ and δ and is currently tested in several clinical trials for hematological malignancies (e.g., NCT02004522, NCT02049515, and NCT02204982). We first confirmed that IPI145 selectively inhibited myeloid cell proliferation and PI3K-mediated Akt phosphorylation in vitro (Figures S5E and S5F). We then started sorafenib treatment of RT2 mice at 13-weeks of age and added IPI145 to the treatment modality at 15-weeks of age when RT2 mice were still responding to sorafenib. After two weeks of combined myeloid PI3K-inhibition and sorafenib treatment we confirmed IPI145 blocked PI3K-activity in CD11b+ myeloid cells (Figure 5B). Consequently, IPI145 treatment maintained the low microvessel density observed in tumors responding to sorafenib (Figure 5D) and impaired tumor regrowth (Figure 5E), which nearly doubled the overall survival of RT2 mice (Figure 5F). Notably, congruent with our observation that myeloid-PI3K activity was low in untreated RT2 PNET, IPI145 alone had no effect on survival of RT2 mice (Figure 5B and 5F). This underscores the notion that in PNET, PI3K activity is switched on in myeloid cells as a distinct step during therapeutic resistance.
To understand how inhibition of PI3K signaling in myeloid cells enables a sustained response to antiangiogenic therapy, we assessed the composition and status of the tumor-associated myeloid cell populations. Myeloid cells from tumors of RT2 mice treated with sorafenib plus IPI145 revealed a reduction in intratumoral Gr1+Ly6CHi monocytes and TAM (Figures 5G and S5G). Importantly, IPI145 inhibited both the repression of immune-stimulating gene expression and the induction of immune-suppressing gene expression observed in tumors undergoing relapse to antiangiogenic therapy (Figures 5H). Furthermore, IPI145 reduced expression of proangiogenic factors and maintained high levels of CXCL14 and CXCL4 (Figures 5I). The changes in gene expression profiles directly reflected the effects of IPI145 on myeloid cells because the proinflammatory and angiostatic expression patterns of whole tumors reflected those of TAM, Gr1+Ly6CHi and Gr1+Ly6GHi monocytes, and TAN (Figures 6A–6D). Moreover, the immune-stimulating nature of these cells was confirmed by an increase in tumor-associated CTLs and their expression of Perforin (Figures 6E and 6F). Altogether, these results confirm that myeloid cells undergo a PI3K-mediated switch to become immune-suppressive and proangiogenic, and thus facilitate tumor relapse during antiangiogenic therapy. They also demonstrate that simultaneous inhibition of PI3K-signaling in all myeloid cell subtypes overcomes the oscillating resistance observed when targeting distinct myeloid cell subpopulations.
Figure 6. IPI145 induces immune stimulatory and angiostatic factors in myeloid cells during antiangiogenic therapy.

(A–D) QPCR analyses of TAM (A), Gr1+Ly6CHi (B) and Gr1+Ly6GHi monocytes (C), and TAN (D) from tumors of RT2 mice treated with sorafenib −/+ IPI145. P values calculated comparing each treatment group to untreated (UT). (E) CD8+ CTLs from tumors of mice treated with sorafenib plus IPI145.(F) Perforin expression in CD8+ CTLs from tumors of mice treated with sorafenib plus IPI145. Dotted lines indicate baseline gene expression in untreated samples. *p≤0.05 versus untreated; #p≤0.05 versus 4 week sorafenib alone. Data from untreated and sorafenib-treated tumors from Figures 2B–2E, 4B–4G are presented in 6A–6F for comparison. Mean±SEM is presented for all quantitation.
PI3K-activated myeloid cells in PyMT tumors limit the efficacy of antiangiogenic therapy
As PNET tumors entail a majority of inert CD11b+ cells in which PI3K is inactive, but induce myeloid-PI3K signaling to curtail angiogenesis inhibition, we hypothesized that the number of PI3K-activated myeloid cells may determine the effects of anti-angiogenic agents in cancers. To test this proposition, we required a model in which anti-VEGF therapy was only partially effective. We employed an orthotopic mouse model of breast cancer driven by the MMTV-PyMT transgene, a system widely used to study the impact of myeloid cells during tumor progression, together with the anti-VEGFR2 antibody DC101 (Barrios et al., 2005; Coussens et al., 2013; Mazzieri et al., 2011; O’Driscoll et al., 2003). In contrast to the PNET model, antiangiogenic therapy did not produce distinct response or relapse phases in PyMT tumors, but slowed down the pace of growth by 33% (Figure 7A). Analysis of tumors after 12 day-treatment revealed DC101 reduced microvessel density by 37% while TAN and Gr1+ monocytes increased leading to a modest elevation of myeloid cells (Figures 7B, 7C, and S7B). Interestingly, like in RT2 PNET, anti-Gr1 reduced the number of TAN and Gr1+Ly6GHi monocytes while increasing intratumoral TAM. Conversely, anti-CSF1 reduced the number of TAM and increased TAN content, displaying again an oscillating pattern of resistance (Figures 7C and S7B). Thus, combinatorial treatment of DC101 with either anti-Gr1 or anti-CSF1 did not reduce tumor growth or vessel density compared to DC101 alone, and neither antibody alone affected tumor growth (Figures 7B and S7A).
Figure 7. Myeloid PI3K limits antiangiogenic therapy in PyMT tumors.

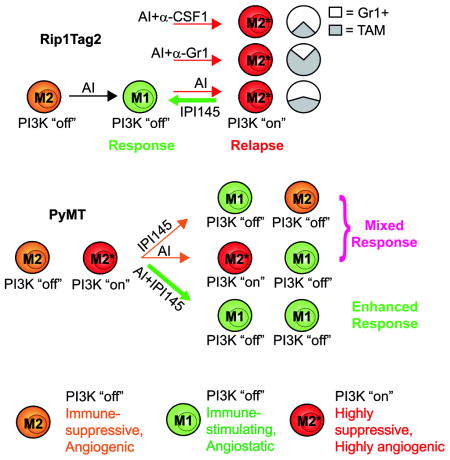
(A) PyMT tumor growth. *p<0.05 for treatment groups versus control at day 12. Data represents at least six mice per group. (B) Microvessel density of tumors at 12 day- treatment.*p< 0.05 versus control. (C) Intratumoral myeloid cell composition at 12day treatment. P< 0.05 for TAM, Gr1+Ly6GHi monocytes, and TAN from DC101 alone versus DC101 plus anti-Gr1; p<0.05 for TAM and TAN from DC101 versus DC101 plus anti-CSF1 at 12 days. (D) CD11b+ Phospho-S6 (pS6) staining and quantification of PyMT tumors. Scale bars, 5 um. (E) Tumor growth in mice treated with IPI145 as indicated. *p< 0.05 for DC101, IPI145 versus control at day 12; #p< 0.05 for DC101 plus IPI145 versus DC101 at day 12. Data represents at least six mice per group. (F and G) Microvessel density (F) and myeloid cell composition (G) in tumors after 12 day- treatment. *p< 0.05 versus control at 12 days. P<0.05 for TAM and TAN in DC101 plus IPI145 versus cognate populations in DC101 alone. (H and I) QPCR analyses of immune- (H) and angiogenesis- (I) modulating genes (H) after 4 and 12 days therapy. *p< 0.05 versus four day control; #p< 0.05 versus 12 day control. Dotted lines indicate baseline gene expression in control samples. Mean±SEM is presented for all quantitations (J and K). Summary of response to antiangiogenic therapy in RT2 (J) and PyMT (K) models. (J) Myeloid cells in naïve RT2 PNET exhibit a “PI3K-off” M2 phenotype. Angiogenesis inhibitors skew myeloid cells towards a CXCL14-expressing M1 phenotype; these promote tumor response by impeding angiogenesis and facilitating anti-tumor immunity. In turn, tumors activate myeloid PI3K-signaling, likely by increasing SDF1α and IL-6. PI3K-activated myeloid cells display a highly angiogenic and immune-suppressive M2* phenotype. M2* myeloid cells convey resistance to therapy by blocking anti-tumor immunity and promoting angiogenesis. IPI145 blocks the acquisition of the M2* phenotype, resulting in sustained tumor response. (K) Myeloid cells within PyMT tumors are both M2 and M2*. Antiangiogenic therapy alone skews M2 cells towards an M1 phenotype without affecting M2* cells, while IPI145 alone converts M2* cells to an M1 phenotype and does not influence M2 cells. Thus, either treatment elicits only partial responses while combining angiogenesis inhibitors and IPI145 results in conversion of both M2 and M2* cells to the M1 phenotype, thus enhancing tumor response. See Discussion for details and also Figures S6 and S7.
We then investigated whether the PI3K-activation status in tumor-associated myeloid cells was responsible for the partial response to antiangiogenic therapy. Indeed, analysis of pS6 kinase+ CD11b+ cells in untreated tumors already revealed PI3K-activation in 46.2% of tumor-associated myeloid cells (Figure 7D), over 2.5 fold higher than what we observed in naïve RT2 tumors (Figure 5B). Therefore, IPI145 alone elicited an intermediate response. DC101 treatment also produced an intermediate response but nearly doubled the population of PI3K-activated myeloid cells (Figure 7D). Congruently, we found that DC101 induced the expression of the immunosuppressive genes IL-10 and TGFβ1 as early as four days after the start of therapy, without affecting expression of the proinflammatory genes IL-23 and TNFα (Figure 7H). Further, like in the PNET model, DC101 induced expression of the proangiogenic genes VEGF and bFGF concomitant with an increase in both CXCL14 and 4 expression (Figure 7I). These data suggest that the immune-suppressive and proangiogenic activities of PI3K-activated myeloid cells appear to outweigh the immune-stimulatory and angiostatic activities of the PI3K-nonactive myeloid cells, thus explaining the intermediate response to antiangiogenic therapy (Figures 7H and 7I). Therefore, blocking PI3K in myeloid cells should be sufficient to increase sensitivity of PyMT tumors to DC101. Notably, like PNET tumor cells, PyMT breast tumor cells did not express detectable levels of PI3K γ or δ and did not respond to IPI145 in vitro (Figure S7D and S7E). Indeed, we found that combining IPI145 with DC101 substantially reduced the number of tumor-associated pS6+ CD11b+ cells to 9% (Figure 7D). This corresponded to a reduction in tumor growth with a further trend in microvessel density reduction concomitant with enhanced immune-stimulatory and reduced immune-suppressive gene expression profiles (Figures 7E–F, 7H–I). IPI145 decreased the levels of intratumoral TAM, Gr1+ monocytes, and TAN compared to both control and DC101-treated tumors (Figures 7G and S7C) and further enhanced CXCL4 and CXCL14 expression, similar to the results observed in the PNET model (Figure 7I). As expected, IPI145 repressed DC101-induced expression of IL-10 and TGFβ1 and induced IL-23 and TNFα expression in TAM, Gr1+ monocytes, and TAN (Figures S6A–S6D) as well as enhanced CXCL4 and CXCL14 expression in these populations and repressed DC101-induced VEGF and bFGF transcription (Figures S6A–S6D). Consequently, IPI145 induced Perforin expression in tumor-associated CTLs (Figures S6E and S6F). Thus, in the PYMT and PNET model, PI3K-activation in myeloid cells hindered the efficacy of antiangiogenic therapy by promoting immune-suppression and limiting the expression of angiostatic factors. These results not only demonstrate that oscillating immune cells activate PI3K-signaling to mediate resistance, but also suggest that the extent of PI3K-activated myeloid cells may predict tumor response to antiangiogenic therapy.
DISCUSSION
In this study we demonstrated that intratumoral myeloid cells dynamically drive responsiveness as well as resistance to angiogenic inhibitors targeting the VEGF/VEGFR pathway. Sorafenib and DC101 skewed different inert CD11b+ myeloid cell populations including TAM, TAN as well as Gr1+Ly6CHi and Ly6GHi monocytes to an immune-stimulatory and angiostatic phenotype in tumors by enhancing their expression of the angiostatic and immune-stimulatory chemokine CXCL14 (and to a lesser extent CXCL4). Surprisingly, the efficacy of VEGF/VEGFR inhibitors depended on the induction of this myeloid CXCL chemokines rather than on direct inhibition of VEGFR signaling in endothelial cells. While depleting CTL cells during antiangiogenic therapy in RT2 mice lowered the apoptotic rate without affecting vessel density, blocking CXCL14 was sufficient to disable both reduction in vessel density and enhanced tumor cell apoptosis thereby completely abrogating the ability of sorafenib to promote tumor stasis. While CXCL4 is believed to impose antiangiogenic activity by interfering with integrins on the surface of endothelial cells (Carmeliet and Jain, 2011; Gabrilovich et al., 1998; Sharpe et al., 1990), less is known about the mechanism of antiangiogenic activity of CXCL14, although CXCL14 is known for its effects on dendritc cell maturation (Gabrilovich et al., 1999). Other CXCL chemokines such as CXCL9 and CXCL10 have also been identified in macrophages that exert angiostatic functions implying a myeloid-specific angiostatic signature (Gabrilovich et al., 1996). Our findings reveal the necessity of myeloid cells in antiangiogenic therapy and elicit that the proangiogenic capacity observed in immunosuppressive myeloid populations does not stem from the enhanced expression of angiogenic factors but rather from the repression of angiostatic CXCL chemokines that also elicit immune-stimulating functions. In order to reinitiate angiogenesis and immune-suppression, tumors induced infiltration of Gr1+ monocytes and neutrophils and produced factors that activated the PI3K signaling axis in all CD11b+ immune cells. This rendered myeloid cells nonresponsive to angiogenesis inhibitors and increased their proangiogenic and immune-suppressive properties (Figures 7J and 7K). Our findings further reveal that the PI3K activation status of myeloid cells in tumors determines the efficacy of anti-angiogenic therapy. While in naïve PNET tumors, most myeloid cells were inactive leading to a pronounced antiangiogenic effect, in PyMT tumors, with more than 40% myeloid PI3K activity, therapy only partially impaired angiogenesis and tumor growth. Interestingly, targeting the enhanced influx of Gr1+ cells was not sufficient to prolong response to angiogenic blockade in both tumor models as it resulted in an increase in TAM concomitant with enhanced intratumoral M-CSF levels, while targeting TAM induced an increase in Gr1+ cells due to elevated G-CSF levels (Figures S2D–S2F) leading to an oscillating pattern of distinct immune cell populations. Chemotherapeutics are commonly given in conjunction with antiangiogenic agents, and have been shown to modulate immune suppression (Coussens et al., 2013), however, addition of the chemotherapeutic temolozolomide (TMZ) did not affect oscillating compensation of myeloid cells when TAM or Gr1+ cells were targeted during antiangiogenic therapy, nor the timing of the angiogenic relapse, though it further reduced tumor burden by about 30% (Figures S2G–S2I). Further evidence for oscillating resistance comes from a previous study demonstrating that genetically depleting TAM in a transgenic mouse model of cervical cancer, which were the major source of the proangiogenic enzyme MMP9, resulted in a compensatory increase in TAN that then became the primary source of MMP9 promoting neovascularization (Voron et al., 2014). Furthermore, various approaches to deplete TAM in models of thyroid, cervical and pancreatic cancers have also shown to increase tumor-associated Gr1+ cells (Quail and Joyce, 2013; Schmid and Varner, 2012). Interestingly, however, Gr1+ cells in the absence of TAM could not compensate for the TAM-dependent maintenance of ALDH+ cancer stem cell fraction in PDAC (Quail and Joyce, 2013) and growth deficiency of Braf-driven thyroid tumors (Schmid and Varner, 2012). Taken together, these results support the notion that innate immune cells share functions in angiogenesis and immune-modulation, likely to maintain tissue homeostasis, but also have distinct and non-compensatory functions in other areas such as stemcellness, invasion and metastasis (Lu and Bergers, 2013; Noy and Pollard, 2014; Quail and Joyce, 2013).
With the certain degree of functional redundancy among tumor-infiltrating myeloid cells to regulate angiogenesis and immunity, we identified two Th2 and simplistically referred to as “M2”-like myeloid phenotypes in tumors that differ in their PI3K activation status (Figures 7J and 7K). While PI3K-inert myeloid cells expressed several proangiogenic factors and lacked immune-stimulating activity (M2 cells), myeloid cells with active PI3K signaling aggravated angiogenic and immune-suppressive activities and thwarted the immune-stimulatory and angiostatic conversion by VEGF/VEGFR inhibitors (M2* cells). Interestingly, M2* cells exhibited an expression profile including increased MMR (MRC1) levels that was in part reminiscent of a TEM signature (Dannull et al., 2005; Lu et al., 2008). TEM are a subpopulation of TAM with exacerbated angiogenic activity that were also found to accumulate in relapsing tumors undergoing anti-VEGFR therapy (Rigamonti et al., 2014). It is therefore intriguing to speculate that the TEM signature is in part regulated by macrophage PI3K activation.
In contrast to epithelial cells and cancer cells, myeloid cells specifically express the γ and δ isoforms of PI3K that can be blocked with IPI145. Although we did not differentiate between PI3Kγ and δ activity, PI3Kγ is highly enriched in myeloid cells, where it transduces GPCR, TLR and RTK signaling to facilitate myeloid cell infiltration to inflammatory sites and evoke inflammatory responses in tumors and other diseases (Anisimov et al., 2013; Mahdipour and Mace, 2012; Mencke et al., 2009) suggesting the γ isoform as the major regulator of tumor inflammation in myeloid cells. Several tumor-derived chemoattractants have been described to activate PI3Kγ signaling (Schmid et al., 2011; Segaliny et al., 2014). Among those we found IL-6 and SDF1α to be upregulated in relapsing PNET tumors. As SDF1α and IL-6 are hypoxia–regulated genes it is conceivable that microenvironmental changes, such as therapy-induced hypoxia, promote such conversion. Indeed, macrophage polarization is regulated in part by intratumoral hypoxia, in which Semaphorin 3A/Neuropilin-1 signaling enables infiltration of myeloid cells into hypoxic areas in which they secrete various immune-suppressive and proangiogenic factors (Chung et al., 2013; Dvorak, 1986; Kasuya and Tokura, 2014).
Blocking VEGF/VEGFR signaling induced the expression of the angiostatic and immunestimulatory chemokine CXCL14 (and to a lesser extent CXCL4) in myeloid cells that was abrogated once myeloid cells exhibited PI3K activation upon tumor relapse.
Finally, these data further implement and support a rational basis for future therapeutic modalities to combine antiangiogenic and immune therapies to generate more durable effects and likely sensitize resistant tumors to antiangiogenic therapy. This is further supported by observations that the abnormal tumor vasculature fosters an immune suppressive microenvironment that enables tumors to evade host immunosurveillance (Baeriswyl and Christofori, 2009). In addition, the proangiogenic factor VEGF not only suppresses the function of various immune cells but also diminishes leukocyte-endothelial interactions and thereby hinders infiltration of immune T-effector cells into the tumor (Jain and Carmeliet, 2012; Kadambi et al., 2001; Snuderl et al., 2013; Tsuzuki et al., 2000). Congruently, VEGF blockade was recently found to result in TAM polarization towards an immune-supporting state and increased T-cell infiltration into tumors by creating an even distribution of patent vessels which reduced tumor hypoxia (Carmeliet and Jain, 2000), while genetic deletion of Rgs5 in pericytes increased T-cell infiltration into tumors and enhanced survival after adoptive T-cell transfer by normalizing the tumor vasculature (Van Cutsem et al., 2011).
The existence of oscillating innate immune cells to convey reneovascularization argues for the idea of modulating the overall immune response to control tumor growth as opposed to targeting distinct myeloid populations, as this approach would not leave behind non-targeted myeloid cells able to foster tumor reneovascularization. Congruently, we found that PI3K inhibition in all myeloid cells generated an enduring angiostatic and immune-stimulatory environment in which anti-angiogenic therapy remained efficient. Other approaches to reverse immune suppression, including blockade of self-tolerance checkpoints, are showing increasing promise, with the anti-CTLA-4 antibody ipilimumab already receiving FDA approval for advanced melanoma. Various antiangiogenic immune-therapies have been tested in the clinic, more recently in combination with checkpoint blockers with encouraging results (Lahl et al., 2007; Wing et al., 2006). Ongoing and future studies will reveal whether combination of anti-angiogenic therapies with these new immune-modulating strategies will more robustly inhibit tumor angiogenesis and promote an enduring immune-stimulatory milieu that leads to prolonged survival benefits in cancer patients.
EXPERIMENTAL PROCEDURES
Detailed methods are provided as Supplemental Experimental Procedures
Tumor models
RT2 mice were previously described (D, 1964) and maintained as heterozygotes in the C57BL/6 background. Sorafenib treatment with or without TMZ was initiated at 13 weeks of age and was administered until mice reached either 15 weeks or 17 weeks. Mice treated with anti-Gr1, anti-CSF1, or IPI145 (Active Biochem, China) received these beginning at 15 weeks, two weeks after initiating sorafenib. For PyMT experiments, syngeneic PyMT breast tumor cells were implanted into the fourth mammary fat pads of female FVB mice at four to five weeks of age. Tumors reached 5 mm in diameter before initiating DC101 treatment alone or in combination with anti-Gr1 or anti-CSF1. All animal studies were reviewed and approved by the UCSF Institutional Animal Care and Use Committee. For more experimental details, see Supplementary Experimental Procedures.
Human PNET specimens
Immune cells were assessed for phospho-S6 staining by a neuropathologist (D. Meyronet) in 34 deidentified tumor samples from Hospices Civils de Lyon, France representing naïve-treated patients, patients treated with 5-FU, or treated with bevacizumab. To confirm this assessment, immunofluorescent staining for CD45 and phospho-S6 was performed blindly on tumor samples corresponding to eight untreated patients, six treated with 5-FU, and two treated with bevacizumab.
Expression analysis
QPCR expression analyses were performed using whole tumor or FACS-sorted cell populations and the indicated primer sets. Relative gene expression was calculated as previously described (I et al., 2014). Briefly, L19 was used as a housekeeping gene to generate ΔCt; ΔCt values from untreated mice were used as reference for treatment groups to generate ΔΔCt, and relative gene expression was calculated as 2(−ΔΔCt). Analyses of sorted cells from RT2 mice were performed using at least eight experimental mouse replicates while whole tumor analyses were performed using five experimental replicates. Analyses of sorted cells from PyMT tumors as well as whole PyMT tumors were performed using at least 6 experimental mouse replicates. All reactions were run in triplicate. For primer sequences, see Supplementary Experimental Procedures.
Statistical analysis
Statistical analyses were performed under the guidance of the Helen Diller Family Comprehensive Cancer Center Biostatistics and Computational Biology Core. For mouse studies, p-values for RT2-PNET and PyMT tumor burdens were calculated using the Wilcoxon rank sum test. P-values for microvessel density, multicolor flow cytometry, CD11b/phospho-S6 and CD45/phospho-S6 immunostaining, and CD8+ T cell content were also calculated using the Wilcoxon rank sum test. This same test was used to evaluate survival (since uncensored). For gene expression analyses, where the transformed signals are more Gaussian and the sample sizes were smaller, comparisons between experimental and control mice for single genes were made using a two-sample t-test. For the purposes of determining whether a signature is present, such as immune response or angiogenesis, p-values were added across genes. For this purpose, Fisher’s inverse chi-square method was utilized.
Supplementary Material
Acknowledgments
We thank Jairo Barreto, Lourdes Adriana Esparza for mouse husbandry, genotyping and drug administration, Bill Hyun for help with multi-color flow cell sorting, Binh Tuyen and Kathrik Kadalabalu Matha for technical assistance, and Drs. Lisa Coussens, Jeff Pollard, Judy Varner and Ruth Ganss for helpful discussions. This work was supported by grants from the NIH (RO1 CA099948 (to G.B.), U54CA163155 (to G.B.), T32CA108462-09 (to L.R.) and the AACR Carcinoid foundation.
Footnotes
Supplemental information includes Supplemental Experimental Procedures and 7 Figures and can be found with this article online at XX.
AUTHOR CONTRIBUTIONS
LBR and GB designed research and wrote and edited manuscript, LBR conducted experiments, DM and VH procured human pancreatic neuroendocrine tumor tissues; MF provided anti-CXCL14 antibodies and EB provided a clinical perspective. GB supervised research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anisimov A, Tvorogov D, Alitalo A, Leppanen VM, An Y, Han EC, Orsenigo F, Gaal EI, Holopainen T, Koh YJ, et al. Vascular endothelial growth factor-angiopoietin chimera with improved properties for therapeutic angiogenesis. Circulation. 2013;127:424–434. doi: 10.1161/CIRCULATIONAHA.112.127472. [DOI] [PubMed] [Google Scholar]
- Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Seminars in cancer biology. 2009;19:329–337. doi: 10.1016/j.semcancer.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Barrios CS, Johnson BD, Henderson J, Fink JN, Kelly KJ, Kurup VP. The costimulatory molecules CD80, CD86 and OX40L are up-regulated in Aspergillus fumigatus sensitized mice. Clinical and experimental immunology. 2005;142:242–250. doi: 10.1111/j.1365-2249.2005.02905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature reviews Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nature medicine. 2013;19:1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–490. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHF Apropos of 6 Cases of the Scalenus Anticus Syndrome Treated by Scalenotomy and Section of the Pectoralis Minor. Annales de chirurgie. 1964;18:205–209. [PubMed] [Google Scholar]
- Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, Vieweg J. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. The Journal of clinical investigation. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierickx R, Fornhoff M, Stas R, DHH, Hanegreefs G, Alleman J. Review of the Activity of the Antwerp Thoracic Center. Acta chirurgica Belgica. 1963;(Suppl 2) SUPPL2:58–63. [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. The New England journal of medicine. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nature medicine. 1996;2:1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clinical cancer research: an official journal of the American Association for Cancer Research. 1999;5:2963–2970. [PubMed] [Google Scholar]
- Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. The Journal of clinical investigation. 2004;114:623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISK, Madeeh Hashmi A, Awais Aftab M, Westermeyer J, TDH Actigraphy in post traumatic stress disorder. Pakistan journal of medical sciences. 2014;30:438–442. [PMC free article] [PubMed] [Google Scholar]
- Jain RK, Carmeliet P. SnapShot: Tumor angiogenesis. Cell. 2012;149:1408–1408. e1401. doi: 10.1016/j.cell.2012.05.025. [DOI] [PubMed] [Google Scholar]
- Kadambi A, Mouta Carreira C, Yun CO, Padera TP, Dolmans DE, Carmeliet P, Fukumura D, Jain RK. Vascular endothelial growth factor (VEGF)-C differentially affects tumor vascular function and leukocyte recruitment: role of VEGF-receptor 2 and host VEGF-A. Cancer research. 2001;61:2404–2408. [PubMed] [Google Scholar]
- Kasuya A, Tokura Y. Attempts to accelerate wound healing. Journal of dermatological science. 2014;76:169–172. doi: 10.1016/j.jdermsci.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. Tumor angiogenesis. The New England journal of medicine. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. The Journal of experimental medicine. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer research. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- Lu K, Lamagna C, Bergers G. Chapter 3. Bone marrow-derived vascular progenitors and proangiogenic monocytes in tumors. Methods in enzymology. 2008;445:53–82. doi: 10.1016/S0076-6879(08)03003-6. [DOI] [PubMed] [Google Scholar]
- Lu KV, Bergers G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS oncology. 2013;2:49–65. doi: 10.2217/cns.12.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdipour E, Mace KA. Analyzing the angiogenic potential of Gr-1(+)CD11b (+) immature myeloid cells from murine wounds. Methods Mol Biol. 2012;916:219–229. doi: 10.1007/978-1-61779-980-8_17. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, De Palma M. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer cell. 2011;19:512–526. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Mencke N, Vobis M, Mehlhorn H, JDH, Rehagen M, Mangold-Gehring S, Truyen U. Transmission of feline calicivirus via the cat flea (Ctenocephalides felis) Parasitology research. 2009;105:185–189. doi: 10.1007/s00436-009-1381-5. [DOI] [PubMed] [Google Scholar]
- Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nature reviews Immunology. 2011;11:702–711. doi: 10.1038/nri3064. [DOI] [PubMed] [Google Scholar]
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll L, Linehan R, SMK, Cronin D, Purcell R, Glynn S, EWM, ADH, NJOH, Parkinson M, Clynes M. Lack of prognostic significance of survivin, survivin-deltaEx3, survivin-2B, galectin-3, bag-1, bax-alpha and MRP-1 mRNAs in breast cancer. Cancer letters. 2003;201:225–236. doi: 10.1016/s0304-3835(03)00518-4. [DOI] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell reports. 2014;8:696–706. doi: 10.1016/j.celrep.2014.06.059. [DOI] [PubMed] [Google Scholar]
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nature reviews Immunology. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- Schaerli P, Willimann K, Ebert LM, Walz A, Moser B. Cutaneous CXCL14 targets blood precursors to epidermal niches for Langerhans cell differentiation. Immunity. 2005;23:331–342. doi: 10.1016/j.immuni.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer cell. 2011;19:715–727. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MC, Varner JA. Myeloid cells in tumor inflammation. Vascular cell. 2012;4:14. doi: 10.1186/2045-824X-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segaliny AI, Mohamadi A, Dizier B, Lokajczyk A, Brion R, Lanel R, Amiaud J, Charrier C, Boisson-Vidal C, Heymann D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. International journal of cancer Journal international du cancer. 2014 doi: 10.1002/ijc.29376. [DOI] [PubMed] [Google Scholar]
- Sharpe RJ, Byers HR, Scott CF, Bauer SI, Maione TE. Growth inhibition of murine melanoma and human colon carcinoma by recombinant human platelet factor 4. Journal of the National Cancer Institute. 1990;82:848–853. doi: 10.1093/jnci/82.10.848. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nature biotechnology. 2007a;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007b;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin JD, Kamoun W, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152:1065–1076. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nature reviews Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki Y, Fukumura D, Oosthuyse B, Koike C, Carmeliet P, Jain RK. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha--> hypoxia response element--> VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer research. 2000;60:6248–6252. [PubMed] [Google Scholar]
- Van Cutsem E, Lambrechts D, Prenen H, Jain RK, Carmeliet P. Lessons from the adjuvant bevacizumab trial on colon cancer: what next? Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:1–4. doi: 10.1200/JCO.2010.32.2701. [DOI] [PubMed] [Google Scholar]
- Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, Terme M. Control of the immune response by pro-angiogenic factors. Frontiers in oncology. 2014;4:70. doi: 10.3389/fonc.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Fehervari Z, Sakaguchi S. Emerging possibilities in the development and function of regulatory T cells. International immunology. 2006;18:991–1000. doi: 10.1093/intimm/dxl044. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.