

Abstract
In a substudy of a clinical trial, we assessed whether activation of latent human immunodeficiency virus (HIV) by the histone deacetylase inhibitor panobinostat had detrimental effects on the central nervous system (CNS). Adults infected with HIV received oral panobinostat 20 mg 3 times per week every other week for 8 weeks. In cerebrospinal fluid (CSF), we assayed panobinostat concentration, HIV RNA, and the level of neuroinflammatory or degenerative biomarkers in 11 individuals before and during study therapy. Neither panobinostat nor HIV RNA was detected in CSF. In addition, there was no change from baseline in CSF biomarkers. Thus, panobinostat administration was not associated with CNS adverse effects as assessed by CSF biomarkers.
Keywords: central nervous system, cerebrospinal fluid, histone deacetylase inhibitors, human immunodeficiency virus, panobinostat
Histone deacetylase inhibitors (HDACi), including the highly potent pan-HDACi panobinostat, are currently evaluated in experimental clinical trials for their ability to induce human immunodeficiency virus (HIV) expression in latently infected cells with the overall aim of eradicating the latent HIV reservoir. The central nervous system (CNS) merits particular consideration in this context. At least in some patients, the CNS may represent a reservoir and/or sanctuary site harboring latent HIV in macrophages, microglia, and astrocytes [1]. However, whether these cell populations allow persistence of replication competent virus in patients on suppressive antiretroviral therapy (ART) on the order of years, which is the proposed practical definition of a reservoir for HIV [2], is unknown. Therefore, the in vivo importance of a potential CNS reservoir for HIV-1 eradication remains controversial. Still, some investigators have cautioned that several of the therapeutic approaches currently being considered, such as activating HIV from latency by HDACi administration, may not achieve HIV eradication from the CNS and could potentially have devastating consequences on the brain [3]. The proposed adverse effects on the CNS include (1) neuronal injury caused by early viral proteins induced by HDACi [4]; (2) CNS immune reconstitution inflammatory syndrome [5]; (3) adverse effects on brain function caused by elimination of latently infected microglia and/or astrocytes [3]; and (4) neuronal injury due to inflammation in the brain caused by activated T cells [3]. Thus, nervous system damage could potentially be caused both directly by HIV and indirectly via HIV-associated immune-inflammatory responses in the CNS, including the transfer of these from the periphery into the CNS. More importantly, each of these 3 processes (CNS viral infection, neuroinflammation, and neurodegeneration) can be assessed by the measurement of specific biomarkers in cerebrospinal fluid (CSF). In a recently conducted clinical trial among HIV-infected adults on suppressive ART, cyclic administration of panobinostat led to significant increases in HIV transcription in CD4+ T cells, as measured by cell-associated unspliced HIV RNA, as well as an increase in HIV viremia, providing evidence that panobinostat activates HIV from latency [6]. Per protocol, CSF was collected before and during panobinostat therapy, and this study thus provided a unique opportunity to investigate whether HDACi-induced HIV-expression was associated with any adverse effects on the CNS as assessed by biomarkers of neuroinflammation and neurodegeneration.
MATERIALS AND METHODS
Between September 2012 and February 2014, we conducted an investigator-initiated, single-arm, phase I/II clinical trial at Aarhus University Hospital, Denmark to evaluate the ability of panobinostat to activate HIV from HIV latency. Details of the study have been published elsewhere [6]. In brief, 15 HIV-infected adults on ART with virological suppression for at least 2 years (plasma HIV RNA <50 copies/mL) and CD4+ T cell counts above 500/µL received oral panobinostat 20 mg 3 times per week every other week for 8 weeks, ie, 4 cycles of treatment, while maintaining combination ART. To address whether viral reactivation induced by panobinostat treatment was associated with adverse effects on the CNS, lumbar puncture and collection of 5 mL CSF was performed as an optional procedure before initiating panobinostat and during the last treatment cycle. Ethics committee approval was obtained in accordance with the principles of the Helsinki Declaration. Each patient provided written informed consent before any study procedures. This trial is registered with ClinicalTrial.gov, number NCT01680094.
Immediately after collection, CSF was stored at −80°C until biomarker analyses were performed. Using enzyme-linked immunosorbent assays (ELISA), we measured the CSF concentration of total tau (t-tau), phosphorylated tau (p-tau), and soluble amyloid precursor protein-β; biomarkers known to be associated with neuronal injury [7, 8]. To assess alterations in the level of neuroinflammation, we measured the CSF concentration of neopterin, C-reactive protein (CRP), soluble CD14 (sCD14), soluble CD163 (sCD163), monocyte chemotactic protein-1 (MCP-1, CCL2), interferon-γ-induced protein-10 (IP-10, CXCL10), macrophage inflammatory protein-1β (MIP-1β, CCL4), and matrix metallopeptidase-9 (MMP-9). All biomarkers of neuroinflammation were assayed using ELISA except CRP, which was determined by a particle-enhanced immunoturbidimetric assay. A validated liquid chromatography-tandem mass spectrometry method using 100 µL sample volume was used for the determination of panobinostat in CSF and was performed by Novartis Institutes for BioMedical Research (East Hanover, NJ). The limit of quantification in CSF with this assay was 0.1 ng/mL (equivalent to 0.3 nM). Finally, using 1 mL CSF, the presence or absence of HIV RNA in CSF was analyzed by a transcription-mediated amplification (TMA)-based assay as described by the manufacturer (Procleix Ultrio Plus, Novartis). This is a validated and US Food and Drug Administration-approved detection method for HIV RNA in plasma with a detection limit of 3.8 copies/mL using 1 mL sample volume [9]. However, limitations in sample material prevented specific validation of this assay for CSF. Changes from baseline in biomarker levels were tested using Wilcoxon signed-rank test. Correlations between CSF biomarkers and measures of viral reactivation and inflammation in the periphery were explored using spearman rank-order correlation. Statistical analyses and illustrations were performed in Stata 13.1 and GraphPad Prism 6.
RESULTS
Of 15 patients included in the clinical trial, 11 consented to lumbar punctures before panobinostat dosing and during the last cycle of panobinostat treatment. The on-panobinostat CSF lumbar puncture was performed approximately 8 hours after receipt of the final panobinostat dose for 10 of the 11 patients. One patient had lumbar puncture performed 24 hours after receipt of the first dose in the final treatment cycle. Panobinostat was below the limit of detection in CSF for all obtained samples. Similarly, all CSF samples were negative for HIV RNA as assessed by the TMA-based assay, both at baseline and during panobinostat treatment. In contrast, plasma HIV RNA was detectable for 6 of the 11 patients at the time of the on-panobinostat lumbar puncture [6]. Paired levels of biomarkers of neurodegeneration and neuroinflammation are shown in Figures 1 and 2, respectively. Overall, we recorded no significant change from baseline to the final panobinostat treatment week in biomarkers of neurodegeneration or in biomarkers of neuroinflammation. A tendency towards a decrease in MCP-1 (P = .067) and an increase in t-tau (P = .065) was noted, but these findings must be interpreted in the context of multiple testing. Similarly, as assessed by ratios of MCP-1/IP-10 (CCL2/CXCL10), MIP-1β/IP-10 (CCL3/CXCL10), and MCP-1/MIP-1β (CCL2/CCL4) before and during panobinostat treatment, we found no evidence of the relative accumulation of chemokines that promote monocyte and neutrophil infiltration into the CSF, as was seen in patients with cryptococcosis-associated immune reconstitution inflammatory syndrome [10]. Finally, CSF biomarker levels, or changes herein, did not show any consistent correlation with either plasma soluble markers of inflammation or flow cytometry-based measures of cellular activation status in circulating T cells and monocytes (data not shown).
Figure 1.
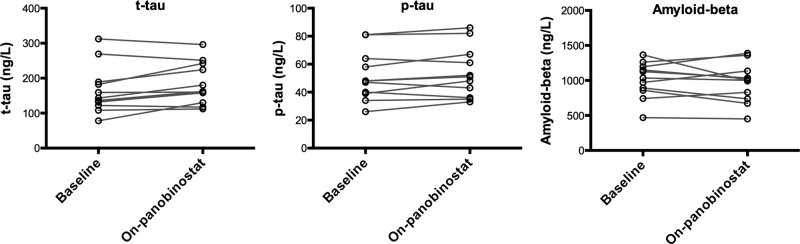
Levels of biomarkers of neurodegeneration at baseline and during the final panobinostat dosing week, shown for each of the 11 patients who consented to lumbar punctures. All biomarkers were determined by enzyme-linked immunosorbent assays. Changes in biomarker levels were tested using Wilcoxon signed-rank test. For phosphorylated tau (p-tau) and soluble amyloid precursor protein-β (amyloid-beta), P > .1; for total tau (t-tau), P = .065.
Figure 2.
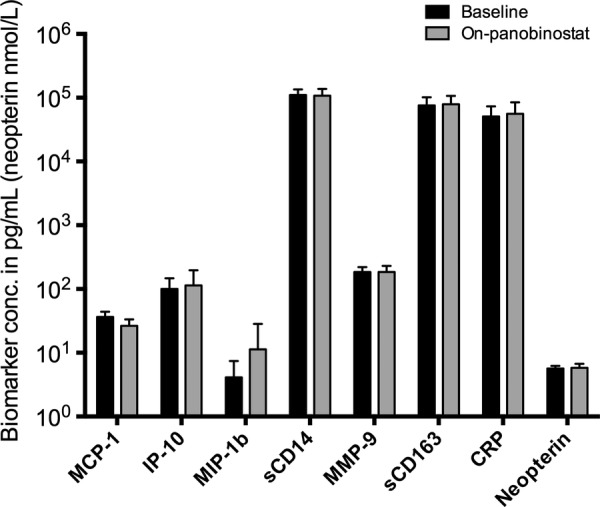
Levels of biomarkers of neuroinflammation at baseline and during the final panobinostat dosing. All biomarkers except C-reactive protein (CRP) were determined by enzyme-linked immunosorbent assays. C-reactive protein was determined by a particle-enhanced immunoturbidimetric assay. Changes in biomarker levels were tested using Wilcoxon signed-rank test. P > .1 for all comparisons except monocyte chemotactic protein-1 (MCP-1) (P = .067). Abbreviations: IP-10, interferon-γ induced protein-10; MIP-1b, macrophage inflammatory protein-1β; MMP-9, matrix metallopeptidase-9; sCD14, soluble CD14; sCD163, soluble CD163.
DISCUSSION
Although caution has been raised that activating HIV from latency by HDACi could potentially have harmful consequences on the brain, this is the first study to assess potential CNS adverse effects of HDACi-induced HIV reactivation. We did this by measuring a range of CSF biomarkers before and during panobinostat treatment. None of the assayed biomarkers changed significantly from pre-panobinostat levels. Moreover, we found no consistent correlation between CSF biomarkers and levels of HIV transcription or soluble and cell-based markers of inflammation in the periphery.
We recognize several limitations of this study. First, although prespecified in the study protocol, the assessment of CSF biomarkers was an exploratory substudy, which may not be sufficiently powered to detect small changes. Therefore, our negative results do not exclude the need for further studies. Second, in the absence of a control group, we were unable to control for longitudinal variation in the studied biomarkers. Third, we did not perform systematic longitudinal neuropsychological testing, which would have provided additional clinical insights. Fourth, limited sample volume prevented more sensitive analyses of CSF HIV viral load or epigenetic changes in the CNS compartment. Finally, although CSF biomarkers represent a valuable approach to assess treatment effects and evolving pathobiology [7], translating findings into clinically relevant interpretation is difficult. Despite their overall categorization into biomarkers of neuronal injury and neuroinflammation, each biomarker may in fact indicate distinctive, to some degree overlapping, biological and/or pathological processes. Thus, t-tau and p-tau are different states of the neuronal tau protein that are elevated in the CSF in Alzheimer's disease, whereas amyloid-β, an amyloid cleavage product, is depressed in the CSF in Alzheimer's disease [11]. Although they do indicate active CNS injury in the setting of HIV infection [12], none of these biomarkers are specific for HIV neuropathology. Therefore, their ability to detect subtle changes in neuronal injury associated with HIV is debatable, and future analyses should ideally include assessment of neurofilament light chain. Neopterin is a pteridine metabolite produced by cells of the monocyte-macrophage lineage and possibly also astrocytes; it is an often-used biomarker of neuroinflammation, readily measurable in the CSF and increases with HIV disease progression [13]. Interferon-γ-induced protein-10 is an important chemokine for CSF lymphocytes, whereas MCP-1 and MIP-1β are chemotactic for a variety of immune cells [14]. Both sCD163 and sCD14 are monocyte activation markers that are increased in HIV [15]. Matrix metallopeptidase-9 is a matrix metallopeptidase involved in the integrity of extracellular matrix.
Obviously, the concern of CNS adverse effects from HIV reactivation is dependent on CNS penetration of the drug in question. Preclinical studies indicate that the HDACi vorinostat and romidepsin to some degree cross the blood-brain barrier [16], but this has not previously been investigated for panobinostat. Our report thus contains the first in vivo data to inform of this issue and shows that panobinostat either does not penetrate the CNS or only reaches very low concentrations (below the limit of detection). Still, induction of HIV transcription and virion production in the periphery may lead to dissemination of viral products or inflammatory processes into the CNS, which is why adverse effects within this compartment might not require CNS penetration of the inducing agent. Finally, despite the lack of significant changes in CSF biomarker levels during panobinostat treatment, we searched extensively for any correlation between CSF biomarkers and measures of both HIV reactivation and peripheral inflammation, but we did not find consistent evidence hereof. In comparison, levels of neurofilament light chain and amyloid-β, both significantly higher in primary HIV infection compared with HIV uninfected controls, did not correlate with plasma viral load in a recent cross-sectional analysis [8].
CONCLUSIONS
In conclusion, repeated, cyclic treatment with the HDACi panobinostat was not associated with CNS adverse effects as measured by CSF biomarkers of inflammation and neurodegeneration in HIV patients on suppressive ART.
Acknowledgements
Author contributions. T. A. R., M. T., L. Ø., C. R. B., R. O., and O. S. S. conceived of and designed the study and developed the study protocol. L. Ø. provided clinical oversight of the study. T. A. R. and O. S. S. enrolled patients in the study and performed lumbar puncture procedures. H. J. M., C. E., and M. T. performed measurement of CSF biomarkers. C. R. B. performed measurement of cell-associated HIV RNA. Novartis Institutes for BioMedical Research (East Hanover, NJ) performed determination of panobinostat in CSF. All authors participated in data interpretation. The manuscript was drafted by T. A. R., M. T., and O. S. S. All authors provided input to the manuscript and approved the final version.
Financial support. This work was primarily funded by The Danish Council for Strategic Research, but it also received contributions from The Lundbeck Foundation, The Danish AIDS Foundation, The American Fund for AIDS Research, The Augustinus Foundation, Frode V. Nyegaard Foundation, Scandinavian Society for Antimicrobial Chemotherapy Foundation, Aarhus University, and Aarhus University Hospital. Novartis supplied panobinostat for use in the study.
Potential conflicts of interest. Aarhus University has filed a patent application covering the use of panobinostat in HIV-infected patients on which T. A. R., M. T., C. R. B., L. Ø., and O. S. S. are inventors. Aarhus University and H. J. M. have received royalties from IQ-Products, the Netherlands.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Churchill MJ, Wesselingh SL, Cowley D, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol 2009; 66:253–8. [DOI] [PubMed] [Google Scholar]
- 2.Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012; 37:377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nath A, Clements JE. Eradication of HIV from the brain: reasons for pause. AIDS 2011; 25:577–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chauhan A, Turchan J, Pocernich C, et al. Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem 2003; 278:13512–9. [DOI] [PubMed] [Google Scholar]
- 5.Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS 2013; 8:165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen TA, Tolstrup M, Brinkmann CR, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single-group, clinical trial. Lancet HIV 2014; 1:e13–21. [DOI] [PubMed] [Google Scholar]
- 7.Price RW, Peterson J, Fuchs D, et al. Approach to cerebrospinal fluid (CSF) biomarker discovery and evaluation in HIV infection. J Neuroimmune Pharmacol 2013;8:1147–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peluso MJ, Meyerhoff DJ, Price RW, et al. Cerebrospinal fluid and neuroimaging biomarker abnormalities suggest early neurological injury in a subset of individuals during primary HIV infection. J Infect Dis 2013; 207:1703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stramer SL, Krysztof DE, Brodsky JP, et al. Comparative analysis of triplex nucleic acid test assays in United States blood donors. Transfusion 2013; 53:2525–37. [DOI] [PubMed] [Google Scholar]
- 10.Chang CC, Omarjee S, Lim A, et al. Chemokine levels and chemokine receptor expression in the blood and the cerebrospinal fluid of HIV-infected patients with cryptococcal meningitis and cryptococcosis-associated immune reconstitution inflammatory syndrome. J Infect Dis 2013; 208:1604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herukka SK, Hallikainen M, Soininen H, et al. CSF Abeta42 and tau or phosphorylated tau and prediction of progressive mild cognitive impairment. Neurology 2005; 64:1294–7. [DOI] [PubMed] [Google Scholar]
- 12.Gisslen M, Krut J, Andreasson U, et al. Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurol 2009; 9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagberg L, Cinque P, Gisslen M, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther 2010; 7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cinque P, Bestetti A, Marenzi R, et al. Cerebrospinal fluid interferon-gamma-inducible protein 10 (IP-10, CXCL10) in HIV-1 infection. J Neuroimmunol 2005; 168:154–63. [DOI] [PubMed] [Google Scholar]
- 15.Kamat A, Lyons JL, Misra V, et al. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr 2012; 60:234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg SL, Stone J, Xiao JJ, et al. Plasma and cerebrospinal fluid pharmacokinetics of depsipeptide (FR901228) in nonhuman primates. Cancer Chemother Pharmacol 2004; 54:85–8. [DOI] [PubMed] [Google Scholar]