

Abstract
Objective
Systemic sclerosis (SSc)–related interstitial lung disease (ILD) is one of the leading causes of mortality. We undertook this study to analyze the gene expression of lung tissue in a prospective cohort of patients with SSc-related ILD and to compare it with that in control lungs and with 2 prospective clinical parameters in order to understand the molecular pathways implicated in progressive lung disease.
Methods
Lung tissue was obtained by open lung biopsy in 28 consecutive patients with SSc-related ILD and in 4 controls. High-resolution computed tomography (HRCT) and pulmonary function testing (PFT) were performed at baseline and 2–3 years after treatment based on lung histologic classification. Microarray analysis was performed, and the results were correlated with changes in the HRCT score (FibMax) and PFT values. Quantitative polymerase chain reaction (qPCR) and immunohistochemistry were used to confirm differential levels of messenger RNA and protein.
Results
Lung microarray data distinguished patients with SSc-related ILD from healthy controls. In the lungs of patients with SSc-related ILD who had nonspecific interstitial pneumonia (NSIP), expressed genes included macrophage markers, chemokines, collagen, and transforming growth factor β (TGFβ)– and interferon (IFN)–regulated genes. Expression of these genes correlated with progressive lung fibrosis defined by the change in FibMax. Immunohistochemistry confirmed increased markers of collagen (COL1A1), IFN (OAS1 and IFI44), and macrophages (CCL18 and CD163), and the positive correlation with the change in FibMax was confirmed by qPCR in a larger group of SSc patients with NSIP. Several genes correlated with both the change in FibMax (r > 0.4) and the change in % predicted forced vital capacity (r < −0.1), including IFN and macrophage markers, chemokines, and heat-shock proteins.
Conclusion
These results highlight major pathogenic pathways relevant to progressive pulmonary fibrosis in SSc-related ILD: macrophage emigration and activation, and up-regulated expression of TGFβ- and IFN-regulated genes.
Systemic sclerosis (SSc) is a chronic autoimmune disease in which pulmonary involvement, particularly interstitial lung disease (ILD), is known to contribute to morbidity and mortality (1-3). The prevalence of SSc-related ILD is high, and ~15% of patients develop severe restrictive lung disease, the major cause of death in SSc. Distinguishing those who develop severe disease from those who develop slow or stable disease remains a great challenge in targeting appropriate therapy (4,5).
Microarray technology has been used to address changes in gene expression in SSc skin and peripheral blood mononuclear cells (PBMCs) and has shown striking differences from gene expression in healthy controls along with specific gene expression signatures that correlate with SSc organ involvement. Skin expression of transforming growth factor β (TGFβ)– and interferon (IFN)–regulated genes correlates with the modified Rodnan skin thickness score (MRSS) (6,7). In addition, PBMCs from patients with limited cutaneous SSc (lcSSc) show increased expression of IFN-regulated genes and a second cluster of genes associated with monocyte/macrophage activation, the macrophage/monocyte activation cluster, which distinguishes patients with pulmonary arterial hypertension (8,9).
Gene expression analysis of lung tissue in SSc-related ILD until now has been restricted to end-stage lung tissue obtained at the time of lung transplantation (10). Such tissue analyses, however, may represent only the final and irreversible phase of lung involvement and may provide only limited insight into the early pathogenic events.
The present study was undertaken to gain insight into potential pathogenic pathways important in prospectively observed patients with progressive SSc-related ILD. Our data from this unique cohort suggest that TGFβ, IFN, and macrophage activation are key to the pathogenesis of this severe complication, as increased expression of these markers is seen primarily in patients who will develop progressive lung fibrosis.
PATIENTS AND METHODS
Study participants
Between January 2002 and July 2004, 28 consecutive patients with SSc-related ILD confirmed by high-resolution computed tomography (HRCT), with associated respiratory symptoms and/or reduced performance on pulmonary function tests (PFTs), underwent an open lung biopsy and were followed up for at least 3 years. The lung biopsies were performed as part of a clinical protocol designed to study the importance of the histologic lung pattern to the prognosis of patients with SSc-related ILD. Most subjects involved in the previous study (11) were selected (7 were not included due to lack of lung material for RNA isolation). The patients included in the current study were classified as having diffuse cutaneous SSc (dcSSc) (n = 11) and lcSSc (n = 10) according to diagnostic (12) and subtype (13) criteria. Histologically healthy lung tissue was obtained from normal lungs of 4 controls during surgical resection for lung cancer.
The Boston University Medical Center and the University of Sao Paulo Institutional Review Boards reviewed and approved the conduct of this study. Disease duration was established from the onset of Raynaud’s phenomenon. The MRSS (14) was used to determine the extent of skin involvement.
Histologic diagnosis
Two pathologists (ERP and VLC) classified the lung specimens based on a distinctive pattern termed centrilobular fibrosis (11,15) and according to the traditional patterns of the latest consensus classification of the idiopathic interstitial pneumonias (16).
HRCT
Two pulmonologists (RBC and CRdeC) evaluated the images in a blinded manner. Briefly, as described previously (17), each lung was divided into the following 3 zones: upper (lung apex to aortic arch), middle (aortic arch to inferior pulmonary veins), and lower (inferior pulmonary veins to lung bases). For baseline and followup of the HRCT scans, the extent of the pulmonary abnormality in each of the 6 zones was scored on a scale of 0–4 as follows: 0 = absent, 1 = 1–25%, 2 = 26–50%, 3 = 51–75%, and 4 = 76–100%. The following HRCT scan findings were recorded: pure ground-glass opacities (increased lung attenuation in the absence of reticular interstitial thickening or architectural distortion), pulmonary fibrosis (reticular intralobular interstitial thickening, traction bronchiectasis, and bronchiolectasis), and honeycomb cysts (clustered air-filled lung cysts with contiguous walls). The sum of all scores in the 6 lung zones is referred to as the FibMax. The change in FibMax (the followup FibMax minus the baseline FibMax) was used to evaluate the progression of lung fibrosis as previously described (17).
PFTs
PFTs were measured by the % predicted value and included forced vital capacity (FVC) and diffusing capacity for carbon monoxide (DLco) using a single-breath technique. The PFTs were performed using a Collins GS PFT System (Collins Medical). A change in the % predicted value of the FVC was also used to gauge the progression of lung fibrosis.
Lung biopsy
Open lung biopsy was performed by formal thoracotomy, avoiding honeycombing areas on the right lower lobes; for some patients, a second sample was also collected from their right middle lobes (11).
Followup
Patients with a pattern of nonspecific interstitial pneumonia (NSIP) on lung biopsy were treated with monthly intravenous cyclophosphamide (0.5–1 gm/m2) for at least 1 year, and patients with isolated centrilobular fibrosis had their antireflux treatment intensified (see ref. 11). HRCT and PFTs were performed before biopsy and repeated 2–3 years after the 1-year treatment (Figure 1A).
Figure 1.
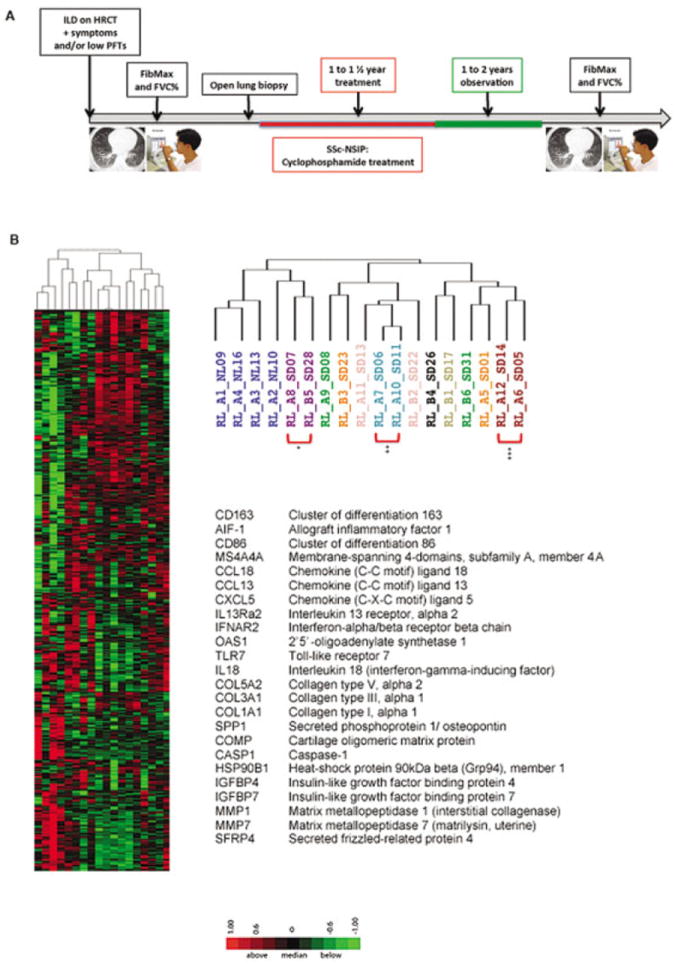
A, Timeline of the study (see Patients and Methods). B, Heatmap showing the expression of genes clustered using complete linkage, hierarchical, unsupervised clustering. Expression values above the mean for each gene are indicated in red; those below the mean are indicated in green. The major dendrogram bifurcation places samples from all 4 healthy controls (represented by dark blue) onto the branch at the left along with 2 samples (from the middle and lower lobes) from 1 systemic sclerosis (SSc) patient with nonspecific interstitial pneumonia (NSIP) (represented by purple) (*). The right branch, including only samples from patients with SSc-related interstitial lung disease (ILD), bifurcates further into a middle sub-branch, which includes biopsy samples from both middle and lower lobes from 2 SSc patients with NSIP (represented by pink and light blue [**]) as well as single-lobe biopsy samples from 2 SSc patients with NSIP (represented by green [lower lobe] and orange [middle lobe]). In contrast, the sub-branch at the far right includes 3 of 6 samples from patients classified as having SSc with centrilobular fibrosis (1 sample represented by light green and 2 samples represented by red, where the patient represented by red had samples from both middle and lower lobes clustered together [***]). The other 3 samples on this branch (represented by black, green, and orange) were obtained from the lower lobes of SSc patients with NSIP. HRCT = high-resolution computed tomography; PFTs = pulmonary function tests; FibMax = sum of all scores in 6 lung zones; FVC = forced vital capacity.
RNA isolation and microarray analysis hybridization
Total RNA from human lung biopsy tissue was extracted using microRNeasy Mini Kits (Qiagen). RNA samples were stored at −80°C.
Microarray data clustering
At the Boston University Microarray Resource Facility, all procedures were performed as described in the Affymetrix GeneChip 3′IVT Express user manual (www.affymetrix.com). Briefly, total RNA was isolated and sample integrity was verified. The biotin-labeled amplified RNA was purified, fragmented, and hybridized to Affymetrix U133 2.0 microarrays. The signal of the samples was amplified, and microarrays were immediately scanned using an Affymetrix GeneArray Scanner 3000 7G Plus. The resulting CEL files were summarized using Affymetrix Expression Console (current version 1.1). A MAS 5 algorithm with global scaling normalization was used to generate gene-level data. The mean target intensity of each array was set at 500. Clustering was performed using Cluster software, version 3.0.
Quantitative polymerase chain reaction (qPCR)
Complementary DNA was synthesized from total RNA using reverse transcriptase (Gibco BRL) and random primers (Applied Biosystems) as described previously (9). Quantitative PCR was performed using an ABI Prism 7700 sequence detection system and a TaqMan assay (both from Applied Biosystems), according to the company’s protocol. Primers for the 18S ribosomal RNA and messenger RNA (mRNA) target genes were obtained from Applied Biosystems.
Histology and immunohistochemistry
The expression of CCL18 and IFN-inducible protein 44 (IFI-44) was assessed on deparaffinized human lung sections. Briefly, tissue sections were rehydrated, steamed with citrate buffer (BioGenex), blocked and washed with Tris buffered saline–Tween, and then incubated with rabbit anti-human IFI-44 (Sigma) and rabbit anti-human CCL18 (LifeSpan Biosciences) antibodies. After washing, the tissue sections were incubated with the secondary anti-rabbit antibody, washed again, and developed (Dako). IFI-44 and CCL18 staining were scored in a blinded manner on a scale from 0 (no staining) to 3+ (strongest staining) across the entire stained lung section.
Statistical analysis
Comparisons of qPCR expression were analyzed by one-way analysis of variance with Bonferroni adjustment for multiple comparisons. Two-group comparisons were analyzed by Student’s t-test. Correlations were tested and presented as Pearson’s correlation coefficient (r) or as coefficient of correlation (r2).
RESULTS
Baseline characteristics of patients with SSc-related ILD
A timeline of the study is summarized in Figure 1A. Demographic data, PFT results, and FibMax values before and after treatment are summarized in Table 1. All patients with SSc-related ILD were female; half (52%) had dcSSc and 48% were positive for anti–Scl-70 (anti–topoisomerase I) antibody. Only 1 patient was an active cigarette smoker and 2 were ex-smokers. There was no significant difference in disease status at the end of the study between patients treated with cyclophosphamide (for NSIP) and those treated with intense anti–gastroesophageal reflux measures (for centrilobular fibrosis) (P > 0.05) (data not shown) (see ref. 11). Despite treatment, most patients with SSc-related ILD developed progressive disease based on the Fib-Max, with a mean ± SD baseline score of 6.88 ± 4.47 compared to a posttreatment score of 12.76 ± 6.59 (P = 0.006). The mean ± SD % predicted FVC in patients with SSc-related ILD was stable from before treatment to after treatment (70.57 ± 10.59 versus 69.33 ± 16.43, respectively; P = 0.77). Samples were obtained from the healthy lungs of 3 female controls and 1 male control; the mean ± SD age of the healthy controls and patients with SSc-related ILD was similar (51 ± 8 years versus 44 ± 9.4 years; P = 0.16). One of the controls was an active cigarette smoker and 3 were ex-smokers. Their mean ± SD % predicted FVC was within the normal range (85.66 ± 6.5).
Table 1.
Clinical features of the patients before lung biopsy and after treatment*
| Disease, patient/age | Clinical features before lung biopsy
|
Clinical features after treatment
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Disease duration, years | Subtype | Anti–topo I | Tobacco use | MRSS | FVC, % predicted | DLco, % predicted | FibMax | FVC, % predicted | FibMax | |
| NSIP | ||||||||||
| 1/42 | 3 | dcSSc | + | No | 23 | 72 | NA | 2 | NA | 5 |
| 2/50 | 15 | dcSSc | + | Former | 15.5 | 88 | NA | 10 | 53 | NA |
| 3/44 | 10 | dcSSc | – | Former | 45 | 68 | 77 | NA | 60 | 13 |
| 4/41 | 8 | dcSSc | – | No | 24 | 70 | 44 | 4 | NA | 5 |
| 5/27 | 1 | lcSSc | – | No | 19 | 84 | 51 | 5 | 73 | 12 |
| 6/55 | 16 | lcSSc | – | No | 12 | 61 | 45 | 9 | 80 | 16 |
| 7/53 | 7 | lcSSc | – | No | 5 | 55 | 38 | 14 | 48 | NA |
| 8/57 | 6 | dcSSc | – | No | 21 | 84 | 59 | NA | 86 | NA |
| 9/43 | 1 | dcSSc | – | No | 46 | 78 | NA | 7 | 73 | 31 |
| 10/46 | 4 | lcSSc | + | No | 10 | 70 | NA | 7 | 38 | 16 |
| 11/45 | 1 | lcSSc | – | No | 0 | 74 | 91 | 7 | 75 | 8 |
| 12/24 | 13 | dcSSc | + | No | 34.5 | 68 | 74 | 2 | 64 | NA |
| 13/30 | 7 | lcSSc | – | Yes | 17 | 74 | NA | 18 | 75 | 14 |
| 14/57 | 9 | lcSSc | + | No | 7 | 61 | NA | 5 | 88 | 9 |
| 15/40 | 6 | dcSSc | + | No | 16 | 70 | 63 | 2 | 51 | 13 |
| 16/41 | 2 | dcSSc | + | No | 28.5 | 65 | 74 | 2 | NA | NA |
| CLF | ||||||||||
| 1/38 | 5 | lcSSc | + | No | 4 | 48 | 39 | NA | NA | NA |
| 2/48 | 3 | dcSSc | + | No | 27 | 54 | 41 | 5 | NA | NA |
| 3/53 | 23 | lcSSc | – | No | 13.5 | 81 | 89 | NA | 105 | NA |
| 4/51 | 13 | lcSSc | – | No | 6.5 | 76 | 66 | 7 | 60 | 10 |
| 5/31 | 3 | dcSSc | + | No | 12 | 81 | 67 | 11 | 86 | 14 |
Anti–topo I = anti–topoisomerase I; MRSS = modified Rodnan skin thickness score; FVC = forced vital capacity; DLco = diffusing capacity for carbon monoxide; FibMax = sum of all scores in 6 lung zones; NSIP = nonspecific interstitial pneumonia; dcSSc = diffuse cutaneous systemic sclerosis; NA= not available; lcSSc = limited cutaneous systemic sclerosis; CLF= centrilobular fibrosis.
Distinct gene expression patterns in patients with SSc-related ILD
Lung microarray data were first analyzed for genes that could differentiate patients with SSc-related ILD from healthy controls. The most informative genes were defined as those with a false discovery rate (FDR) of <0.05 (18) (13,732 genes in total). Genes were hierarchically clustered for both samples and genes using complete linkage and unsupervised clustering. Genes with a range of expression >500 (by Affymetrix expression algorithm) were analyzed in order to focus the analysis on genes showing relatively large changes in expression and to minimize the chance of false-positive or nonspecific results (total of 9,528 genes).
The major dendrogram bifurcation (Figure 1B) placed samples from all healthy controls (represented by dark blue) onto the branch at the left along with 2 samples (from the middle and lower lobes) from 1 SSc patient with NSIP (represented by purple). The right branch, including only samples from patients with SSc-related ILD, bifurcated further into a middle sub-branch, which included biopsy samples from both middle and lower lobes from 2 SSc patients with NSIP (represented by pink and light blue) as well as single-lobe biopsy samples from 2 SSc patients with NSIP (represented by green [lower lobe] and orange [middle lobe]). In contrast, the sub-branch at the far right included 3 of 6 samples from patients classified as having SSc with centrilobular fibrosis (1 sample represented by light green and 2 samples represented by red, where the patient represented by red had samples from both middle and lower lobes clustered together). The other 3 samples on this branch (represented by black, green, and orange) were obtained from the lower lobes of SSc patients with NSIP.
Figure 1B shows some up-regulated genes selected by potential biologic relevance and/or previously described to be up-regulated in SSc, including genes associated with macrophage activation, such as CD163 (cluster of differentiation 163), AIF-1 (allograft inflammatory factor 1), CD86 (cluster of differentiation 86), and MS4A4A (membrane-spanning 4-domains, subfamily A, member 4A). Several chemokines potentially contributing to leukocyte infiltration were also up-regulated in SSc-related ILD, including CCL18 (chemokine [C-C motif] ligand 18), CCL13 (chemokine [C-C motif] ligand 13), and CXCL5 (chemokine [C-X-C motif] ligand 5). Genes related to IFN signaling included IFNAR2 (IFN-α/β receptor β-chain), OAS1 (2′,5′-oligoadenylate synthetase 1, 40/46 kd), IL18 (interleukin 18 [IFN-γ–inducing factor]), and TLR7 (Toll-like receptor 7). Several genes associated with fibrosis and TGFβ regulation were also up-regulated in SSc-related ILD, including several collagen genes (COL5A2 [collagen type V, α2], COL3A1 [collagen type III, α1], and COL1A1 [collagen type I, α1]) as well as SPP1 (secreted phosphoprotein 1/osteopontin) and COMP (cartilage oligomeric matrix protein). SSc patients with centrilobular fibrosis were not analyzed further because the pathogenesis of centrilobular fibrosis has been postulated to be distinct from that of NSIP (11). A list of the top 200 genes with higher expression in SSc patients with NSIP than in healthy controls is available online at http://www.bu.edu/cort/supplementarytables/.
Prospective cohort of patients with SSc-related ILD
Two of the most important questions facing clinicians treating SSc-related ILD are whether lung disease is active and whether the patient’s clinical disease is likely to progress or remain stable. Therefore, we evaluated the association of gene biomarkers with changes in the FibMax values, the sum of all fibrosis scores in the 6 lung zones (see Patients and Methods). The change in the FibMax value (the followup FibMax minus the baseline FibMax) for each patient, when available, was correlated with the same patient’s lung gene expression profile. A similar analysis was performed with changes in the FVC for each patient (change in % predicted FVC).
Correlation of lung gene expression with change in FibMax
Genes with an FDR of <0.05 (18) (13,732 genes in total) were further analyzed using Pearson’s correlation, comparing lung gene expression from the microarray data with the corresponding change in Fib-Max for each SSc patient with NSIP. We preselected genes with at least 2-fold greater induction in SSc patients with NSIP than in healthy controls, both to focus the analysis on genes showing relatively large changes in expression and to minimize the chance of false-positive or nonspecific results. A list of the 100 genes with the highest positive correlation between expression and change in FibMax in SSc patients with NSIP is available online at http://www.bu.edu/cort/supplementarytables/.
Strong correlation between clusters of genes and progression of SSc with NSIP based on change in FibMax
To better explore the several pathways relevant to the progression of SSc with NSIP, the same genes with an FDR of <0.05 (18) that showed greater expression in SSc patients with NSIP than in healthy controls and whose expression had a Pearson’s correlation coefficient of >0.4 with the change in FibMax (1,859 genes), were further selected and clustered using complete linkage, hierarchical, supervised clustering, grouping healthy subjects and SSc patients with NSIP. This analysis showed 3 main clusters that distinguished the 2 groups. These were classified as collagen, IFN, and macrophage clusters.
Collagen signature
Several collagen genes clustered together (Figure 2A). Microarray results were confirmed by qPCR in a larger group of SSc patients with NSIP (n = 25) overexpressing COL1A1 compared to healthy controls (n = 4) (Figure 2C). Figure 2F shows a positive linear correlation (r2 = 0.39, P < 0.01) between COL1A1 mRNA expression by qPCR and the change in FibMax.
Figure 2.
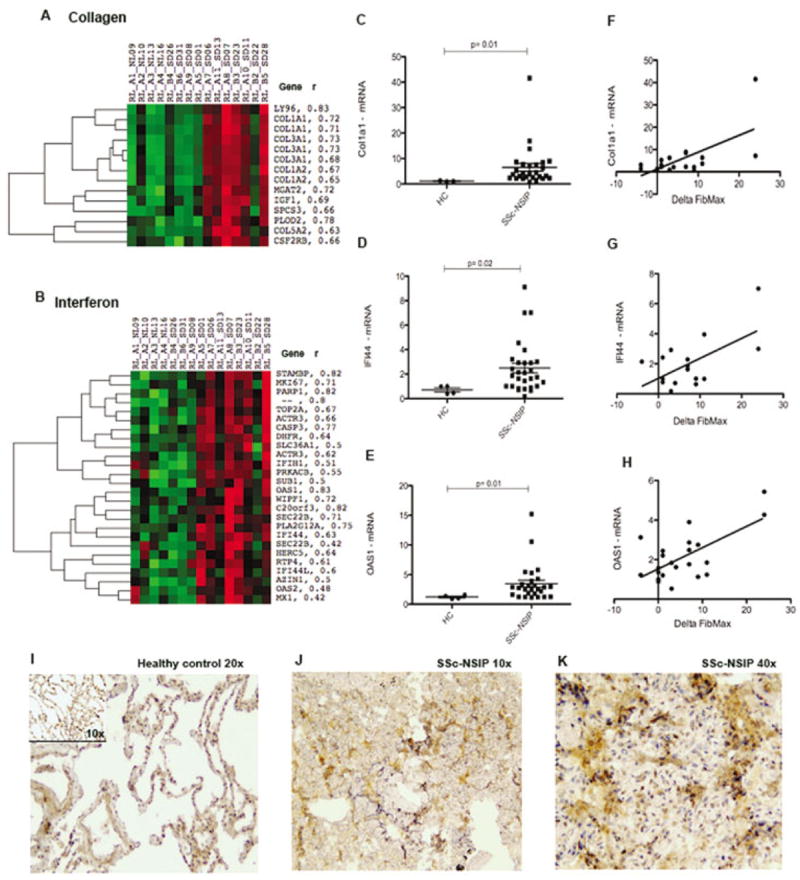
Collagen and interferon (IFN) gene clusters highly expressed in SSc patients with NSIP. A and B, Heatmaps showing the expression of genes clustered using complete linkage, hierarchical, supervised clustering. Names of genes and Pearson correlation coefficients (r) are shown at the right of the collagen (A) and IFN (B) clusters. C–E, Messenger RNA expression of COL1A1 (C), IFI44 (D), and OAS1 (E) in samples obtained from lungs of healthy controls (HC) (n = 4) and SSc patients with NSIP (n = 25) using a larger population. Data are expressed as the fold change normalized to mRNA expression in a single sample from a healthy control. Symbols represent individual samples. Bars show the mean ± SD. F–H, Linear regression analysis of the relationship between the change in FibMax and lung mRNA expression of COL1A1 (r2 = 0.39, P = 0.005) (F), IFI44 (r2 = 0.36, P = 0.009) (G), and OAS1 (r2 = 0.44, P = 0.007) (H). I, Cells staining positive for IFI44 (brown) in a healthy control lung. J and K, Strong staining for IFI44 (brown) within the interstitium and peribronchiolar areas of the lung of an SSc patient with NSIP. See Figure 1 for other definitions.
IFN signature
An IFN gene signature has already been shown in the skin and blood of SSc patients (8,9,19). Surprisingly, in our cohort, many IFN-regulated genes correlated strongly with the change in FibMax and clustered together (Figure 2B). The genes found in this cluster include many well-known IFN-regulated genes: IFI44, OAS2 (2′,5′-oligoadenylate synthetase 2, 69/71 kd), MX1 (myxovirus [influenza virus] resistance 1, IFN-inducible protein p78 [mouse]), and OAS1. The IFN-regulated genes IFI44 and OAS1 were further confirmed by qPCR in a larger group of patients (Figures 2D and E). The genes IFI44 (r2 = 0.44, P < 0.01) and OAS1 (r2 = 0.36, P < 0.01) correlated positively with the change in FibMax (Figures 2G and H).
IFI-44 protein is highly expressed in SSc patients with NSIP
We confirmed robust expression of IFI-44 protein in SSc patients with NSIP (Figures 2J and K) compared to healthy controls (Figure 2I). Lung samples from 5 SSc patients with NSIP and 4 healthy controls were analyzed and scored from 0 (no staining) to 3+ (bright and/or diffuse). Samples from SSc patients with NSIP had stronger IFI-44 staining than did those from healthy controls (mean ± SD 2.66 ± 0.57 versus 0.66 ± 0.57; P = 0.04). An isotype control for IFI-44 staining confirmed specificity (data not shown).
Activated macrophages
Several alternatively activated macrophage markers correlated strongly with the progression of SSc with NSIP and were highly upregulated (further data available online at http://www.bu.edu/cort/supplementarytables/). Most of these genes clustered together, including CD86, CD163, MS4A4A, CCR1 (chemokine [C-C motif] receptor 1; the main receptor for macrophage chemoattractive chemokines), and the chemokine CCL18 (Figure 3A), which is mainly expressed by macrophages (20). Two genes associated with alternatively activated macrophages were further confirmed by qPCR in a larger group of patient lung biopsy samples. CD163 (Figure 3B) and CCL18 (Figure 3C) were highly up-regulated in SSc patients with NSIP compared to healthy controls. A positive linear correlation with the change in FibMax was observed for CD163 (r2 = 0.24, P = 0.03) and CCL18 (r2 = 0.20, P = 0.05) (Figures 3D and E).
Figure 3.
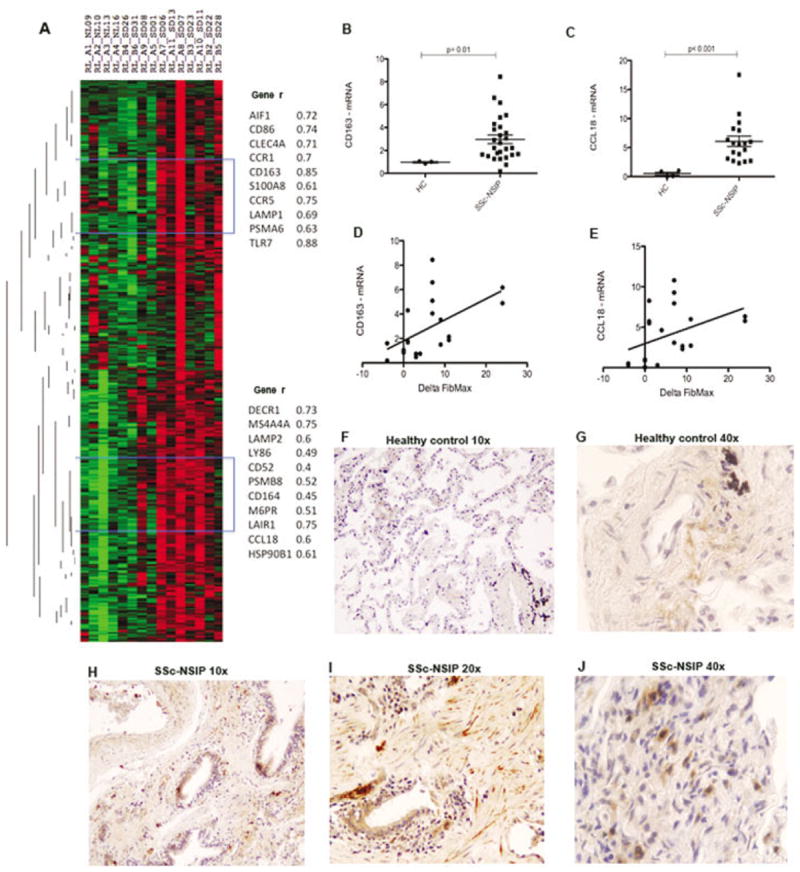
Macrophage gene cluster highly expressed in SSc patients with NSIP. A, Heatmap showing the expression of genes clustered using complete linkage, hierarchical, supervised clustering. Names of selected genes and Pearson correlation coefficients (r) are shown at the right. Blue brackets represent the position of genes described at right. B and C, Messenger RNA expression of CD163 (B) and CCL18 (C) in samples obtained from lungs of healthy controls (HC) (n = 4) and SSc patients with NSIP (n = 25) in a larger population. Data are expressed as the fold change normalized to mRNA expression in a single sample from a healthy control. Symbols represent individual samples. Bars show the mean ± SD. D and E, Linear regression analysis of the relationship between the change in FibMax and lung mRNA expression of CD163 (r2 = 0.24, P = 0.03) (D) and CCL18 (r2 = 0.20, P = 0.05) (E). F and G, Few cells staining positive for CCL18 (brown) in a healthy control lung. H–J, Cells staining positive for CCL18 (brown) within the interstitium and peribronchiolar areas of the lung of an SSc patient with NSIP. See Figure 1 for other definitions.
Strong up-regulation of protein expression of the activated macrophage marker–regulated gene CCL18 in SSc patients with NSIP
We confirmed robust protein expression of CCL18 in SSc patients with NSIP (Figures 3H–J) compared to healthy controls (Figures 3F and G). Bronchiolar cells (Figures 3H and I), fibroblast-like cells within the interstitium (Figure 3I), and immune cells (Figure 3I) were strongly positive for CCL18 in SSc patients with NSIP. Lung samples from 5 SSc patients with NSIP and 4 healthy controls were analyzed and scored from 0 (no staining) to 3+ (bright and/or diffuse). Samples from SSc patients with NSIP showed stronger CCL18 staining than those from healthy controls (mean ± SD 2.25 ± 0.88 versus 1.12 ± 0.25; P = 0.01). An isotype control for CCL18 was used to confirm specificity (data not shown).
Correlation of lung gene expression with change in % predicted FVC
We also examined the correlation between the change in % predicted FVC (the followup % predicted FVC minus the baseline % predicted FVC) of each patient, when available, with the same patient’s lung gene expression profile. We analyzed genes with an FDR of <0.05 (18) with at least a 2-fold greater induction in SSc patients with NSIP than in healthy controls. A list of the top 50 highly expressed genes correlating negatively with change in % predicted FVC (r < −0.1) in SSc patients with NSIP is available online at http://www.bu.edu/cort/supplementarytables/. Compared to the numbers of genes whose expression correlated strongly with the change in FibMax, the expression of relatively few genes had a strong negative correlation (r < −0.4) with change in % predicted FVC. Relatively small changes in % predicted FVC were detected during the study period in these patients, technically limiting the ability to detect statistically significant correlations. The expression of several genes correlated inversely, although not strongly, with the change in % predicted FVC (further data available online at http://www.bu.edu/cort/supplementarytables/).
Correlation of common gene expression with progression of SSc with NSIP
A gene signature that correlates with both a loss of lung function and progression of fibrosis on HRCT would likely best represent the degree of lung injury. We found several genes from the microarray analyses that correlated with both the change in FibMax (Pearson’s r > 0.4) and the change in % predicted FVC (Pearson’s r < −0.1) (Table 2). IFN and macrophage markers, chemokines, interleukins, and heat-shock proteins were among this group of genes (Table 2).
Table 2.
Common genes correlated with both change in FibMax and change in % predicted FVC*
| Gene symbol | Gene name | r with change in FibMax | r with change in % predicted FVC |
|---|---|---|---|
| OAS1 | 2′,5′-oligoadenylate synthetase 1, 40/46 kd | 0.77 | −0.13 |
| AOX1 | Aldehyde oxidase 1 | 0.58 | −0.39 |
| CD163 | CD163 molecule | 0.85 | −0.20 |
| CCL18 | Chemokine (C-C motif) ligand 18 (pulmonary- and activation-regulated) | 0.60 | −0.14 |
| CFI | Complement factor I | 0.69 | −0.14 |
| FAM20B | Family with sequence similarity 20, member B | 0.73 | −0.12 |
| HSP90B1 | Heat-shock protein 90 kd β (Grp94), member 1 | 0.61 | −0.22 |
| HIST1H2AC | Histone cluster 1, H2ac | 0.89 | −0.29 |
| IL18 | Interleukin 18 (interferon-γ–inducing factor) | 0.56 | −0.32 |
| IDH1 | Isocitrate dehydrogenase 1 (NADP+), soluble | 0.74 | −0.53 |
| MS4A4A | Membrane-spanning 4-domains, subfamily A, member 4A | 0.75 | −0.24 |
| NCK1 | NCK adaptor protein 1 | 0.72 | −0.28 |
| PTPRO | Protein tyrosine phosphatase, receptor type, O | 0.77 | −0.16 |
| RETN | Resistin | 0.62 | −0.22 |
| RARRES1 | Retinoic acid receptor responder (tazarotene induced) 1 | 0.84 | −0.26 |
See Table 1 for definitions.
Highly expressed genes in SSc with NSIP not correlated with progression
In the lungs, several genes were highly up-regulated in SSc patients with NSIP compared to healthy controls (further data available online at http://www.bu.edu/cort/supplementarytables/). These included COL17A1 (collagen type XVII, α1), which had a mean fold increase of 9.79 but whose expression showed no correlation with the change in FibMax (r = −0.15, P > 0.05). MMP7 (matrix metallo-peptidase 7) was highly up-regulated in SSc patients with NSIP compared to controls (mean fold increase 6.9), although its expression was not correlated with the change in FibMax (r = 0.01, P > 0.05). In addition, COMP, a TGFβ-regulated gene highly expressed in SSc skin (21), was also up-regulated in the lungs (mean fold increase 4.72); however, its expression did not show a significant correlation with progressive lung disease as measured by the change in FibMax (r = −0.06, P > 0.05).
DISCUSSION
Our unique cohort of SSc patients with NSIP who had lung gene expression in an early fibrotic stage along with prospective clinical parameters revealed 3 main pathways associated with progressive lung disease: macrophage activation, IFN-regulated gene expression, and profibrotic/TGFβ-regulated gene expression. Since the lung tissues analyzed here were not from SSc patients with end-stage lung disease and the samples were obtained avoiding macroscopic honeycombing sections (11), they provide valuable information regarding the molecular mechanisms involved in progressive SSc pulmonary fibrosis, which is well known to be strongly related to mortality (1-3).
ILD is still the main cause of mortality in SSc, with ~42% of patients with SSc-related ILD dying of progressive lung disease within 10 years of diagnosis (22). Cyclophosphamide therapy for SSc-related ILD, while showing some efficacy in 2 prospective randomized clinical trials, provides only modest benefit, highlighting the need for better therapies based on improved understanding of pathogenesis (23,24). In addition, it is currently difficult to distinguish patients with SSc-related ILD whose disease will progress from those with slowly progressive or stable disease. Our unique cohort of SSc patients with NSIP, despite its small numbers, enabled us to identify 3 main pathways strongly involved in the advance of lung fibrosis. Moreover, our findings strongly implicate innate immune activation in this process, which is consistent with the already described IFN signature and alternatively activated macrophage markers in the blood and skin of SSc patients that strikingly distinguish them from healthy controls (8,9,19,20).
Macrophage markers showed increased expression and correlated strongly with progressive lung fibrosis in SSc patients with NSIP. CCL18 was also up-regulated in the lungs of SSc patients with NSIP, and expression correlated with worsening of lung disease. Alternatively activated macrophages are known to release CCL18, which attracts immune cells including monocytes (25) and was also shown to directly stimulate collagen production in fibroblasts (26,27). Previous studies have shown that serum CCL18 levels predict the progression of idiopathic pulmonary fibrosis (28), and they have been suggested as a biomarker for SSc patients at high risk of progressive SSc-related ILD (29). Other studies have implicated macrophages indirectly in the development and progression of SSc-related ILD. Gene expression profiles of bronchoalveolar lavage fluid from patients with SSc-related ILD showed increased expression of chemokines and their receptors, which is consistent with activation of alveolar macrophages (30). These activated macrophages have been shown to spontaneously produce elevated levels of chemokines and increased expression of CD206 (macrophage mannose receptor 1), an alternatively activated macrophage marker, in several fibrotic lung diseases including SSc-related ILD (31). In addition, monocytes from the blood of patients with SSc-related ILD display a fibrogenic phenotype characterized by expression of CD163, another alternatively activated macrophage marker, and by enhanced secretion of CCL18 and interleukin-10 in response to proinflammatory activation (32). Taken together, consistent with previous findings, our data show that alternatively activated macrophages are heavily recruited to the lungs of patients with SSc-related ILD and that the monocyte chemoattractant CCL18 is highly up-regulated in these lungs, strongly implicating alternatively activated macrophages and CCL18 directly in progressive SSc-related ILD.
In SSc patients with NSIP, we also observed high levels of IFN-regulated genes that correlated with progressive lung fibrosis, suggesting a pathogenic role of IFN. Other studies have implicated type I IFNs indirectly in SSc-related ILD, showing that sera containing anti–topoisomerase I antibody, which is known to be associated with dcSSc and with ILD (33), induce IFNα in cultures of human PBMCs (34). More importantly, IFN-inducing activity was observed more frequently in SSc patients with ILD (34). Plasmacytoid dendritic cells were the cells responsible for this high IFNα production in SSc patients with ILD (35), and high levels of IFNα, associated with high expression of CCL2, were associated with the presence of lung fibrosis (35). Despite several in vitro studies showing that IFNα reduces the expression of TGFβ and procollagen type I mRNA in fibrosis and that IFNα also inhibits macrophage TGFβ mRNA expression (36,37), low-dose IFNα has been shown to have a profibrotic effect in the intratracheal bleomycin lung fibrosis model (38,39). Overall, our findings in lung tissue from SSc patients with NSIP are consistent with an important role of IFN in the progression of fibrotic lung involvement in SSc. Based on the paradoxical effect of IFN reported in the literature, we speculate that type I IFN may have a complex effect on lung fibrosis but could be involved in macrophage activation and in other profibrotic pathways.
Collagens compose a third cluster of genes showing increased expression in progressive SSc with NSIP. This finding was not surprising, since it is well described that collagens, mainly type I collagen, are highly induced in several types of fibrosis, including lung fibrosis (40). Increased collagen might have 2 possible explanations. It might reflect just a relatively late and irreversible final phase of organ fibrosis, or it might reflect an event crucial to progressive lung disease. Since we observed that high levels of type I collagen correlated positively with worsening of the HRCT score, it appears that collagen expression is a marker of progressive lung disease. More importantly, both macrophage and collagen signatures might reinforce a novel paradigm suggested in lung fibrosis, where collagen-producing leukocytes, mainly cells expressing monocyte/macrophage markers, mediate fibrosis (41,42). In addition, using a specific TGFβ1-transgenic mouse model (41) and the adenoviral TGFβ lung fibrosis model (43), macrophage deletion has been found to have remarkable effects on collagen deposition, fibrosis score, and myofibroblast hyperplasia. Taken together, these signatures might represent a common event, the activation of macrophages that ultimately develop a profibrotic phenotype.
The only other study examining gene expression in fibrotic lungs of SSc patients showed a distinct lung gene signature in patients compared to healthy controls (10), with most of the top 20 highly expressed genes, such as collagens, metalloproteinases, and growth factors, involved in extracellular matrix deposition. Our results also showed similar strong up-regulation of several collagens, MMP7, osteopontin (SPP1), and growth factors. This profibrotic signature in fibrotic lungs is well described, indicating an active tissue remodeling program (44). On the other hand, we observed strong expression of several genes involved in immune activation, with a spectrum that includes interleukins, chemokines, macrophage markers, and IFN-regulated genes, suggesting that immune activation could represent an earlier process that is not as easily detectable in the later stages of SSc lung fibrosis (10).
Baseline gas transfer (DLco) and FVC levels have traditionally been used as measures of disease severity, and reduction of both parameters has been associated with increased mortality in SSc-related ILD (45). However, the wide range of normal PFT values (80–120% of predicted) is a major constraint. Therefore, it has been shown more recently that the extent of fibrosis appears to be the most important prognostic variable for SSc-related ILD (46). More importantly, changes in the visual fibrosis score of pulmonary fibrosis alone were shown to be predictive of the therapeutic response to cyclophosphamide (17,23) and correlated with physiologic lung measures (FVC and total lung capacity) as well as with improvement in dyspnea and skin thickness (17). The lung fibrosis score used in our cohort was adapted from Goldin et al (17). The HRCT images available from our SSc patients with NSIP were complete and accurately showed all 3 defined zones; therefore, the sum of all 3 scores (ground-glass opacities, fibrosis, and honeycomb cysts) from both lungs was used and correlated with gene expression. With this approach, we took into account the entire spectrum of lung damage, as there is still uncertainty regarding the structural basis of ground-glass opacities, which along with pulmonary fibrosis, are the most common CT findings in symptomatic SSc patients (17).
In conclusion, our results provide rare insight into gene expression in patients with SSc-related ILD before the end stage and highlight the several most prominent pathogenic pathways associated with progressive pulmonary fibrosis in SSc-related ILD: macrophage emigration and activation, and up-regulated expression of TGF- and IFN-regulated genes. Taken together, our findings further support the notion that blocking one or more of these pathways may provide the best approach to treating this life-threatening complication.
Acknowledgments
We thank Yuriy Alekseyev, PhD (Department of Pathology and Laboratory Medicine) and Marc Lenburg, PhD (Section of Computational Biomedicine) of Boston University Microarray Resource Core Facility for performing all microarray-based experiments and help with data analysis.
Supported by the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases [NIAMS] Center of Research Translation grant 1-P50-AR-060780-01 and NIAMS grant 2-R01-AR-051089-06A1), the Fundação de Amparo à Pesquisa do Estado de São Paulo, and the Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Christmann had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Christmann, Sampaio-Barros, Stifano, Borges, de Carvalho, Kairalla, Parra, Spira, Simms, Capellozzi, Lafyatis.
Acquisition of data. Christmann, Sampaio-Barros, Stifano, de Carvalho, Kairalla, Parra, Capellozzi, Lafyatis.
Analysis and interpretation of data. Christmann, Sampaio-Barros, Stifano, Parra, Capellozzi, Lafyatis.
References
- 1.Ioannidis JP, Vlachoyiannopoulos PG, Haidich AB, Medsger TA, Jr, Lucas M, Michet CJ, et al. Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med. 2005;118:2–10. doi: 10.1016/j.amjmed.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 2.Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69:1809–15. doi: 10.1136/ard.2009.114264. [DOI] [PubMed] [Google Scholar]
- 3.Sampaio-Barros PD, Bortoluzzo AB, Marangoni RG, Rocha LF, Del Rio AP, Samara AM, et al. Survival, causes of death, and prognostic factors in systemic sclerosis: analysis of 947 Brazilian patients. J Rheumatol. 2012;39:1971–8. doi: 10.3899/jrheum.111582. [DOI] [PubMed] [Google Scholar]
- 4.Assassi S, Sharif R, Lasky RE, McNearney TA, Estrada-Y-Martin RM, Draeger H, et al. the GENISOS Study Group. Predictors of interstitial lung disease in early systemic sclerosis: a prospective longitudinal study of the GENISOS cohort. Arthritis Res Ther. 2010;12:R166. doi: 10.1186/ar3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth MD, Tseng CH, Clements PJ, Furst DE, Tashkin DP, Goldin JG, et al. for the Scleroderma Lung Study Research Group. Predicting treatment outcomes and responder subsets in scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63:2797–808. doi: 10.1002/art.30438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clements P, Lachenbruch P, Seibold J, White B, Weiner S, Martin R, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol. 1995;22:1281–5. [PubMed] [Google Scholar]
- 7.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3:e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pendergrass SA, Hayes E, Farina G, Lemaire R, Farber HW, Whitfield ML, et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS One. 5:e12106. doi: 10.1371/journal.pone.0012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christmann RB, Hayes E, Pendergrass S, Padilla C, Farina G, Affandi AJ, et al. Interferon and alternative activation of mono cyte/macrophages in systemic sclerosis–associated pulmonary arterial hypertension. Arthritis Rheum. 2011;63:1718–28. doi: 10.1002/art.30318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63:783–94. doi: 10.1002/art.30159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Souza RB, Borges CT, Capelozzi VL, Parra ER, Jatene FB, Kavakama J, et al. Centrilobular fibrosis: an underrecognized pattern in systemic sclerosis. Respiration. 2009;77:389–97. doi: 10.1159/000156958. [DOI] [PubMed] [Google Scholar]
- 12.Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma) Arthritis Rheum. 1980;23:581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 13.LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 14.Furst DE, Clements PJ, Steen VD, Medsger TA, Jr, Masi AT, D’Angelo WA, et al. The modified Rodnan skin score is an accurate reflection of skin biopsy thickness in systemic sclerosis. J Rheumatol. 1998;25:84–8. [PubMed] [Google Scholar]
- 15.De Carvalho ME, Kairalla RA, Capelozzi VL, Deheinzelin D, do Nascimento Saldiva PH, de Carvalho CR. Centrilobular fibrosis: a novel histological pattern of idiopathic interstitial pneumonia. Pathol Res Pract. 2002;198:577–83. doi: 10.1078/0344-0338-00305. [DOI] [PubMed] [Google Scholar]
- 16.American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. published erratum appears in Am J Respir Crit Care Med 2002;166:426. [DOI] [PubMed] [Google Scholar]
- 17.Goldin JG, Lynch DA, Strollo DC, Suh RD, Schraufnagel DE, Clements PJ, et al. High-resolution CT scan findings in patients with symptomatic scleroderma-related interstitial lung disease. Chest. 2008;134:358–67. doi: 10.1378/chest.07-2444. [DOI] [PubMed] [Google Scholar]
- 18.Pawluk-Kolc M, Zieba-Palus J, Parczewski A. Application of false discovery rate procedure to pairwise comparisons of refractive index of glass fragments. Forensic Sci Int. 2006;160:53–8. doi: 10.1016/j.forsciint.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 19.York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and Toll-like receptor agonists. Arthritis Rheum. 2007;56:1010–20. doi: 10.1002/art.22382. [DOI] [PubMed] [Google Scholar]
- 20.Schutyser E, Richmond A, Van Damme J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. 2005;78:14–26. doi: 10.1189/jlb.1204712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farina G, Lemaire R, Pancari P, Bayle J, Widom RL, Lafyatis R. Cartilage oligomeric matrix protein expression in systemic sclerosis reveals heterogeneity of dermal fibroblast responses to transforming growth factor β. Ann Rheum Dis. 2009;68:435–41. doi: 10.1136/ard.2007.086850. [DOI] [PubMed] [Google Scholar]
- 22.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66:940–4. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655–66. doi: 10.1056/NEJMoa055120. [DOI] [PubMed] [Google Scholar]
- 24.Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclo-phosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–70. doi: 10.1002/art.22204. [DOI] [PubMed] [Google Scholar]
- 25.Schraufstatter I, Takamori H, Sikora L, Sriramarao P, DiScipio RG. Eosinophils and monocytes produce pulmonary and activation-regulated chemokine, which activates cultured monocytes/macrophages. Am J Physiol Lung Cell Mol Physiol. 2004;286:L494–501. doi: 10.1152/ajplung.00323.2002. [DOI] [PubMed] [Google Scholar]
- 26.Atamas SP, Luzina IG, Choi J, Tsymbalyuk N, Carbonetti NH, Singh IS, et al. Pulmonary and activation-regulated chemokine stimulates collagen production in lung fibroblasts. Am J Respir Cell Mol Biol. 2003;29:743–9. doi: 10.1165/rcmb.2003-0078OC. [DOI] [PubMed] [Google Scholar]
- 27.Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173:781–92. doi: 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 28.Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:717–23. doi: 10.1164/rccm.200808-1201OC. [DOI] [PubMed] [Google Scholar]
- 29.Tiev KP, Hua-Huy T, Kettaneh A, Gain M, Duong-Quy S, Toledano C, et al. Serum CC chemokine ligand-18 predicts lung disease worsening in systemic sclerosis. Eur Respir J. 2011;38:1355–60. doi: 10.1183/09031936.00004711. [DOI] [PubMed] [Google Scholar]
- 30.Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol. 2002;26:549–57. doi: 10.1165/ajrcmb.26.5.4683. [DOI] [PubMed] [Google Scholar]
- 31.Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin Immunol. 2010;137:89–101. doi: 10.1016/j.clim.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 32.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 2010;90:812–23. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catoggio LJ, Bernstein RM, Black CM, Hughes GR, Maddison PJ. Serological markers in progressive systemic sclerosis: clinical correlations. Ann Rheum Dis. 1983;42:23–7. doi: 10.1136/ard.42.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA, et al. Induction of interferon-α by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon-α activity with lung fibrosis. Arthritis Rheum. 2008;58:2163–73. doi: 10.1002/art.23486. [DOI] [PubMed] [Google Scholar]
- 35.Eloranta ML, Franck-Larsson K, Lovgren T, Kalamajski S, Ronnblom A, Rubin K, et al. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis. 2010;69:1396–402. doi: 10.1136/ard.2009.121400. [DOI] [PubMed] [Google Scholar]
- 36.Jimenez SA, Freundlich B, Rosenbloom J. Selective inhibition of human diploid fibroblast collagen synthesis by interferons. J Clin Invest. 1984;74:1112–6. doi: 10.1172/JCI111480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castilla A, Prieto J, Fausto N. Transforming growth factors β1 and α in chronic liver disease: effects of interferon alfa therapy. N Engl J Med. 1991;324:933–40. doi: 10.1056/NEJM199104043241401. [DOI] [PubMed] [Google Scholar]
- 38.Berkman N, Goldstein RH, Breuer R. Bleomycin-induced lung injury is enhanced by interferon-α. Life Sci. 1997;60:PL415–21. doi: 10.1016/s0024-3205(97)00332-9. [DOI] [PubMed] [Google Scholar]
- 39.Berkman N, Kremer S, Or R, Lossos IS, Christensen TG, Goldstein RH, et al. Human recombinant interferon-α2a and interferon-αA/D have different effects on bleomycin-induced lung injury. Respiration. 2001;68:169–77. doi: 10.1159/000050488. [DOI] [PubMed] [Google Scholar]
- 40.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–50. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, et al. TGF-β driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol. 2011;43:154–62. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 42.Peng X, Mathai SK, Murray LA, Russell T, Reilkoff R, Chen Q, et al. Local apoptosis promotes collagen production by monocyte-derived cells in transforming growth factor β1-induced lung fibrosis. Fibrogenesis Tissue Repair. 2011;4:12. doi: 10.1186/1755-1536-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569–81. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 44.Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2006;173:188–98. doi: 10.1164/rccm.200504-644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan C, Knight C, Lunt M, Black CM, Silman AJ. Predictors of end stage lung disease in a cohort of patients with scleroderma. Ann Rheum Dis. 2003;62:146–50. doi: 10.1136/ard.62.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177:1248–54. doi: 10.1164/rccm.200706-877OC. [DOI] [PubMed] [Google Scholar]