

Abstract
The application of non-viral gene delivery vectors is often accompanied with the poor correlation between transfection efficiency and the safety profiles of vectors: vectors with high transfection efficiencies often suffer from high toxicities, making it unlikely to improve their efficiencies by increasing the DNA dosage. In the current study, we developed a ternary complex system which consisted of a highly membrane-active cationic helical polypeptide (PVBLG-8), a low-toxic, membrane-inactive cationic helical polypeptide (PVBLG-7) capable of mediating mannose receptor targeting, and DNA. The PVBLG-7 moiety notably enhanced the cellular uptake and transfection efficiency of PVBLG-8 in a variety of mannose receptor-expressing cell types (HeLa, COS-7, and Raw 264.7), while it did not compromise the membrane permeability of PVBLG-8 or bring additional cytotoxicities. Because of the simplicity and adjustability of the self-assembly approach, optimal formulations of the ternary complexes with a proper balance between membrane activity and targeting capability were easily identified in each specific cell type. The optimal ternary complexes displayed desired cell tolerability and markedly outperformed the PVBLG-8/DNA binary complexes as well as commercial reagent Lipofectamine™ 2000 in terms of transfection efficiency. This study therefore provides an effective and facile strategy to overcome the efficiency-toxicity poor correlation of non-viral vectors, which contributes insights into the design strategy of effective and safe non-viral gene delivery vectors.
Keywords: non-viral gene delivery, α-helical polypeptide, self-assembly, membrane penetration, mannose targeting, cytotoxicity
1. Introduction
Gene therapy has shown great potentials in treating various genetic diseases, such as cystic fibrosis, diabetes, arthritis, immunological deficiency, and cancer [1–6].3 Compared to viral vectors, non-viral gene delivery vectors allow safe delivery of genetic materials with less inherent immunogenicity and oncogenicity. Polycations, capable of condensing the anionic nucleic acids to facilitate their intracellular uptake, are one of the most widely explored non-viral vectors. Although the cationic charge of polycations features strong membrane binding of delivery vehicles and facilitates cellular uptake and transfection of the gene cargos, it meanwhile causes severe associated cytotoxicities [7–9]. Excessive positive charges can ultimately undermine the transfection efficiency [10]. Therefore, it is of particular importance to balance the charge related membrane activity and cytotoxicity in the design of non-viral vectors such that the gene delivery efficiency could be maximized. One promising approach towards this goal is the combinatorial/parallel synthesis that creates a large library of materials and allows identification of the best-performing candidate via screening [11, 12]. This technology, although promising, requires tedious task and often suffers from high cost. Alternatively, covalent modification of existing polycations with various charge-reducing moieties—including saccharides [13], hydrocarbons [14], and poly(ethylene glycol) (PEG) [15–17], stands as an effective tool to reduce their toxicities. While the modified polycations benefit from improved safety profiles, they typically suffer from diminished gene delivery capabilities [10]. All these challenges thus necessitate a facile and effective strategy for the development of non-viral vectors which can properly balance the membrane activity and toxicity towards maximized gene delivery efficiency.
Cell penetrating peptides (CPPs), exemplified by HIV-TAT, Arg9, penetratin, and melittin, are sequence-specific short oligopeptides that mediate effective membrane penetration and translocation via either energy-dependent endocytosis or energy-independent transduction [18]. Due to their excellent membrane activities, CPPs are able to facilitate the cellular delivery of a variety of exogenous materials, including metals, macromolecules (e.g., proteins and nucleic acids), and nanoparticles [19, 20]. However, when used as gene transfer agents, CPPs are often too short (fewer than 25 amino acid residues) and lack sufficient cationic charge density, which raises great challenges for CPPs to condense and deliver genes by themselves. As such, they often act as membrane-active ligands incorporated or conjugated to existing delivery vehicles to enhance their delivery efficiencies [19, 21, 22]. To address the dearth of CPP-mediated non-viral gene delivery, we recently developed a cationic polypeptide, poly(γ-(4-(((2-(piperidin-1-yl) ethyl) amino) methyl) benzyl-L-glutamate) (PVBLG-8), via a controlled ring-opening polymerization method (Fig. 1A) [23–25] and used PVBLG-8 or its analogues in gene and siRNA delivery [26–28]. PVBLG-8 with stabilized helical structure exhibited desired membrane activity and thus triggered effective cellular uptake as well as gene transfection, which rendered it a better gene delivery vector than traditional oligo-CPPs [23]. However, the appreciable cytotoxicity of PVBLG-8 at higher concentrations makes it unlikely to strength the gene delivery capabilities by increasing the amount used [26], which thus necessitates alternative approaches to maximize its gene transfer efficiencies without causing additional cytotoxicities.
Fig. 1.

PVBLG-8 allows effective transfection while induces appreciable cytotoxicity. (A) Reaction scheme of PVBLG-8. (B) CD spectrum of PVBLG-8 in DI water (0.1 mg/mL) at pH 7. (C) FITC-Tris uptake level in HeLa cells following co-incubation with PVBLG-8 for 2 h at 37 °C. (D) DNA condensation by PVBLG-8 at different PVBLG-8/DNA weight ratios as evaluated by the gel retardation assay. N represents naked DNA. (E) Particle size and zeta potential of PVBLG-8/DNA complexes. (F) Uptake level of PVBLG-8/YOYO-1-DNA complexes in HeLa cells following incubation at 37 °C for 4 h (n=3). (G) In vitro transfection efficiency of PVBLG-8/pEGFP complexes in HeLa cells at different weight ratios (n=3). (H) In vitro cytotoxicity of PVBLG-8/pEGFP complexes in HeLa cells at different weight ratios as determined by the MTT assay (n=3).
With an attempt to balance the transfection efficiency and cytotoxicity, we first developed a PVBLG-8-based random copolypeptide (PVBLG-8-r-7) (Fig. 2A) which contains glucosamine side chains that allow mannose receptor targeting as well as reduce the material cytotoxicity via saccharide-mediated charge shielding. Although the cytotoxicity was slightly decreased, this approach shares the similar disadvantage of PEGylation and the resulting copolypeptide demonstrated decreased membrane activity and transfection efficiency compared to PVBLG-8, possibly due to the diminished cationic charge density. Based on such findings, we thus seek alternative strategies which could endow PVBLG-8 with cellular targeting functionality while maintain the membrane activity as well as the gene delivery capability [29]. To this end, poly(γ-glucosamine methyl) benzyl-L-glutamate) (PVBLG-7) [25], a helical polypeptide bearing glucosamine residues, was incorporated to form the PVBLG-8/PVBLG-7/DNA ternary complexes via self-assembly instead of constructing the PVBLG-8-r-7 copolypeptide. We hypothesized that incorporation of PVBLG-7, a cationic helical polypeptide with minimized membrane activity, can strengthen the gene delivery capabilities of PVBLG-8 via mannose-receptor-mediated cellular targeting while will not generate additional cytotoxicities. In various mammalian cell types (HeLa, COS-7, and Raw 264.7) that express mannose receptors [30–32], the cellular uptake level, intracellular kinetics, transfection efficiency, and cytotoxicity of ternary complexes were explored and compared to PVBLG-8/DNA binary complexes. Upon an optimized combination between membrane activity and cellular targeting, the top-performing formulation with optimal transfection/toxicity balance is identified. This study thus provides insights into the design strategy of safe and effective non-viral vectors for gene delivery.
Fig. 2.

PVBLG-8-r-7 displays diminished transfection efficiency while improved cell tolerability compared to PVBLG-8. (A) Reaction scheme of PVBLG-8-r-7. (B) In vitro transfection efficiencies of PVBLG-8-r-7/pEGFP complexes and PVBLG-8/pEGFP complexes in HeLa cells (n=3). (C) Cytotoxicity of PVBLG-8-r-7 and PVBLG-8 towards HeLa cells as determined by the MTT assay (n=3). (D) Zeta potential of PVBLG-8-r-7/DNA complexes and PVBLG-8/DNA complexes.
2. Experimental
2.1. Materials and cell lines
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used as received unless otherwise specified. Anhydrous tetrahydrofuran (THF), hexane, and dimethylformamide (DMF) were dried by a column packed with 4Å molecular sieves and stored in a glovebox. Dry nitrobenzene (NB) was prepared by treating regular NB with CaH2 followed by distillation under reduced pressure. Hexamethyldisilazane (HMDS) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were used for controlled ring-opening polymerization of amino acid N-carboxyanhydrides developed by us [25].γ-(4-Vinylbenzyl)-L-glutamate N-carboxyanhydride (VB-L-Glu-NCA) was prepared as previously reported [23, 33]. Pierce BCA assay kit was purchased from ThermoFisher Scientific (Rockford, IL, USA). Plasmid DNA (pDNA) encoding enhanced green fluorescence protein (EGFP) (pEGFP) was purchased from Elim Biopharm (Hayward, CA, USA). Lipofectamine™ 2000 (LPF), 3-(4,5-dimethylthiahiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), and YOYO-1 were purchased from Invitrogen (Carlsbad, CA, USA).
HeLa (human cervix adenocarcinoma cells), COS-7 (African green monkey kidney cells), and Raw 264.7 (mouse leukemic monocyte macrophage cells) were purchased from the American Type Culture Collection (Rockville, MD, USA) and were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
2.2. Synthesis and characterization of polypeptides
2.2.1. Synthesis of PVBLG-8
VB-L-Glu-NCA (58 mg, 0.2 mmol) was dissolved in a mixture of DMF (0.9 mL) and nitrobenzene (30 μL) in a glove box, followed by addition of HMDS (13.3 μL, 0.1 M, M/I=150) and TBD solution (13.3 μL, 0.01 M) in DMF. FTIR was used to monitor the polymerization until the conversion reached 99% (within 48 hours) to obtain poly(γ-(4-vinylbenzyl)-L-glutamate) (PVBLG). 1H NMR (500 MHz, CDCl3/TFA-d (85:15, v/v), δ, ppm): 7.34 (d, 2H, ArH), 7.20 (d, 2H, ArH), 6.65 (m, 1H, –CH=CH2), 5.72 (d, 2H, –CH=CH2), 5.24 (d, 2H, –CH=CH2), 5.04 (m, 2H, ArCH2O-), 4.59 (m, 1H, α-H), 2.45 (t, 2H, –COCH2CH2–), 2.11 (m, 1H, –COCH2CH2–), 1.93 (m, 1H, –COCH2CH2–) (Supplementary Fig. S1). Tetrabutylammonium fluoride solution (100 μL, 1 M), benzyl chloroformate (50 μL), and N,N-diisopropylethylamine (DIEA, 50 μL) were added and stirred for 3 h to cleave the N-Si bond and protect the amino end groups. DMF was removed under vacuum, and the resulting polymer was precipitated from cold ethyl ether (45 mL), washed with cold ethyl ether (45 mL_× 3), and collected by centrifugation at 4000 rpm. PVBLG was then dissolved in chloroform (30 mL) and oxidized by O3 at −78 °C. Dimethyl sulfide (1 mL) was added and the solution was stirred at RT overnight before the solvent was removed under vacuum. The product poly(γ-(4-aldehydebenzyl-L-glutamate) (PABLG) was washed with methanol (45 mL × 3) to remove unreacted dimethyl sulfide and other impurities, and collected by centrifugation. 1H NMR (500 MHz, CDCl3/TFA-d (85:15, v/v)): δ 9.86 (s,1H, ArCHO), 7.95–7.45 (br m, 4H, ArH), 5.16 (br s, 2H, ArCH2O–), 4.61 (m, 1H, α-H), 2.55 (br m, 2H, –COCH2CH2–), 2.16 (m, 1H, –COCH2CH2–), 1.98 (m, 1H, –COCH2CH2–) (Supplementary Fig. S2). The obtained PABLG (30 mg) was dissolved in DMF (2 mL), into which 1-(2-aminoethyl)piperidine (150 μL, 10 molar equivalents relative to the Glu repeating unit) was added. After reaction at 50 °C for 24 h, borane pyridine as the reducing agent (133 μL, 10 molar equivalents relative to the Glu repeating unit) was added, and the solution was further stirred at 50 °C for 24 h. HCl (5 M, 1 mL) was added to protonate the amine groups, and the final product PVBLG-8 was dialyzed against water (MWCO = 1 kDa) and lyophilized. 1H NMR (500 MHz, TFA-d): δ 7.53 (m, 4H, ArH), 5.32 (br s, 2H, ArCH2O–), 4.86 (br s, 2H, ArCH2NH), 4.53 (s, 1H, a–H), 4.01 (s, 4H, –OCH2CH2–), 3.94–3.80 (br m, 6H, –HNCH2CH2N– and –NCH2CH2CH2CH2CH2–), 3.13 (m, 2H, –HNCH2CH2N–), 2.78 (s, 2H, –COCH2CH2–), 2.40 (br m, 2H, –COCH2CH2–), 2.14–1.52 (br m, 6H, –NCH2CH2CH2CH2CH2–) (Supplementary Fig. S3).
2.2.2. Synthesis of PVBLG-7
PABLG (30 mg) was dissolved in DMF (2 mL). Glucosamine hydrochloride (200 mg dissolved in DMSO, 10 molar equivalents relative to the Glu repeating unit) was added. After the solution was stirred at 50 °C for 72 h, the polypeptide was reduced by borane pyridine, protonated with HCl, and dialyzed against water using the same method as described above. 1H NMR (500 MHz, TFA-d): δ 8.35-7.75 (br m, 4H, ArH), 5.54 (m, 2H, OHCHCHCHOH), 5.04 (br m, 2H, ArCH2O–), 4.77 (s, 1H, α–H), 4.49 (br s, 2H, ArCH2NH–), 4.34-3.66(br m, 5H, OHCH2CHCHCHCHCHO-), 2.99 (s, 2H, –COCH2CH2–), 2.32 (br m, 2H, –COCH2CH2–) (Supplementary Fig. S4).
2.2.3. Synthesis of PVBLG-8-r-7
PABLG (30 mg) was dissolved in DMF (2 mL). Glucosamine hydrochloride (60 mg dissolved in DMSO, 3 molar equivalents relative to the Glu repeating unit) was added. After stirring at 50 °C for 24 h, 1-(2-aminoethyl)piperidine (150 μL, 10 molar equivalents relative to the Glu repeating unit) was added, and the solution was stirred at 50 °C for another 24 h. The polypeptide was then reduced by borane pyridine, protonated with HCl, and dialyzed against water in the same method as described above. 1H NMR (500 MHz, TFA-d): δ 8.10-7.50 (m, 4H, ArH), 5.31 (s, 2H, ArCH2O–), 4.88 (m, 2H, OHCHCHCHOH) 4.52-4.26 (br m, 3H, ArCH2NH– and α–H), 4.09-3.81 (br m, 5H, OHCH2CHCHCHCHCHO-), 3.92–3.60 (br m, 6H, –HNCH2CH2N– and –NCH2CH2CH2CH2CH2–), 3.08 (m, 3H, –HNCH2CH2N– and –NHCH), 2.75 (m, 4H, –COCH2CH2–), 2.40 (br m, 2H, –COCH2CH2–), 2.04–1.52 (br m, 6H, –NCH2CH2CH2CH2CH2–) (Supplementary Fig. S5).
2.3. Characterization of polypeptides
1H NMR spectra were recorded on a Varian UI500NB MHz spectrometer. Chemical shifts were reported in ppm and referenced to the solvent proton impurities. Gel permeation chromatography (GPC) experiments were performed on a system equipped with an isocratic pump (Model 1100, Agilent Technology, Santa Clara, CA, USA), a DAWN HELEOS multi-angle laser light scattering (MALLS) detector (Wyatt Technology, Santa Barbara, CA, USA), and an Optilab rEX refractive index detector (Wyatt Technology, Santa Barbara, CA, USA). Separations were performed using serially connected size exclusion columns (100 Å, 500 Å, 103 Å, and 104 Å Phenogel columns, 5 μm, 300 × 7.8 mm, Phenomenex, Torrance, CA, USA) at 60 °C with DMF containing 0.1 M LiBr as the mobile phase. The detection wavelength was set at 658 nm, and the MALLS detector was calibrated using pure toluene, which allowed determination of the absolute molecular weights (MWs) instead of calibration using polymer standards. The MWs of polypeptides were calculated according to the dn/dc value of each polymer using the internal calibration system processed by the ASTRA V software (version 5.1.7.3, Wyatt Technology, Santa Barbara, CA, USA). Circular dichroism (CD) measurements were carried out on a JASCO J-700 CD spectrometer (Oklahoma City, OK, USA). Polypeptide was dissolved in DI water at a concentration of 0.1 mg/mL, and was placed in a quartz cell with a pathlength of 0.1 cm. The mean residue molar ellipticity and helicity of each polymer were calculated based on the measured apparent ellipticity following the reported formula [25]:
2.4. Formulation of binary and ternary complexes
Polypeptides and DNA were dissolved in DI water at 0.2 mg/mL. To form binary complexes, polypeptide was added into the DNA solution at various weight ratios followed by vortex for 30 sec and incubation at RT for 20 min. While for the ternary complexes, PVBLG-8 and PVBLG-7 were mixed at determined weight ratios before they were added to the DNA solution at a fixed PVBLG-8/DNA weight ratio of 15. The mixture was vortexed for 30 sec and incubated at RT for 20 min to obtain the ternary complexes.
2.5. Characterization of binary and ternary complexes
A gel retardation assay was first adopted to evaluate the DNA condensation by cationic polymers. Freshly prepared complexes were loaded on a 1% agarose gel at 100 ng DNA/well followed by electrophoresis at 100 V for 30 min. Naked DNA was used as a control, and DNA migration in the agarose gel was visualized by a Gel Doc imaging system (Biorad, Herclues, CA, USA) following staining with ethidium bromide (EB). To quantitatively measure the DNA condensation level, the EB exclusion assay was performed as follows [17]. DNA was first stained with EB at the DNA/EB weight ratio of 10 and RT for 1 h. PVBLG-8, PVBLG-7, or a mixture of them were added into the DNA/EB solution followed by further incubation at RT for 30 min before quantification of the fluorescence intensity (λex = 510 nm, λem = 590 nm). The DNA condensation efficiency (%) was calculated according to the following equation:
Where FEB, F, and F0 denote the fluorescence intensity of pure EB solution, DNA/EB solution with polypeptide, and DNA/EB solution without any polypeptide, respectively.
Particle size and zeta potential of freshly prepared complexes at various weight ratios were also evaluated by dynamic laser scanning (DLS) on a Malvern Zetasizer (Herrenberg, Germany). To evaluate the complex stability, ternary complexes were diluted with PBS (pH 7.0) by 10, 30, 50, and 100 folds, respectively, incubated at 37 °C for 1 h, and subject to DLS measurement.
2.6. Cell uptake
To allow visualization and quantification of the cellular internalization, DNA (1 mg/mL) was labeled with YOYO-1 (20 μM) at one dye molecule per 50 bp DNA [34] and used to form complexes as described above. Cells were seeded on 24-well plates at 5×104 cells/well and cultured until they reached confluence. The cell culture medium was replaced with Opti-MEM (500 μL/well) into which complexes were added at 0.5 μg YOYO-1-DNA/well. After incubation at 37 °C for 4 h, cells were washed with PBS containing heparin (20 U/mL) for 3 times to remove the surface-bound cationic complexes [35] and lysed with RIPA lysis buffer (500 μL/well) at RT for 20 minutes. The YOYO-1-DNA content in the lysate was monitored by spectrofluorimetry (λex = 485 nm, λem = 530 nm) and the protein level was measured using the BCA kit. Uptake level was expressed as ng DNA associated with 1 mg cellular protein. To explore the PVBLG-7-mediated targeting via mannose-receptor recognition, cells were pre-incubated with Opti-MEM (500 μL/well) supplemented with mannose at different concentrations (100 μM, 200 μM, 400 μM, and 800 μM) for 30 minutes prior to the addition of complexes and throughout the 4-h uptake experiment at 37 °C.
The internalization and intracellular distribution of complexes were also observed by confocal laser scanning microscopy (CLSM). HeLa cells cultured on coverslips in 6-well plate were incubated with complexes in Opti-MEM (2 mL) at 1 μg DNA/well. Following incubation for different time (0.5, 1, 2, and 4 h), cells were washed three times with PBS containing heparin (20 U/mL), fixed with 4% paraformaldehyde, and stained with DAPI (10 μg/mL) before observation by confocal laser scanning microscopy (CLSM, LSM700, Zeiss, Germany).
2.7. Intracellular kinetics
To explore the cellular uptake pathways of complexes, we performed the cell uptake study at low temperature (4 °C) or in the presence of endocytic inhibitors. To block the energy-dependent endocytosis, the cell uptake study was performed at 4 °C during the 2-h period. Otherwise, cells were pre-incubated with endocytic inhibitors including genistein (100 μg/mL), methyl-β-cyclodextrin (mβCD, 5 mM), wortmannin (10 μg/mL), and chlorpromazine (10 μg/mL) for 30 minutes prior to polypeptide application and throughout the 2-h uptake experiment at 37 °C. The cellular uptake level was determined as described above, and results were expressed as percentage uptake of the control cells which were incubated with complexes at 37 °C for 2 h in the absence of endocytic inhibitors.
To explore the endosomal/lysosomal entrapment, HeLa cells were treated with YOYO-1-DNA-containing complexes (1 μg/mL) for 4 h at 37 °C, stained with Hoechst 33258 (5 μg/mL) and Lysotracker® Red (200 nM), and observed by CLSM.
2.8. Membrane activity
The capability of polypeptides to induce membrane disruption was evaluated in terms of the cell uptake level of a hydrophilic, membrane-impermeable dye, fluorescein isothiocyanate (FITC) in its non-reactive form (fluorescein-tris(hydroxymethyl)methanethiourea, FITC-Tris) [18]. Briefly, HeLa cells were seeded on 96-well plates at 1×104 cells/well and cultured for 24 h. The medium was replaced with Opti-MEM (100 μL/well), into which polypeptides and FITC-Tris were added at 2 μg/well and 0.2 μg/well, respectively. Free FITC-Tris without polypeptides was added as a control. After incubation at 37 °C for 2 h, cells were washed with PBS containing heparin (20 U/mL) for 3 times and then lysed with the RIPA lysis buffer (100 μL/well). The FITC-Tris content in the lysate was quantified using spectrofluorimetry (λex = 485 nm, λem = 530 nm) and the protein level was determined using the BCA kit. Uptake level was expressed as ng FITC-Tris associated with 1 mg cellular protein.
2.9. In vitro transfection
Cells were seeded on 24-well plates at 5×104 cells/well and cultured in serum-containing media for 24 h before reaching confluence. The culture medium was changed to Opti-MEM (500 μL/well) into which binary or ternary complexes were added at 0.5 μg DNA/well. After incubation at 37°C for 4 h, the medium was replaced by DMEM containing 10% FBS (500 μL/well) and cells were further incubated for 48 h before assessment of EGFP expression by flow cytometry. The transfection efficiency was expressed as percentage of EGFP-positive cells (%), and the EGFP expression was also observed by fluorescence microscopy. To future explore the PVBLG-7-mediated targeting effect, cells were incubated with complexes in mannose-supplemented Opti-MEM for 4 h as described above and then cultured in serum-containing media for another 48 h before flow cytometry assessment.
2.10. Cytotoxicity
Cells were seeded on 96-well plates at 1×104 cells/well and cultured in serum-containing media for 24 h. The medium was replaced with Opti-MEM (100 μL/well), into which polypeptides or polypeptide/DNA complexes were added at the PVBLG-8 final concentrations of 100, 50, 20, and 10 μg/mL, respectively. After incubation at 37°C for 4 h, the medium was changed to serum-containing DMEM and cells were further cultured for 48 h before viability assessment by the MTT assay. Results were represented as percentage viability of control cells that did not receive polypeptide or complex treatment.
2.11. Statistical analysis
Statistical analysis was performed using Student’s t-test and differences between test and control groups were judged to be significant at *p < 0.05 and very significant at **p < 0.01.
3. Results
3.1. Synthesis and characterization of PVBLG-8 and PVBLG-8/DNA binary complex
PVBLG-8 was synthesized via HMDS-initiated ring-opening polymerization (ROP) of VB-L-Glu-NCA and subsequent side-chain amination [23, 36]. HMDS allowed a well-controlled ROP of VB-L-Glu-NCA, as evidenced by the monomodal peaks in the GPC curves (Supplementary Fig. S6), well-defined MW (degree of polymerization 160 as calculated by GPC), and narrow PDI (<1.2). The conjugation efficiency of 1-(2-aminoethyl)piperidine in PVBLG-8 was determined to be 90% based on 1H NMR analysis (Supplementary Fig. S3). PVBLG-8 exhibited excellent solubility in water at pH lower than 9, and it adopted typical α-helical structure with 99% helicity as evidenced by the characteristic double minima at 208 nm and 222 nm in the CD spectrum (Fig. 1B). The helical structure of PVBLG-8 notably contributed to its desired membrane activity via the pore formation mechanism, leading to appreciable cellular internalization of the membrane impermeable FITC-Tris, (Fig. 1C). The helicity of the PVBLG-8 was remarkably stable against pH change between 1 and 10, indicating that the polypeptide was able to maintain its helicity-dependent membrane activities at both the neutral extracellular pH and the acidic endosomal/lysosomal pH, which thus allowed it to trigger effective intracellular internalization as well as endosomal escape (Supplementary Fig. S7A) [23]. The helicity of PVBLG-8 was also stable against salt concentration increment up to 0.8 M, suggesting that it could remain stable helical structures in the physiological fluids with ionic strength of approximately 0.15 M (Supplementary Fig. S7B). Because of its high molecular weight and cationic charge density, PVBLG-8 was able to condense DNA through electrostatic interactions to form 150-nm polyplexes at the weight ratio higher than 5 (Fig. 1D and 1E). After condensing DNA to form the binary complex, the helical structure of PVBLG-8 was well maintained (Supplementary Fig. S8), which thus allowed the complexes to mediate helicity-dependent membrane penetration to trigger cellular internalization and transfection of the gene cargo (Fig. 1F and 1G). However, sharing the same drawback as other polycations, PVBLG-8 exhibited dose-dependent cytotoxicity due to its extravagant membrane activity (Fig. 1H). As a result, the cellular uptake level and gene transfection efficiency were reduced when excessive polypeptide was utilized (weight ratio higher than 15, Fig. 1F and 1G).
3.2. Synthesis and characterization of PVBLG-8-r-7 and PVBLG-8-r-7/DNA complexes
In attempts to potentiate the gene delivery capability of PVBLG-8 while reducing the cytotoxicity, as our first strategy, we developed a PVBLG-8-based random copolypeptide, PVBLG-8-r-7 that contained glucosamine on its side-chain terminal (Fig. 2A). We hypothesized that the glucosamine residue would allow active targeting to mannose receptors on specific cell types to promote the gene transfer efficiency. However, in contrary to our hypothesis, the transfection efficiency of PVBLG-8-r-7 was decreased compared to the original PVBLG-8 (Fig. 2B), which was likely due to saccharide-mediated shielding of the positive charges of the PVBLG-8 segment (Fig. 2D). Such strategy shared the similar shortcoming of PEGylation [37], although the cell tolerability was slightly improved (Fig. 2C) due to the charge shielding effect (Fig. 2D).
3.3. Synthesis and characterization of PVBLG-7 and PVBLG-7/DNA binary complex
Since the copolypeptide strategy did not work, we thus sought alternative approaches which can endow PVBLG-8 with cellular targeting functionality while do not compromise its membrane activity. The self-assembly strategy was adopted which allows convenient preparation of nanostructured complexes from molecular building blocks [29, 38, 39]. Targeting moiety can be incorporated via non-covalent molecular recognition rather than covalent conjugation, such that the integral PVBLG-8 structure can be maintained and the lead complexes can be easily identified via combinatorial selection of substrates [38, 39]. To realize this approach, the targeting building block needs to meet two requirements. First, it should have non-covalent interactions with PVBLG-8 or DNA, allowing it to be integrated into the PVBLG-8/DNA complexes. Second, it should display weak or minimal membrane activity so that it will not induce additional cytotoxicity. With regards to these requirements, PVBLG-7 was developed as a targeting component towards mannose receptors on specific cell types, and the PVBLG-8/PVBLG-7/DNA ternary complexes were constructed via self-assembly to attain non-viral gene delivery (Fig. 4A). PVBLG-7 was also synthesized through polymerization of VB-L-Glu-NCA and side-chain amination [25] (Fig. 3A), and thus it shared the same molecular weight and PDI as PVBLG-8 (Supplementary Fig. S6). The conjugation efficiency of glucosamine in PVBLG-7 was determined to be 75% by 1H NMR (Supplementary Fig. S4), and the helicity of PVBLG-7 was lower than PVBLG-8 (58%, Fig. 3B), presumably due to the lower cationic charge density on the side chain terminal that reduced the intramolecular repulsion, the driving force for the stabilization of helical structure. Because of its lower cationic charge density, PVBLG-7 showed much weaker DNA condensation capacity, as evidenced by the observation that PVBLG-7 condensed only 60% of DNA at a much higher weight ratio of 30 (80% DNA condensed at PVBLG-8/DNA weight ratio higher than 5, Supplementary Fig. S9A and Fig. 3D) and the PVBLG-7/DNA complexes showed lower zeta potential (~20 mV, Fig. 3E) than PVBLG-8/DNA complexes (~40 mV, Fig. 1E). In accordance with its lower cationic charge density and helical content, PVBLG-7 exhibited weaker membrane activity (Fig. 3C) and cytotoxicity (Fig. 3F) than PVBLG-8, which indicated that addition of PVBLG-7 may not induce additional toxicities. We then evaluated the transfection efficiency of PVBLG-7/DNA complexes at different weight ratios. The results revealed that the optimal transfection efficiency of PVBLG-7 (~15% EGFP positive cells) was notably lower than that of PVBLG-8 (~50% EGFP positive cells, Fig. 3G).
Fig. 4.

(A) Schematic representation of PVBLG-8/PVBLG-7/DNA ternary complexes. (B) SEM image of ternary complexes at the PVBLG-8/PVBLG-7/DNA weight ratio of 15/7/1 (bar = 500 nm). (C) DNA condensation by polypeptides as evaluated by the gel retardation assay. (D) Particle size and zeta potential of PVBLG-8/PVBLG-7/DNA ternary complexes (PVBLG-8/DNA weight ratio = 15). (E) Stability of ternary complexes following dilution with PBS at different folds.
Fig. 3.

PVBLG-7 bears low cationic charge density than PVBLG-8 and thus exhibits weaker DNA condensation capability, lower membrane permeability, and lower cytotoxicity. (A) Reaction scheme of PVBLG-7. (B) CD spectrum of PVBLG-7 in DI water (0.1 mg/mL) at pH 7. (C) FITC-Tris uptake level of HeLa cells following co-incubation with PVBLG-7 for 2 h at 37 °C. (D) DNA condensation by PVBLG-7 at different weight ratios as evaluated by the gel retardation assay. N represents naked DNA. (E) Particle size and zeta potential of PVBLG-7/DNA complexes. (F) In vitro cytotoxicity of PVBLG-7 in HeLa cells as determined by the MTT assay (n=3). (G) In vitro transfection efficiency of PVBLG-7/pEGFP complexes in HeLa cells at different weight ratios (n=3).
3.4. Characterization of ternary complexes
Because PVBLG-8/DNA binary complexes showed maximal cell uptake level and transfection efficiency at the weight ratio of 15 (Fig. 1F and 1G), we thus prepared a series of PVBLG-8/PVBLG-7/DNA ternary complexes by fixing the PVBLG-8/DNA weight ratio at 15 while changing the PVBLG-8/PVBLG-7 weight ratio from 1 to 13. Gel retardation assay showed retarded DNA migration in the agarose gel for all the test ternary complexes, suggesting that DNA could be effectively co-condensed by cationic PVBLG-8 and PVBLG-7 (Fig. 4D). An increase in the PVBLG-7 content in the ternary complexes did not significantly alter the DNA condensation level (Supplementary Fig. S9B), indicating that PVBLG-8 with much higher cationic charge density contributed to majority of the DNA condensation in the ternary complexes. Consistently, an increase in the PVBLG-7 content resulted in unappreciable alteration in the particle size and zeta potential (Fig. 4C), and the ternary complexes at the PVBLG-8/PVBLG-7/DNA weight ratio of 15/7/1 revealed spherical morphology and diameter of 150–200 nm as shown in the SEM image (Fig. 4B). Upon dilution with PBS up to 100 folds, the particle size of ternary complexes (PVBLG-8/PVBLG-7/DNA weight ratio of 15/7/1) maintained unaltered, which indicated their desired stability against salt and dilution (Fig. 4E).
3.5. Cell uptake and intracellular mechanism
The capability of ternary complexes to deliver DNA intracellularly was evaluated and compared to binary complexes in three different mammalian cell types that express mannose receptors, HeLa, COS-7, and Raw 264.7 [30–32]. PVBLG-8/DNA binary complexes remarkably promoted the internalization level of YOYO-1-DNA, peaking at the PVBLG-8/DNA weight ratio of 15:1 and outperforming LPF/DNA complexes by 3–4 fold (Fig. 1F and Fig. 5). The excellent membrane activity of PVBLG-8 promoted the interaction between complexes and cell membranes, thus triggering efficient cellular internalization that increased with the PVBLG-8/DNA weight ratio up to 15. However, further increase in the PVBLG-8/DNA weight ratio to 20 and 30 decreased the cell uptake level, mainly due to the excessive membrane activity of PVBLG-8 that caused irreversible cell damage. Therefore, at the optimized PVBLG-8/DNA weight ratio of 15, PVBLG-7 was incorporated to form the ternary complexes. As shown in Fig. 5, addition of PVBLG-7 did lead to further improvement in the cell uptake level, mainly attributed to its targeting effect via recognition of mannose receptors. The cell uptake level peaked at the PVBLG-8/PVBLG-7 weight ratio of 15:7, 15:3, and 15:3 for HeLa, COS-7, and Raw 264.7 cells, respectively, which was 1.5–2 fold higher than the PVBLG-8/DNA binary complexes (Fig. 5). Further increase of the PVBLG-7 amount decreased rather than increased the cell uptake level, which could result from the competitive binding between mannose receptors and excessive PVBLG-7 that was not associated with the complexes. To further verify the targeting effect, we performed the uptake study in the presence of free mannose at various concentrations. As shown in Fig. 6A, Supplementary Fig. S11A and S11B, an increase in the mannose concentration (6–50 molar folds of glucosamine in the ternary complexes, PVBLG-8/PVBLG-7/DNA=15/7/1) led to significantly decreased cell uptake level of the ternary complexes but not the PVBLG-8/DNA binary complexes. Such findings thereby substantiated that PVBLG-7 promoted the cellular internalization of ternary complexes by targeting to cell membranes via recognition of mannose receptors that could be competitively occupied by free mannose.
Fig. 5.
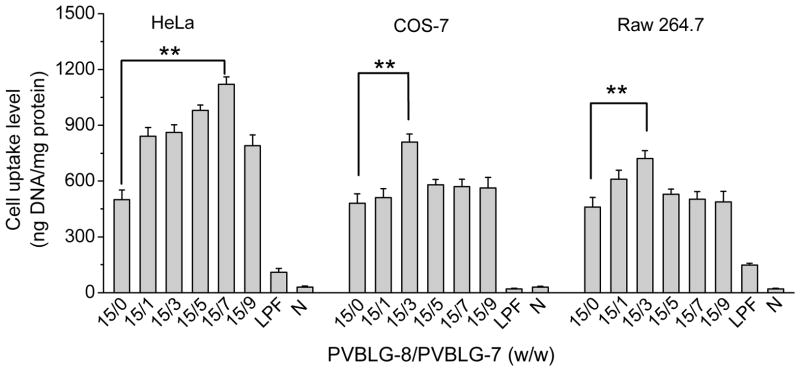
Uptake level of ternary complexes containing YOYO-1-DNA in HeLa, COS-7, and Raw 264.7 cell following 4-h incubation at 37 °C (n=3). PVBLG-8/DNA weight ratio was maintained constant at 15. N represents naked DNA.
Fig. 6.

Ternary complexes mediate efficient intracellular delivery of DNA via direct translocation and mannose receptor mediated endocytosis. (A) Uptake level of ternary complexes in HeLa cells in the presence of free mannose at various concentrations (n=3). (B) Uptake level of ternary complexes in HeLa cells at 4 °C or in the presence of various endocyt ic inhibitors. (C) FITC-Tris uptake level of HeLa cells following co-incubation with PVBLG-8 and PVBLG-7 at different weight ratios for 2 h at 37 °C (n=3). (D) CLSM images showing the cellular internalization and distribution of PVBLG-8/DNA binary complexes and PVBLG-8/PVBLG-7/DNA ternary complexes in HeLa cells following incubation at 37 °C for 4 h (bar = 20μm).
The gene transfection efficiency of non-viral vectors is closely related to their intracellular kinetics, such as the internalization pathway and endosomal escape mechanism [40]. We thus mechanistically probed the intracellular kinetics of the ternary complexes in HeLa, COS-7, and Raw 264.7 cells. By performing the cell uptake study at lower temperature (4 °C) or in the presence of various endocytic inhibitors, we first elucidated the internalization pathway of ternary complexes. Energy-dependent endocytosis was completely blocked at 4 °C; chlorpromazine inhibited clathrin-mediated endocytosis (CME) by triggering the dissociation of the clathrin lattice; genisteine and mβCD inhibited caveolae by inhibiting tyrosine kinase and depleting cholesterol, respectively; wortmannin inhibited macropinocytosis by suppressing phosphatidyl inositol-3-phosphate [40]. Lowering the temperature resulted in 70% reduction in the cell uptake level, implying that majority of the complexes were internalized via energy-dependent endocytosis. The cell uptake level was also significantly inhibited by genistein, mβCD, and chlorpromazine in all the three cell lines (Fig. 6B), which indicated that both the caveolae- and clathrin-mediated pathway were involved during endocytosis of ternary complexes. Wortmannin exerted inhibitory effect in HeLa and COS-7 cells rather than in Raw 264.7 cells, indicating that the macropinocytosis mechanism for ternary complexes was cell line-dependent (Fig. 6B and Supplementary Fig. S11C). Apart from the endocytosis pathway, we also probed the capability of ternary complexes to induce pore formation on cell membranes, an important non-endocytosis mechanism mediated by cationic helical polypeptides [23]. The uptake level of FITC-Tris, a hydrophilic and membrane-impermeable dye, was monitored after co-incubation with ternary complexes to represent the pore formation levels. As shown in Fig. 6C, ternary complexes notably enhanced the FITC-Tris uptake level, which was comparable to that of the PVBLG-8/DNA binary complexes at all test PVBLG-8/PVBLG-7 ratios. It therefore indicated that PVBLG-8, after condensing DNA to form either binary complexes or ternary complexes, maintained its membrane activity that was not counteracted by the incorporation of PVBLG-7. With the optimal formulation of PVBLG-8/PVBLG-7/DNA weight ratio of 15/7/1, we further evaluated the endosomal escape and DNA nuclear transport of ternary complexes in HeLa cells by CLSM. As shown in Fig. 6D, YOYO-1-DNA was extensively taken up by HeLa cells post 2-h treatment with ternary complexes, and it was largely separated from Lysotracker® Red-stained endosomes/lysosomes, indicating that they were able to mediate effective endosomal escape. The internalized YOYO-1-DNA was also noted to be distributed to the Hoechst 33258-stained nuclei, suggesting that the ternary complexes could trigger nuclear transport of DNA to initiate gene transcription (Fig. 6D).
3.6. In vitro transfection
The gene transfection efficiencies of ternary complexes in HeLa, COS-7, and Raw 264.7 cells were monitored by flow cytometry and compared to those of binary complexes. For the PVBLG-8/DNA binary complexes, maximal transfection efficiency was noted at the PVBLG-8/DNA weight ratio of 15, which was consistent with their cell uptake level. Such result further substantiated our statement that transfection efficiency cannot be improved by keeping increasing the amount of PVBLG-8, largely due to its excessive membrane activity that caused irreversible cell damage (Fig. 1G). By fixing the PVBLG-8/DNA ratio at such optimized value, we then evaluated the transfection efficiency of ternary complexes containing various PVBLG-7 contents. In the three cell lines that expressed mannose receptors, all the test ternary complexes exhibited significantly higher gene expression level than binary complexes, peaking at the PVBLG-8/PVBLG-7 weight ratios of 15/7, 15/3, 15/3 in HeLa, COS-7, and Raw 264.7 cells, respectively (Fig. 7A, 7B, 8A and 8B). Fluorescent images of HeLa cells also revealed higher percentage of GFP-positive cells and higher green fluorescence intensities for ternary complexes compared to binary complexes (Fig. 7C). Such transfection results accorded well with the cell uptake level, again validating our design strategy to improve the transfection efficiency of PVBLG-8 via PVBLG-7-mediated cellular targeting. To further demonstrate the PVBLG-7-mediated mannose receptor targeting effect, we monitored the transfection efficiency of ternary complexes in the presence of free mannose. As shown in Fig. 7D and Supplementary Fig. S12, transfection efficiencies of the ternary complexes were largely compromised in the presence of mannose, and when the mannose concentration was increased up to 800 μM (50 molar ratios to glucosamine groups in ternary complex, PVBLG-8/PVBLG-7/DNA = 15/7/1), the ternary complexes exhibited comparable transfection efficiency to binary complexes, suggesting that the PVBLG-7-mediated targeting effect was completely blocked. As a control, binary complexes deprived of the targeting moiety (PVBLG-7) were not influenced by free mannose in terms of transfection efficiency, which served as another evidence for the targeting effect of PVBLG-7 (Fig. 7D and Supplementary Fig. S12).
Fig. 7.

Ternary complexes mediate higher transfection efficiencies than binary complexes. (A) Transfection efficiency of ternary complexes in HeLa cells at different PVBLG-8/PVBLG-7 weight ratios (n=3). PVBLG-8/DNA weight ratio was kept constant at 15, and for the PVBLG-7/DNA binary complexes, the optimal PVBLG-7/DNA weight ratio of 20 was used. (B) Representative flow cytometry spectra showing the transfection efficiencies of binary and ternary complexes in HeLa cells. (C) Fluorescent images of HeLa cells transfected with binary complexes (PVBLG-8/DNA weight ratio = 15) and ternary complexes (PVBLG-8/PVBLG-7/DNA weight ratio = 15:7:1) (bar= 100 μm). (D) Transfection efficiency of binary and ternary complexes in HeLa cells in the presence of free mannose at different concentrations (n=3).
Fig. 8.
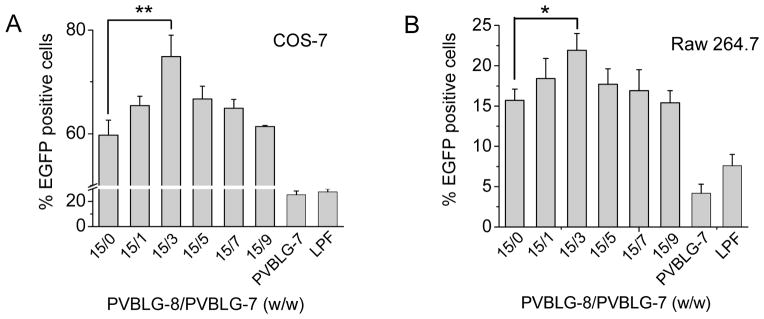
Ternary complexes transfect various mammalian cell lines expressing mannose receptors. Transfection efficiency of ternary complexes in COS-7 cells (A) and Raw 264.7 cells (B) at different PVBLG-8/PVBLG-7 weight ratios (n=3). PVBLG-8/DNA weight ratio was kept constant at 15. For the PVBLG-7/DNA binary complexes, the optimal PVBLG-7/DNA weight ratios of 15 and 20 were used for COS-7 and Raw 264.7 cells, respectively.
3.7. Cytotoxicity
The cytotoxicity of complexes towards HeLa, COS-7, and Raw 264.7 cells were evaluated by the MTT assay following 24-h treatment. As shown in Fig. 9, ternary complexes exhibited a dose-dependent cytotoxicity comparable to that of the binary complexes. Such case verified that PVBLG-7 did not induce additional toxicities to the complexes and it can serve as a non-toxic targeting building block to potentiate the gene transfection. Compared to the free polymers at equivalent concentrations (Supplementary Fig. S13), ternary complexes showed lower cytotoxicity, which was attributed to the partial neutralization of their positive charges by the anionic DNA molecule. More noteworthy was that at the transfection dose (equal to 1.5 μg PVBLG-8/well), the ternary complexes showed low cytotoxicity (higher than 80% cell viability), which can ensure their safe application towards non-viral gene delivery.
Fig. 9.
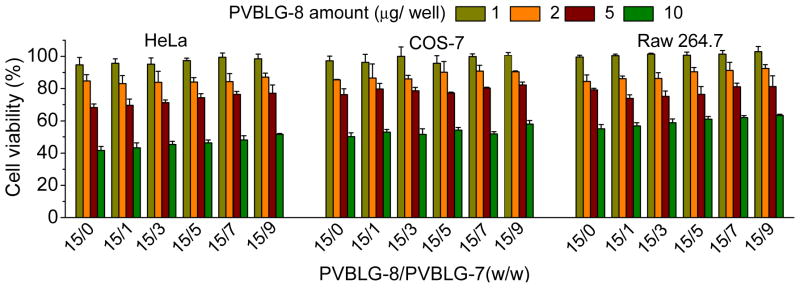
In vitro cytotoxicity of binary and ternary complexes in HeLa, COS-7, and Raw 264.7 cells following 24-h treatment as evaluated by the MTT assay (n=3). PVBLG-8/DNA weight ratio was kept constant at 15.
4. Discussion
Synthetic non-viral vectors (lipids and polycations) are widely recognized as promising vectors for gene delivery. However, the poor correlation between the delivery efficiency and safety profiles, especially with regard to chemotoxicity, renders the non-viral vectors with limited applications [10]. Vectors with high transfection efficiency often show high toxicity, while those with low toxicity frequently suffer from low transfection efficiency. One of the main reasons for such poor correlation is probably that different, even conflicting functionalities are required at different stages of the gene delivery processes. For instance, high content of amine moieties in the vectors plays important roles in overcoming the endosomal barrier via a combination of membrane destabilization and “proton-sponge” effect [41]. On the other hand, the cationic charges may induce nonspecific interactions with negatively charged serum components to form thrombi in the capillary, and they may perturb the integral structure of the plasma membranes to induce cytotoxicity or immune responses [42]. It is therefore of great demand to achieve a proper balance between efficiency and toxicity towards the design strategy of non-viral vectors.
Recently, there have been many reports in developing non-viral gene delivery vectors using a combinatorial/parallel synthesis approach to construct large libraries of materials with unique molecular structures. For instance, Anderson et al. [12] created a library of over two thousand unique molecules via Michael addition reactions between varieties of diacrylates and amines. In a similar approach, Barua et al. [43] generated a library of eighty compounds via ring-opening polymerization of diglycidyl ethers by amines. Often, the only rational design in the library approach is the inclusion of cationic amines in the molecular building block that allows DNA condensation, and the success of it mainly relies on the screening process to identify leading materials with appropriate balance between gene transfection efficiencies and cytotoxicities. Despite the success, the library approach is labor intensive and leads to high cost. In comparison, rational modification of existing star materials to impart additional functionalities against the tansfection barriers and reduce the cytotoxicities would display lower risk and higher likelihood of success. Multi-functionalization via multi-step conjugation is one of the most commonly used strategies, among which PEGylation of polycations serves as a practical solution to reduce the material toxicity by partly shielding the surface positive charges [17, 37]. However, significant reduction of the transfection efficiency is inevitable, mainly because of the reduced cellular uptake level and the impaired capacity to aid endosomal escape. Charge-conversion materials, which can undergo the charge conversion from positive to neutral/negative post-transfection, demonstrates a promising strategy in realizing the full potential of cationic materials while minimizing the cytotoxicity post-transfection [10, 44–46]. Despite the success of the aforementioned approaches, they all involve complex chemistries, which may raise difficulties in quality control and significant batch-to-batch variation. With diverse functionalities incorporated in a single material, it was also difficult to modulate the relative amount of each functional moiety towards the optimal combination.
In comparison to these reported approaches, we in the current contribution developed a membrane-permeable and mannose-receptor targeting ternary complex via a facial and modular self-assembly strategy. The complexes were based on effective cationic helical polypeptides we recently identified through a library approach3 [23]. PVBLG-8 was capable of inducing effective membrane destabilization/disruption, which can thus facilitate cellular internalization and endosomal escape to mediate effective gene transfection. Because excessive membrane activity of PVBLG-8 at high doses would cause irreversible cell damage, it is unlikely to strength its transfection capabilities by keeping increasing the PVBLG-8 amount. As such, PVBLG-7, an analogue to PVBLG-8 that bears glucosamine residues and displays minimal membrane activities, was introduced to the ternary complexes to allow mannose receptor-mediated cellular targeting and correspondingly potentiate the transfection efficiency of PVBLG-8. Such hypothesis was substantiated by the enhanced cellular uptake level and transfection efficiencies in various mannose receptor-expressing cell types; such effect was completely inhibited in the presence of free mannose that competitively occupied the mannose receptors, which further verified the targeting capability of PVBLG-7. The incorporation of PVBLG-7 did not compromise the membrane activity of PVBLG-8, and it did not bring additional cytotoxicities to PVBLG-8 due to its low membrane activity. These results collectively indicated that within the self-assembled ternary complexes, each individual building block worked synergistically and performed its intended roles without impairing the functionalities of remaining components. In a direct comparison, a random copolypeptide PVBLG-8-r-7 demonstrated decreased rather than increased transfection efficiency compared to PVBLG-8, which was attributed to the compromised membrane permeability of the PVBLG-8 moiety by the PVBLG-7 segment. As such, the self-assembly approach adopted herein is advantageous over the strategy that diverse functionalities are assembled in a single polymer where they influence each other. Because of the simplicity and adjustability of the self-assembly approach, a proper balance between the membrane activity and targeting capability can also be easily determined by modulating the relative amount of each functionality, thus identifying a cell-line specific formulation for the ternary complexes towards maximized gene transfection efficiency yet minimized cytotoxicity.
During the design of the self-assembled complexes, the second component used together with PVBLG-8 should meet the following three requirements. First, it should have non-covalent interactions with PVBLG-8 or DNA, allowing it to be integrated into the PVBLG-8/DNA complexes. Second, it should display weak or minimal membrane activity so that it will not induce additional cytotoxicity. Third, it should carry a specific functionality that PVBLG-8 does not have, such as cell targeting or the nuclear transport capability. PVBLG-7 perfectly meets such requirements. It bears proper cationic charge densities so that it can co-condense DNA with PVBLG-8 to form stable ternary complexes; the low membrane activity of PVBLG-7 will not cause addition damage to the cell membranes. The glucosamine side chains on PVBLG-7 allow active targeting to mannose receptor expressing cells to potentiate the cellular internalization level. It is desired that PVBLG-7 should adopt α-helical conformation, because polypeptides with helical structure have stronger DNA binding affinity than the random-coiled analogues [46]. Our results showed that the optimal combination of membrane activity and targeting capacity was different in various mannose receptor-expression cell types (HeLa, COS-7, and Raw 264.7). To this regard, the self-assembly approach was more effective and less labor-consuming than multi-step chemical reactions in terms of varying the relative amount of each functionality. As a result, the top-performing ternary complexes with maximal synergistic effect were identified in the three test cell types, which represented a dramatic improvement over the commercial reagent LPF. Using the self-assembly strategy, polypeptides with various side chains can also be incorporated to a single complex to exhibit multifunctionalities and thus overcome the multiple cellular barriers against transfection. For instance, polypeptides containing imidazole side groups can further promote endosomal escape; polypeptides containing SV40 and TAT side chains can potentially facilitate the nuclear targeting and transport; other targeting ligands (e.g., folate, transferrin, biotin) can also be used to target different cell types.
5. Conclusion
We demonstrated a convenient, flexible, and modular self-assembly approach in developing effective and safe non-viral gene delivery vectors, which was achieved by combining a highly membrane-penetrating while toxic cationic helical polypeptide (PVBLG-8) with a less cationic, relatively inert, and cell-targeting polypeptide (PVBLG-7). Within the PVBLG-8/PVBLG-7/DNA ternary complexes, the membrane activity of PVBLG-8 and the cellular targeting capability of PVBLG-7 worked synergistically without compromising each other. As such, a proper balance between the membrane activity and the targeting efficiency was easily identified in each certain cell type by modulating the relative amount of each individual component, and thus the gene transfection efficiency of PVBLG-8 was maximized without inducing additional cytotoxicities. Such approach described here would serve as an effective tool in overcoming the efficiency-toxicity inconsistency of cationic non-viral gene delivery vectors; it is easy to handle, does not involve complex synthetic chemistry, and can be easily optimized by changing the building blocks (e.g. incorporation of another polypeptide bearing nuclear localization signals), which we believe will provide an important addition to the existing efforts in identifying cell-specific gene delivery vectors.
Supplementary Material
Acknowledgments
J.C. acknowledges support from the NSF (CHE-0809420), the NIH (NIH Director’s New Innovator Award 1DP2OD007246, 1R21EB013379).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Candolfi M, Xiong WD, Yagiz K, Liu CY, Muhammad AKMG, Puntel M, et al. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. P Natl Acad Sci USA. 2010;107:20021–6. doi: 10.1073/pnas.1008261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–10. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paul A, Binsalamah ZM, Khan AA, Abbasia S, Elias CB, Shum-Tim D, et al. A nanobiohybrid complex of recombinant baculovirus and Tat/DNA nanoparticles for delivery of Ang-1 transgene in myocardial infarction therapy. Biomaterials. 2011;32:8304–18. doi: 10.1016/j.biomaterials.2011.07.042. [DOI] [PubMed] [Google Scholar]
- 4.Tzeng SY, Guerrero-Cázares H, Martinez EE, Sunshine JC, Quiñones-Hinojosa A, Green JJ. Non-viral gene delivery nanoparticles based on poly(β-amino esters) for treatment of glioblastoma. Biomaterials. 2011;32:5402–10. doi: 10.1016/j.biomaterials.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He C, Yin L, Tang C, Yin C. Multifunctional polymeric nanoparticles for oral delivery of TNF-α siRNA to macrophages. Biomaterials. 2013;34:2843–54. doi: 10.1016/j.biomaterials.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 6.Xiang S, Su J, Tong H, Yang F, Tong W, Yuan W, et al. Biscarbamate cross-linked low molecular weight PEI for delivering IL-1 receptor antagonist gene to synoviocytes for arthritis therapy. Biomaterials. 2012;33:6520–32. doi: 10.1016/j.biomaterials.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 7.Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- 8.Nishikawa M, Huang L. Nonviral vectors in the new millennium: delivery barriers in gene transfer. Hum Gene Ther. 2001;12:861–70. doi: 10.1089/104303401750195836. [DOI] [PubMed] [Google Scholar]
- 9.Ko IK, Ziady A, Lu S, Kwon YJ. Acid-degradable cationic methacrylamide polymerized in the presence of plasmid DNA as tunable non-viral gene carrier. Biomaterials. 2008;29:3872–81. doi: 10.1016/j.biomaterials.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Lee Y, Miyata K, Oba M, Ishii T, Fukushima S, Han M, et al. Charge-conversion ternary polyplex with endosome disruption moiety: A technique for efficient and safe gene delivery. Angewe Chem Intl Ed. 2008;47:5163–6. doi: 10.1002/anie.200800963. [DOI] [PubMed] [Google Scholar]
- 11.Han M, Bae Y, Nishiyama N, Miyata K, Oba M, Kataoka K. Transfection study using multicellular tumor spheroids for screening non-viral polymeric gene vectors with low cytotoxicity and high transfection efficiencies. J Control Release. 2007;121:38–48. doi: 10.1016/j.jconrel.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Anderson DG, Lynn DM, Langer R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angewe Chem Intl Ed. 2003;42:3153–8. doi: 10.1002/anie.200351244. [DOI] [PubMed] [Google Scholar]
- 13.McLendon PM, Fichter KM, Reineke TM. Poly(glycoamidoamine) vehicles promote pDNA uptake through multiple routes and efficient gene expression via caveolae-mediated endocytosis. Mol Pharmaceut. 2010;7:738–50. doi: 10.1021/mp900282e. [DOI] [PubMed] [Google Scholar]
- 14.Oskuee RK, Dehshahri A, Shier WT, Ramezani M. Alkylcarboxylate grafting to polyethylenimine: a simple approach to producing a DNA nanocarrier with low toxicity. J Gene Med. 2009;11:921–32. doi: 10.1002/jgm.1374. [DOI] [PubMed] [Google Scholar]
- 15.Venkataraman S, Ong WL, Ong ZY, Loo SCJ, Ee PLR, Yang YY. The role of PEG architecture and molecular weight in the gene transfection performance of PEGylated poly(dimethylaminoethyl methacrylate) based cationic polymers. Biomaterials. 2011;32:2369–78. doi: 10.1016/j.biomaterials.2010.11.070. [DOI] [PubMed] [Google Scholar]
- 16.Deshpande MC, Davies MC, Garnett MC, Williams PM, Armitage D, Bailey L, et al. The effect of poly(ethylene glycol) molecular architecture on cellular interaction and uptake of DNA complexes. J Control Release. 2004;97:143–56. doi: 10.1016/j.jconrel.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 17.Yin L, Song Z, Kim KH, Zheng N, Tang H, Lu H, et al. Reconfiguring the architectures of cationic helical polypeptides to control non-viral gene delivery. Biomaterials. 2013;34:2340–9. doi: 10.1016/j.biomaterials.2012.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ter-Avetisyan G, Tuennemann G, Nowak D, Nitschke M, Herrmann A, Drab M, et al. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284:3370–8. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon EJ, Liong S, Pun SH. A truncated HGP peptide sequence that retains endosomolytic activity and improves gene delivery efficiencies. Mol Pharmaceut. 2010;7:1260–5. doi: 10.1021/mp1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakase I, Akita H, Kogure K, Graslund A, Langel U, Harashima H, et al. Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Accounts Chem Res. 2012;45:1132–9. doi: 10.1021/ar200256e. [DOI] [PubMed] [Google Scholar]
- 21.Saw PE, Ko YT, Jon S. Efficient liposomal nanocarrier-mediated oligodeoxynucleotide delivery involving dual use of a cell-penetrating peptide as a packaging and intracellular delivery agent. Macromol Rapid Comm. 2010;31:1155–62. doi: 10.1002/marc.200900861. [DOI] [PubMed] [Google Scholar]
- 22.Schellinger JG, Pahang JA, Johnson RN, Chu DSH, Sellers DL, Maris DO, et al. Melittin-grafted HPMA-oligolysine based copolymers for gene delivery. Biomaterials. 2013;34:2318–26. doi: 10.1016/j.biomaterials.2012.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabrielson NP, Lu H, Yin LC, Li D, Wang F, Cheng JJ. Reactive and bioactive cationic α-helical polypeptide template for nonviral gene delivery. Angew Chem Int Ed. 2012;51:1143–7. doi: 10.1002/anie.201104262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabrielson NP, Lu H, Yin LC, Kim KH, Cheng JJ. A cell-penetrating helical polymer for siRNA delivery to mammalian cells. Mol Ther. 2012;20:1599–609. doi: 10.1038/mt.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu H, Wang J, Bai YG, Lang JW, Liu SY, Lin Y, et al. Ionic polypeptides with unusual helical stability. Nat Commun. 2011;2:206. doi: 10.1038/ncomms1209. [DOI] [PubMed] [Google Scholar]
- 26.Yen J, Zhang YF, Gabrielson NP, Yin LC, Guan L, Chaudhury I, et al. Cationic, helical polypeptide-based gene delivery for IMR-90 fibroblasts and human embryonic stem cells. Biomaterials Science. 2013;1:719–727. doi: 10.1039/C3BM00006K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin LC, Song ZY, Kim KH, Zheng N, Gabrielson NP, Cheng JJ. Non-viral gene delivery via membrane-penetrating, mannose-targeting supramolecular self-assembled nanocomplexes. Advanced materials. 2013;25:3063–70. doi: 10.1002/adma.201205088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin LC, Song ZY, Qu Q, Kim KH, Zheng N, Yao C, et al. Supramolecular self-assembled nanoparticles mediate oral delivery of therapeutic TNF-alpha siRNA against systemic inflammation. Angewe Chem Intl Ed. 2013;52:5757–61. doi: 10.1002/anie.201209991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Chen KJ, Wang S, Ohashi M, Kamei K, Sun J, et al. A small library of DNA-encapsulated supramolecular nanoparticles for targeted gene delivery. Chem Commun. 2010;46:1851–3. doi: 10.1039/b923711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas FJ, Perales JC, Frank M, Hanson RD. Receptor-mediated gene transfer into macrophages. Proc Natl Acad Sci. 1996;93:101–5. doi: 10.1073/pnas.93.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park IY, Kim IY, Yoo MK, Choi YJ, Cho MH, Cho CS. Mannosylated polyethylenimine coupled mesoporous silica nanoparticles for receptor-mediated gene delivery. Int J Pharmaceut. 2008;359:280–7. doi: 10.1016/j.ijpharm.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, Bonifacino JS. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 2001;292:1712–6. doi: 10.1126/science.1060750. [DOI] [PubMed] [Google Scholar]
- 33.Lu H, Cheng JJ. Hexamethyldisilazane-mediated controlled polymerization of α-amino acid N-carboxyanhydrides. J Am Chem Soc. 2007;129:14114–5. doi: 10.1021/ja074961q. [DOI] [PubMed] [Google Scholar]
- 34.Gabrielson NP, Cheng JJ. Multiplexed supramolecular self-assembly for non-viral gene delivery. Biomaterials. 2010;31:9117–27. doi: 10.1016/j.biomaterials.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. P Natl Acad Sci USA. 2009;106:6111–6. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu H, Cheng JJ. Controlled ring-opening polymerization of amino acid N-carboxyanhydrides and facile end group functionalization of polypeptides. J Am Chem Soc. 2008;130:12562–3. doi: 10.1021/ja803304x. [DOI] [PubMed] [Google Scholar]
- 37.Liu XQ, Du JZ, Zhang CP, Zhao F, Yang XZ, Wang J. Brush-shaped polycation with poly(ethylenimine)-b-poly(ethylene glycol) side chains as highly efficient gene delivery vector. Int J Pharmaceut. 2010;392:118–26. doi: 10.1016/j.ijpharm.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Wang ST, Su H, Chen KJ, Armijo AL, Lin WY, et al. A supramolecular approach for preparation of size-controlled nanoparticles. Angew Chem Int Ed. 2009;48:4344–8. doi: 10.1002/anie.200900063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Liu K, Chen KJ, Lu YJ, Wang ST, Lin WY, et al. A rapid pathway toward a superb gene delivery system: programming structural and functional diversity into a supramolecular nanoparticle library. Acs Nano. 2010;4:6235–43. doi: 10.1021/nn101908e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. The effect of particle design on cellular internalization pathways. P Natl Acad Sci USA. 2008;105:11613–8. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wattiaux R, Laurent N, Wattiaux-De Coninck S, Jadot M. Endosomes, lysosomes: their implication in gene transfer. Adv Drug Deliver Rev. 2000;41:201–8. doi: 10.1016/s0169-409x(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 42.Hunter AC. Molecular hurdles in polyfectin design and mechanistic background to polycation induced cytotoxicity. Adv Drug Deliver Rev. 2006;58:1523–31. doi: 10.1016/j.addr.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Barua S, Joshi A, Banerjee A, Matthews D, Sharfstein ST, Cramer SM, et al. Parallel synthesis and screening of polymers for nonviral gene delivery. Mol Pharmaceut. 2009;6:86–97. doi: 10.1021/mp800151j. [DOI] [PubMed] [Google Scholar]
- 44.Prata CAH, Zhao Y, Barthelemy P, Li Y, Luo D, McIntosh TJ, et al. Charge-reversal amphiphiles for gene delivery. J Am Chem Soc. 2004;126:12196–7. doi: 10.1021/ja0474906. [DOI] [PubMed] [Google Scholar]
- 45.Liu XH, Yang JW, Lynn DM. Addition of “charge-shifting” side chains to linear poly(ethyleneimine) enhances cell transfection efficiency. Biomacromolecules. 2008;9:2063–71. doi: 10.1021/bm800291v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin L, Tang H, Kim KH, Zheng N, Song Z, Gabrielson NP, et al. Light-responsive helical polypeptides capable of reducing toxicity and unpacking DNA: toward nonviral gene delivery. Angew Chem Int Ed. 2013;52:9182–6. doi: 10.1002/anie.201302820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.