

Abstract
The rational design of effective and safe non-viral gene vectors is largely dependent on the understanding of the structure-property relationship. We herein report the design of a new series of cationic, α-helical polypeptides with different side charged groups (amine and guanidine) and hydrophobicity, and mechanistically unraveled the effect of polypeptide structure on the gene delivery capability. Guanidine-containing polypeptides displayed superior membrane activities to their amine-containing analogues via the pore formation mechanism, and thus possessed notably higher transfection efficiencies. Elongating the hydrophobic side chain also potentiated the membrane activities of the polypeptides, while at the meantime caused higher cytotoxicities. Upon an optimal balance between membrane activity and cytotoxicity, maximal transfection efficiency was achieved which outperformed commercial reagent Lipofectamine™ 2000 (LPF2000) by 3–6 folds. This study thus provides mechanistic insights into the rational design of non-viral gene delivery vectors, and the best-performing materials identified also serve as a promising addition to the existing systems.
Keywords: non-viral gene delivery, α-helical polypeptide, guanidine, hydrophobicity, structure-function relationship
1. Introduction
The advance of molecular biology and genetic engineering has identified many disease-associated genes and their molecular regulators which provide potential targets for disease treatment. Gene therapy, mediated by the delivery of generic materials into target cells to promote or rectify the expression of specific gene, is a promising clinical modality to treat various human diseases, including cancer, infectious diseases, and immunodeficiency [1–4]. The key challenge towards gene therapy is the development of effective yet biocompatible delivery methods or vectors. Viral vectors, although highly efficient, often suffer from severe safety concerns such as carcinogenicity, immunogenicity and insertional mutagenesis [5]. Non-viral vectors, exemplified by cationic lipids and polymers, possess desired biocompatibility and minimal mutagenesis, and thus serve as desired alternatives to viral vectors for gene delivery [6].
Cell penetrating peptides (CPPs) are sequence-specific oligopeptides with distinguished membrane penetrating properties. A large number of CPPs, such as Pep-1, MPG, TP10, and melittin, adopt inherent helical structures or form helices in the cell membranes. Mechanistic simulation also unravels that the formation of a trans-membrane helix presents a rigid amphiphilic structure to stabilize the membrane interactions and promote the membrane permeation [7, 8]. Because of their desired membrane permeability, CPPs are able to facilitate the intracellular delivery of various cargos, such as proteins, peptides, nucleic acids, metals, and even nanoparticles. However, when used as gene delivery vectors, CPPs are often too short (fewer than 25 amino acid residues) and lack sufficient cationic charge density. Therefore, they are often unable to function as stand-alone vectors to independently condense and deliver genes, and in most cases, they were incorporated or conjugated to existing delivery vehicles as membrane-active ligands to enhance the cellular internalization and endosomal escape of the gene cargo [9–11]. In comparison, polypeptides with sufficient backbone length, such as poly-L-lysine (PLL) and poly-L-arginine (PLR), can independently condense and deliver genes, while the gene transfection efficiency remains low [12]. This is mainly because they adopt random coil conformation in the aqueous solution or when associated with phospholipid membranes due to the strong side chain charge repulsion, which thus greatly compromised the membrane activities of these high MW polypeptides [13].
To address the drawbacks of both short CPPs and polypeptides toward gene transfer, we recently developed a strategy to stabilize the helical structure of polypeptides by maintaining a minimum separation of 11-σ bond between the polypeptide backbone and the side charged groups, such that the side-chain charge repulsion can be minimized and the helical structure can be stabilized [14]. A library of cationic polypeptides containing different amine side groups was thus synthesized and screening for their gene delivery capabilities. PVBLG-8 was identified to be top-performing material which notably outperformed traditional CPPs and polypeptides [15]. Although such screening process allows the identification of desired candidates, rational design over the polymer structure and mechanistic study on the structure-property relationship would render additional functionalities and features to maximize the gene delivery efficiency [16–21].
Arginine (Arg) residues are often rich in their primary structures of CPPs, and the guanidine groups of the Arg residues are crucial to the penetration efficiencies of CPPs because of their interactions with the sulfate groups of glycosaminoglycans localized on cell membranes [22]. The penetrating efficiency of the guanidine-rich CPPs can also be activated by hydrophobic counterions that complex around the guanidine-rich backbone to coat the highly cationic structure with lipophilic moieties and thus facilitate the membrane translocation. This also holds true for other synthetic polymers where incorporation of optimal hydrophobicity often leads to enhanced membrane activities [23, 24]. Motivated by these understandings, we herein report our efforts in developing a new series of cationic, α-helical polypeptides with different side charged groups (amine and guanidine) and hydrophobicity, attempting to elucidate the effect of polymer structure and functionality on the gene delivery efficiency. We hypothesized that the incorporation of helical structures, guanidine groups, and elongated hydrophobic side chains would endow the polypeptides with considerable advantages over traditional CPPs and polypeptides, and an optimal combination thereof would thus lead to the maximization of the gene delivery capabilities of cationic helical polypeptides. In two different mammalian cell lines (HeLa and COS-7), polypeptides with diverse structures were comprehensively explored and compared in terms of their membrane activities, intracellular DNA delivery efficiencies, intracellular kinetics, transfection efficiencies, and cytotoxicities. This fundamental study hence provides insights into the design strategy of non-viral gene delivery vectors.
2. Experimental
2.1. Materials and cell lines
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received unless otherwise indicated. Anhydrous tetrahydrofuran (THF), hexane, and dimethylformamide (DMF) were dried by a column packed with 4Å molecular sieves and stored in a glovebox. Hexamethyldisilazane (HMDS) was dissolved in DMF in a glovebox and subsequently used to initiate the controlled ring-opening polymerization (ROP) of N-carboxylanhydride (NCA). Plasmid DNA encoding luciferase (pCMV-Luc) was purchased from Elim Biopharmaceutics (Hayward, CA, USA). YOYO-1, Lipofectamine™ 2000 (LPF2000), and 3-(4,5-dimethylthiahiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Invitrogen (Carlsbad, CA, USA).
HeLa (human cervix adenocarcinoma) and COS-7 (African Green Monkey SV40-transf’d kidney fibroblast) were purchased from the American Type Culture Collection (Rockville, MD, USA) and were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS).
2.2. Instrumentation
1H NMR spectra were recorded on a Varian U500 MHz or a VXR-500 MHz spectrometer. Gel permeation chromatography (GPC) experiments were conducted on a system equipped with an isocratic pump (Model 1100, Agilent Technology, Santa Clara, CA, USA), a DAWN HELEOS multi-angle laser light scattering (MALLS) detector (Wyatt Technology, Santa Barbara, CA, USA), and an Optilab rEX refractive index detector (Wyatt Technology, Santa Barbara, CA, USA). The detection wavelength of HELEOS was set at 658 nm. Separations were performed using serially connected size exclusion columns (100 Å, 500 Å, 103Å and 104 Å Phenogel columns, 5 μm, 300 × 7.8 mm, Phenomenex, Torrance, CA, USA) at 60 °C using DMF containing 0.1 M LiBr as the mobile phase. The MALLS detector was calibrated using pure toluene and can be used for the determination of the absolute molecular weights (MWs). The MWs were determined based on the dn/dc value of each polymer sample calculated offline by using the internal calibration system processed by the ASTRA V software (version 5.1.7.3, Wyatt Technology, Santa Barbara, CA, USA). Circular dichroism (CD) experiments were performed on a JASCO J-815 CD spectrometer. Polypeptides were dissolved in deionized (DI) water at the concentrations of 0.025–0.2 mg/mL unless otherwise indicated. The solution was placed in a quartz cell with a light path of 1 or 10 mm. The mean residue molar ellipticity of each polypeptide was calculated based on the measured apparent ellipticity by following equations reported in literature: Ellipticity ([θ] in deg·cm2·dmol−1) = (millidegrees × mean residue weight)/(pathlength in millimeters × concentration of polypeptide in mg mL−1). The helicity of the polypeptides were calculated by the following formula: helicity = (−[θ222] + 3000)/39000 [25]. In order to examine the helical stability of the polypeptides against pH, the pH of the polypeptide solution was accordingly adjusted with 1 M NaOH or 1 M HCl. The polypeptide concentration was fixed at 0.1 mg/mL for the pH and salt-dependent analyses.
2.3. Synthesis of the γ-(4-propargyloxybenzyl)-L-glutamic acid N-carboxyanhydride (POB-L-Glu-NCA) monomer
K2CO3 (15.2 g, 0.11 mol) and 4-hydroxybenzyl alcohol (9.3 g, 0.075 mol) were suspended in acetone (150 mL) into which propargyl bromide solution (80 wt% in toluene, 10 mL, 0.09 mol) and 18-crown-6 (0.1 mL) were added. The reaction mixture was refluxed at 75 °C for 12 h before removal of acetone by evaporation. Water (200 mL) was then added to the residue, and the aqueous layer was extracted with CH2Cl2 (DCM) (3 × 30 mL). The organic solutions were combined, washed with 15% NaOH solution (200 mL) and water (200 mL), and dried over Na2SO4. The product propargyloxybenzyl alcohol was obtained by removal of the solvent to yield clear yellow oil (9.7 g, yield 80%). 1H NMR (CDCl3): δ 7.28 (d, 2H, ArH), 6.94 (d, 2H, ArH), 4.67 (d, 2H, ArCH2-), 4.60 (s, 2H, CH2O-), 2.50 (t, 1H, HC≡C-).
The obtained propargyloxybenzyl alcohol (8.5 g, 52 mmol) was dissolved in DCM on an ice bath, and thionyl chloride (5 mL, 68 mmol) was added dropwise into the solution. The mixture was stirred at room temperature with the protection of nitrogen for 3.5 h, and water (100 mL) was then added to quench thionyl chloride. The organic layer was washed with water (3 × 50 mL) and dried over MgSO4. The product propargyloxybenzyl chloride was obtained by removal of the solvent to yield clear yellow oil (7.0 g, yield 75%). 1H NMR (CDCl3): δ 7.31 (d, 2H, ArH), 6.94 (d, 2H, ArH), 4.68 (d, 2H, ArCH2-), 4.55 (s, 2H, CH2O-), 2.51 (t, 1H, HC≡C-).
L-Glutamic acid copper (II) complex (3.29 g, 6.7 mmol) and L-glutamic acid (1.99 g, 13.4 mmol) were suspended in a mixture of DMF (12 mL) and water (2 mL) into which 1,1,3,3-tetramethylguanidine (3.4 mL, 27 mmol) was added. The mixture was stirred at 40 °C for 2 h until all compounds were dissolved. DMF (10 mL) and the obtained propargyloxybenzyl chloride (6.5 g, 36 mmol) were added to the solution which was stirred at room temperature for 48 h. Acetone (200 mL) was then added, and the mixture was stirred overnight. The crude product was isolated and washed with acetone (4 times) and water (3 times), and ethylenediaminetetraacetic acid disodium salt (EDTA-Na2) solution (0.45 M) twice. The product γ-(4-propargyloxybenzyl)-L-glutamic acid (POB-L-Glu) was obtained via recrystallization from isopropanol/water (2:1, v/v) (3.2 g, yield 80%). 1H NMR (DMSO-d6/DCl-D2O (20 wt %), 9:1, v/v): δ 7.30 (d, 2H, ArH), 6.96 (d, 2H, ArH), 5.00 (s, 2H, ArCH2-), 4.77 (d, 2H, ArOCH2-), 3.91 (m, 1H, α-H), 3.55 (t, 1H, HC≡C-), 2.54 (m, 2H, -CH2CH2COO-), 2.04 (m, 2H, -CH2CH2COO-).
POB-L-Glu (1.15 g, 4.0 mmol) was dissolved in dry THF (25 mL) followed by addition of the phosgene solution (15 wt% in toluene, 4.0 mL, 5.6 mmol). The mixture was refluxed at 50 °C for 2 h. The solvent was removed under vacuum, and the crude product was recrystallized three times (THF/hexane, 1:5, v/v) to give γ-(4-propargyloxybenzyl)-L-glutamic acid N-carboxyanhydride (POB-L-Glu-NCA) as white crystals (770 mg, yield 61%). 1H NMR (CDCl3): δ 7.29 (d, 2H, ArH), 6.98 (d, 2H, ArH), 6.3 (s, 1H, NH), 5.08 (s, 2H, ArCH2-), 4.70 (d, 2H, ArOCH2-), 4.36 (t, 1H, α-H), 2.55 (m, 3H, -COCH2CH2-, HC≡C-), 2.04 (m, 2H, -CH2CH2COO-), 2.29-2.10 (m, 2H, -CH2CH2COO-).
2.4. Polypeptide synthesis
2.4.1. Polymerization of poly(γ-(4-propargyloxybenzyl)-L-glutamate) (PPOBLG)
In a glovebox, POB-L-Glu-NCA (100 mg, 0.32 mmol) was dissolved in DMF (1.5 mL), followed by the addition of the HMDS solution (64 μL, 0.1 mol/L, M/I = 50). The mixture was stirred at room temperature for 48 h (monomer conversion > 99% as monitored by FTIR), and most DMF was removed under vacuum. The final product PPOBLG was precipitated with cold methanol and collected as white solid. 1H NMR (CDCl3): δ 7.20 (d, 2H, ArH), 6.86 (d, 2H, ArH), 5.00-4.93 (d, 2H, ArCH2-), 4.58 (s, 2H, ArOCH2-), 3.98 (s, 1H, α-H), 2.62-2.49 (br, 3H, -CH2CH2COO-, HC≡C-), 2.29-2.12 (br, 2H, -CH2CH2COO-).
2.4.2. Synthesis of azido amines/guanidines
To obtain 3-azidopropylamine, sodium azide (5.85 g, 90 mmol) was added to a solution of 3-chloropropylamine hydrochloride (5.22 g, 45 mmol) in water (50 mL), and the mixture was heated at 80 °C for 24h. The pH was adjusted to 12 using 1 M NaOH, and the solution was extracted with diethyl ether (3 ×15 mL). The organic layer was dried over Na2SO4 and the solvent was removed under vacuum to obtain colorless oil (1.0 g, yield 30%). 1H NMR (CDCl3): δ 3.28 (t, 2H, -CH2N3), 2.71 (t, 2H, -CH2NH2), 1.61 (m, 2H, -CH2CH2N3).
To obtain N,N-dimethyl-3-azidopropylamine, sodium azide (1.3 g, 20 mmol) was added to an aqueous solution of 3-dimethylamino-1-propyl chloride (1.58 g, 10 mmol, 20 mL), and the mixture was stirred at 70 °C for 12 h. KOH (5.0 g) was then added, and the aqueous solution was extracted with ether (3 × 15 mL). The combined organic phases was dried over Na2SO4 and concentrated to give colorless oil (0.98 g, yield 77%). 1H NMR (CDCl3): δ 3.34 (t, 2H, -CH2N3), 2.35 (t, 2H, -CH2N(CH3)2), 2.25 (s, 6H, -N(CH3)2), 1.75 (m, 2H, -CH2CH2N3).
To obtain 3-azidopropylguanidine, azidopropylamine (1.0 g, 10 mmol), H-pyrazole-1-carboxamidine hydrochloride (1.47 g, 10 mmol), and DIEA (1.74 mL, 10 mmol) were dissolved in dry DMF (15 mL) which was stirred at room temperature overnight. Ether (150 mL) was added to precipitate the crude product which was collected, washed with ether, and dried under vacuum to obtain yellow liquid (1.0 g, yield 60%). 1H NMR (CDCl3): δ 3.33 (t, 2H, -CH2NHC(NH)NH2), 3.18 (t, 2H, -CH2N3), 1.76 (m, 2H, -CH2CH2N3).
The synthesis of 6-azidohexylguanidine was carried out using the same method with 6-azidohexylamine as the starting material. 1H NMR (CDCl3): δ 3.35 (t, 2H, -CH2NHC(NH)NH2), 3.20 (t, 2H, -CH2N3), 1.77 (m, 2H, -CH2CH2NHC(NH)NH2), 1.67 (m, 2H, -CH2CH2N3), 1.32 (m, 4H, -(CH2)2(CH2)2N3).
The synthesis of 8-azidooctylguanidine was carried out using the same method with 8-azidooctylamine as the starting material. 1H NMR (CDCl3): δ 3.35 (t, 2H, -CH2NHC(NH)NH2), 3.20 (t, 2H, -CH2N3), 1.79 (m, 2H, -CH2CH2NHC(NH)NH2), 1.70 (m, 2H, -CH2CH2N3), 1.53-1.02 (m, 8H, -(CH2)4(CH2)2N3).
2.4.3. Synthesis of amine/guanidine functionalized polypeptides
In a glovebox, PPOBLG (20 mg, 0.072 mmol alkyne groups) was dissolved in DMF (1.0 mL) into which various azido amines/guanidines (0.144 mmol) and N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, 30 μL, 0.144 mmol) were added. CuBr (20.8 mg, 0.144 mmol) was then added, and the reaction mixture was stirred at room temperature for 24 h before addition of 1M HCl (1 mL). The final polypeptides were purified by dialysis against water for 3 days (MWCO = 3 kDa) and obtained after lyophilization. The nomenclature of polypeptides was summarized in Table 1.
Table 1.
Structures of amine/guanidine functionalized polypeptides
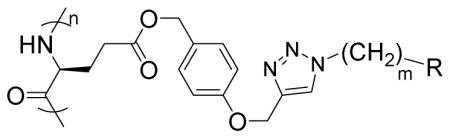
| |||
|---|---|---|---|
| Polypeptide | m | R | Helicity (%) |
| G3 | 3 |
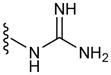
|
52 |
| G6 | 6 |
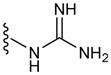
|
59 |
| G8 | 8 |
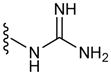
|
55 |
| P3 | 3 |
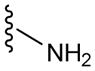
|
60 |
| T3 | 3 |
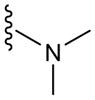
|
71 |
2.5. Polyplex formation and characterization
Polypeptide and pCMV-Luc were separately dissolved in water at 0.2 mg/mL and mixed at various nitrogen/phosphate (N/P) ratios. The mixture was vortexed for 5 s and incubated at 37 °C for 30 min to allow polyplex formation. The resultant polyplexes were subject to electrophoresis in 1% agarose gel at 100 V for 45 min to qualitatively evaluate DNA condensation by the polypeptides. To quantitatively monitor the DNA condensation level, the ethidium bromide (EB) exclusion assay [26] was conducted. Basically, EB solution was mixed with DNA at the DNA/EB ratio of 10:1 (w/w) and incubated at room temperature for 1 h. Polypeptide was then added to the mixture at various N/P ratios, and the mixture was further incubated at room temperature for 30 min before quantification of the fluorescence intensity on a microplate reader (λex = 510 nm, λem = 590 nm). A pure EB exclusion and the DNA/EB solution without any polypeptide were used as negative and positive controls, respectively. The EB exclusion efficiency (% DNA condensed) was defined as the following:
Where FEB, F, and F0 denote the fluorescence intensity of pure EB solution, DNA/EB solution with polypeptide, and DNA/EB solution without any polypeptide, respectively.
Particle size and zeta potential of polyplexes freshly prepared either in DI water or phosphate buffered saline (PBS) were further evaluated by dynamic laser scattering (DLS) on a Malvern Zetasizer (Herrenberg, Germany). To evaluate their stability against dilution, polyplexes were diluted with PBS for 50 times, incubated at room temperature for different time, and subjected to particle size measurement.
2.6. In vitro gene transfection
Cells were seeded on 96-well plates at 1×104 cells/well and cultured in DMEM containing 10% FBS for 24 h. The medium was then replaced by opti-MEM (100 μL/well), into which polyplexes were added at 0.1 μg DNA/well. After incubation at 37 °C for 4 h, the medium was replaced by DMEM containing 10% FBS and cells were further incubated for another 20 h. Luciferase expression was assayed in terms of luminescence intensity using a Bright-Glo Luciferase assay kit (Promega, Madison, WI, USA), and the cellular protein level was determined using a BCA kit (Pierce, Rockford, IL, USA). Results were expressed as relative luminescence unit (RLU) associated with 1 mg of cellular protein (RLU/mg protein). In order to evaluate the transfection efficiency of polyplexes in the presence of serum, cells were incubated with polyplexes in DMEM supplemented with 10% FBS for 4 h. To further probe the temperature-dependent transfection capabilities, cells were incubated with polyplexes in opti-MEM at 4 °C for a 4-h uptake period before further incubation at 37 °C for 20 h. LPF2000 was used as a control according to the manufacture’s protocol in all the above-mentioned studies.
2.7. Membrane activity
The membrane activity of the polypeptides was evaluated by measuring the cellular uptake level of a membrane-impermeable dye, fluorescein isothiocyanate (FITC) in its non-reactive form (fluorescein-tris(hydroxymethyl)methanethiourea, FITC-Tris) [27]. Cells were seeded on 96-well plates at 1×104 cells/well and cultured for 24 h. The medium was replaced by opti-MEM (100 μL/well), into which FITC-Tris and the polypeptide were added at 1 μg/well and 2 μg/well, respectively. After incubation at 37 °C for 2 h, cells were washed with PBS containing heparin (20 U/mL) for three times and then lysed with the RIPA lysis buffer (100 μL/well). The amount of FITC-Tris in the cell lysate was quantified by spectrofluorimetry (λex = 488 nm, λem = 530 nm), and the protein content was determined by the BCA kit. The uptake level was represented as ng FITC associated with 1 mg of cellular protein (ng FITC/mg protein). Cells incubated with free FITC-Tris in the absence of polypeptides were included as a negative control. Commercial CPPs, such as HIV-TAT, Arg9, and poly-L-arginine (PLR) were used as internal controls.
2.8. Intracellular kinetics
DNA (1 mg/mL) was labeled with YOYO-1 (20 μM) at one dye molecule per 50 bp DNA in order to allow the quantification of the cellular uptake level [28]. YOYO-1-DNA was then allowed to form polyplexes with polypeptides at various N/P ratios as described above. Cells were seeded on 96-well plates at 1×104 cells/well and cultured for 24 h. The medium was replaced by opti-MEM followed by addition of the polyplexes at 0.1 μg YOYO-1-DNA/well. After incubation at 37 °C for 4 h, cells were washed with PBS containing heparin (20 U/mL) for four times to remove membrane-bound polyplexes, and were subsequently lysed with the RIPA lysis buffer (100 μL/well). YOYO-1-DNA content in the lysate was quantified by spectrofluorimetry (λex = 485 nm, λem = 530 nm), and the protein content was determined using the BCA kit. Cellular uptake level was expressed as ng YOYO-1-DNA associated with 1 mg of cellular protein (ng YOYO-1-DNA/mg protein).
To probe the internalization mechanism of the polyplexes, the cellular uptake study was also performed at 4 °C or in the presence of various endocytic inhibitors. Briefly, cells were incubated with polyplexes (N/P = 10) at 4 °C for 2 h wherein the energy-dependent endocytosis was blocked. Otherwise, cells were incubated with various endocytic inhibitors including chlorpromazine (10 μg/mL), methyl-β-cyclodextrin (mβCD, 5 mM) and wortmannin (10 μg/mL) for 30 min prior to the addition of the polyplexes and throughout the 2-h uptake study. Results were represented as percentage uptake level of control cells that were incubated with the polyplexes in the absence of inhibitors at 37 °C for 2 h.
The cellular internalization and distribution of polyplexes were further visualized by confocal laser scanning microscopy (CLSM, LSM 700, Zeiss, Germany). HeLa cells were seeded on the coverslip in 6-well plate at 1×105 cells/well and cultured for 24 h. The medium was replaced with opti-MEM (1.5 mL/well), and polypeptide/YOYO-1-DNA polyplexes (N/P = 10) were added at 1 μg YOYO-1-DNA/well before incubation at 37 °C for 4 h. Cells were then washed with heparin-PBS for four times, fixed with 4% paraformaldehyde (PFA), stained with DAPI (2 μg/mL) and Lysotracker®-Red (200 nM) before CLSM observation.
2.9. Cytotoxicity
Cells were seeded on 96-well plates at 1×104 cells/well and cultured for 24 h. The medium was replaced by opti-MEM (100 μL/well) into which polyplexes were added at 0.1, 0.2, 0.4, 1 and 2 μg DNA/well (N/P = 10) or at N/P ratios of 2.5, 5, 10, 15 and 20 (0.1 μg DNA/well). After incubation at 37 °C for 4 h, the medium was replaced by serum-containing DMEM and cells were further incubated for another 20 h in consistence with the transfection process. Cell viability was then evaluated by the MTT assay. Cells without polyplex treatment served as the control and results were represented as percentage viability of control cells.
2.10. Statistical analysis
Statistical analysis was performed using Student’s t-test and differences between test and control groups were judged to be significant at *p < 0.05 and very significant at **p < 0.01.
3. Results
3.1. Synthesis and characterization of the polypeptides
PPOBLG was polymerized via ROP of POB-L-Glu-NCA initiated by HMDS followed by side-chain functionalization via the azide-alkyne Huisgen cycloaddition, the so-called “click” chemistry [29]. HMDS allowed a controlled ROP, yielding well-defined polypeptides with narrow molecular weight distributions (MWDs, ~1.05) and desired degree of polymerization (DP = 49 at the M/I ratio of 50) as evidenced by the GPC analyses (Supplementary Fig. S1). Owing to the high efficiency of the “click” chemistry, the conjugation efficiencies of amine- or guanidine-containing side chains reached over 99% as determined by 1H NMR spectra (Supplementary Fig. S4–S10). All the polypeptides were well soluble in aqueous solutions at pH < 9, and adopted typical α-helical conformations as depicted by the characteristic double-minima ellipticity at 208 and 222 nm in the CD spectra, distinctively different from that of PLR with random coil conformation (Fig. 1A and Supplementary Fig. S11). The calculated helicity was similar among all guanidine-containing polypeptides (52~59%) and slightly higher for the polypeptides containing amine side chains (60% and 71%). Represented by the ellipticities at 222 nm, the helicities of all tested polymers remained unaltered within the concentration range of 0.025–0.2 mg/mL (Fig. 1B), indicating that they remained non-aggregated and monomeric in the aqueous solution despite the hydrophobic contents on side chains [14]. The helicities were ultra-stable within the pH range of 1–9 (Fig. 1C), which allowed these polypeptides to maintain membrane activities that were directly associated with the helical secondary structure at both the neutral extracellular pH and the acidic endosomal/lysosomal pH. As such, when used as gene delivery vectors, polypeptides with such unique helical stability would be able to trigger effective intracellular internalization as well as endosomal escape of the gene cargo by destabilizing/disrupting the cellular/endosomal membranes [15]. The helicities were also stable towards ionic strength increment up to 0.3 M (Fig. 1D), indicating that helical structure can be maintained for all test polypeptides under physiological conditions with ionic strength of approximately 0.15 M.
Fig. 1.

(A) CD spectra of polypeptides (0.1 mg/mL) in the aqueous solution (pH 7.0). Helical stability against polypeptide concentration (B), pH (C) and NaCl concentration (D) as indicated by the molar ellipticity at 222 nm.
3.2. Polyplex formation and characterization
DNA condensation by the polypeptides was characterized by the gel retardation assay (Fig. 2A), which showed that all test polypeptides were able to effectively condense DNA at the N/P ratio higher than 2, evidenced by the restricted DNA migration in the agarose gel. In consistence, a quantitative EB exclusion assay further revealed that more than 70% of the DNA was condensed by the cationic polypeptides at N/P ratios ≥ 5 (Fig. 2B). Guanidine-containing polypeptides displayed slightly higher DNA binding affinities than their amine-containing analogues, presumably because guanidine residues can form bidentate hydrogen bonding with phosphates on DNA molecules to provide additional binding affinities [30]. The obtained polyplexes were further characterized by DLS. As shown in Fig. 3A and 3B, all the polypeptides were able to condense DNA in water at the N/P ratio higher than 5 to form 100–200 nm polyplexes with positive surface charges. Polyplexes formed by guanidine-containing polypeptides displayed higher zeta potentials (~40 mV) than those formed by amine-containing polypeptides (~25 mV), which was probably due to the higher pKa value of the guanidine group. When prepared in buffer solution (0.2 M PBS), higher N/P ratio up to 20 was required to form polyplexes with comparable diameters (except T3), largely attributed to the charge screening effect induced by the excessive ions in the solution that diminished the electrostatic interactions between oppositely charged polypeptide and DNA (Fig. 3C). When polyplexes prepared in PBS were further diluted with PBS for 50 folds and incubated for up to 1.5 h, negligible increment of particle size was noted, indicating the desired stability of polyplexes against ionic strength and dilution (Fig. 3D).
Fig. 2.

DNA condensation by polypeptides at various N/P ratios as evaluated by the gel retardation assay (A) and EB exclusion assay (B). N represents naked DNA.
Fig. 3.

Particle size (A) and zeta potential (B) of polyplexes in DI water at various N/P ratios as determined by DLS measurement. (C) Particle size of polyplexes at various N/P ratios prepared in PBS. (D) Alteration of particle size of polyplexes (N/P = 20, prepared in PBS) upon dilution with PBS (x 50 fold).
3.3. In vitro gene transfection
The transfection efficiencies of polypeptides containing fixed side chain length yet different side charged groups (P3, T3, G3) were first evaluated in HeLa and COS-7 cells in serum-free media. As shown in Fig. 4, polypeptide bearing guanidine side chains exhibited remarkably higher transfection efficiency than their amine-containing counterparts, suggesting that guanidine groups played important roles in mediating effective gene delivery presumably due to their membrane activities. G3 exhibited notably higher transfection efficiency by two-order-of-magnitude than PLR with the random coil conformation, which further confirmed the importance of helicity.
Fig. 4.
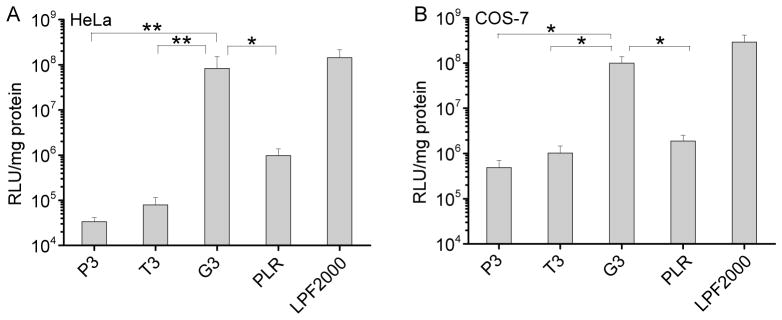
In vitro transfection efficiencies of polyplexes (N/P = 10) in HeLa (A) and COS-7 (B) cells. PLR and LPF2000 were included as positive controls.
The membrane activity of guanidine groups can be “self-activated” by hydrophobic moieties, and we thus compared the transfection efficiencies of polypeptides containing guanidine side terminal groups yet different side chain length (G3, G6, G8). As shown in Fig. 5A and 5B, at lower N/P ratios (2.5 and 5), polypeptides with longer hydrophobic side chains displayed higher transfection efficiencies, which substantiated our design strategy to strength the gene delivery capabilities of helical polypeptides by introducing hydrophobic domains. When the N/P ratio was further elevated to 10 and 15, G6 showed maximal transfection efficiency in HeLa cells while in COS-7 cells, G6 and G8 demonstrated the highest and comparable efficiencies. The transfection efficiency decreased rather than kept increasing when the N/P ratio was further increased up to 20, mainly attributed to the cytotoxicity of the materials at higher dosages. Side chains of polypeptide containing primary amine groups was also elongated, while they all suffered from relatively low gene delivery efficiencies although increment in the transfection efficiency was also obviously noted (Supplementary Fig. S12). With the synergistic effect of guanidine and hydrophobic domains, G6 and G8 outperformed the commercial reagent, LPF2000, by 3~6 fold in terms of the in vitro transfection efficiency.
Fig. 5.

In vitro transfection efficiencies of polyplexes at various N/P ratios in HeLa (A) and COS-7 (B) cells in the serum-free medium. Transfection efficiencies in HeLa (C) and COS-7 (D) cells in the presence of serum.
Compromised transfection efficiency by serum is one major challenge of polycation based non-viral gene vectors [31, 32]. We thus further evaluated the gene trasnsfection efficiencies of polyplexes in 10% FBS. At the same DNA amount (0.1 μg/well) and N/P ratio, greatly compromised gene transfection efficiency was noted for all test materials (Fig. 5C and 5D), presumably because the anionic proteins in the serum can bind and destabilize the polyplexes. However, when we increased the total amount of DNA or the N/P ratio, the transfection efficiency can be largely recovered, indicating that excessive amount of free polypeptides might counteract the serum binding. G8 displayed higher resistance against serum than G3 and G6 at lower DNA amount (0.1 μg/well), probably because the extra hydrophobicity mediated stronger interactions between DNA and the polypeptide, thus partially compensating competitive DNA replacement by serum. When the DNA amount was increased to 0.2 μg/well, G6 reached maximal transfection efficiencies in HeLa cells while G6 and G8 showed comparably high efficiencies in COS-7 cells, similar to the transfection capabilities in the absence of serum.
3.4. Intracellular kinetics
The performance of non-viral gene delivery vectors is closely related to their intracellular kinetics, such as the cellular uptake level, internalization pathway, and endosomal escape mechanism [33]. Hence, we probed the intracellular kinetics of the polypeptide/DNA polyplexes, attempting to provide mechanistic understandings on their gene delivery capabilities.
As shown in Fig. 6 and Supplementary Fig. S13, all tested polypeptides were able to notably promote the cellular uptake of YOYO-1-DNA, significantly outperforming LPF2000 by 4–8 fold. Polypeptides containing guanidine and primary amine side groups (G3 and P3) showed comparable cell uptake level which outperformed that of the tertiary amine-containing polypeptide (T3). In consistent with the transfection efficiencies, an obvious increment in the cell uptake level was also noted when the hydrophobic side chain length was elongated (G3, G6, and G8), which indicated that hydrophobic domain activated the guanidine groups to aid the polypeptide with higher membrane activities.
Fig. 6.
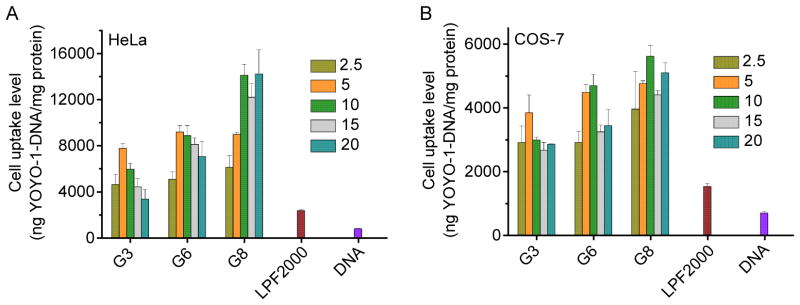
Cellular uptake levels of polypeptide/YOYO-1-DNA polyplexes in HeLa (A) and COS-7 (B) cells at various N/P ratios.
Guanidine-rich CPPs have been reported to facilitate the cellular internalization via a non-endocytic, direct translocation pathway [22], and the internalization pathway also dominates the intracellular fate and ultimately the transfection capacity. As such, we next performed the cell uptake studies under various conditions that are known to inhibit specific uptake pathway. Energy-dependent endocytosis was completely blocked at low temperature (4 °C); chlorpromazine inhibited clathrin-mediated endocytosis (CME) by inducing the dissociation of the clathrin lattice; methyl-β-cyclodextrin (mβCD) inhibited caveolae by depleting cholesterol; wortmannin inhibited macropinocytosis by inhibiting phosphatidyl inositol-3-phosphate [33]. As shown in Fig. 7A, the cellular uptake level was only inhibited by 30% at 4 °C, suggesting that majority of the polyplexes entered the cells via an energy-independent and non-endocytosis pathway. The cell uptake level was also reduced by mβCD (~40%) but not chlorpromazine (except G3) or wortmannin, indicating that polyplexes were internalized mainly via caveolae rather than CME. Because caveolar uptake is a non-acidic and non-digestive route of internalization, cargos in the caveosomes can be directly transported to the Golgi and/or endoplasmic reticulum. Thereby, they would not experience endosomal entrapment and lysosomal degradation, which greatly contributed to the relatively high gene transfection efficiencies of the guanidine-terminated helical polypeptides [33]. Such statement was also supported by the observation that internalized polyplexes (green fluorescence) greatly separated from the Lysotracker-stained endosomes/lysosomes (red fluorescence, Fig. 7E), which suggested that they did not experience severe endosomal entrapment that served as one of the major intracellular barriers against effective gene transfection [34]. In consistence with the internalization mechanism, transfection efficiency at 4 °C still remained a relatively high level despite a notable decrease compared to that at 37 °C (Fig. 7B).
Fig. 7.

Intracellular kinetics of polypeptide/YOYO-1-DNA polyplexes. (A) Cellular uptake of polyplexes (N/P = 10) in COS-7 cells at 4 °C or in the presence of various endocytic inhibitors. (B) Transfection efficiencies of polyplexes (N/P = 10) at 4 °C and 37 °C in COS-7 cells. (C) Cell uptake level of FITC-Tris in HeLa (C) and COS-7 (D) cells following co-incubation with the polypeptides for 2 h at 37 °C. (E) CLSM images of HeLa cells following incubation with G6/YOYO-1-DNA polyplexes (N/P = 10) at 37 °C for 4 h. Bar represented 20 μm.
Because a large amount of the polyplexes was internalized via direct translocation rather than endocytosis, we then investigated the pore formation on cell membranes, an important membrane penetration mechanism induced by the helical polypeptide [15, 28, 32, 35]. FITC-Tris, a membrane-impermeable fluorescent dye in the non-reactive form of FITC after reaction of FITC with Tris, was used as a biomarker, and the pore formation level on cell membranes can be represented by the cellular uptake level of FITC-Tris after co-incubation with the polypeptides. As shown in Fig. 7C and 7D, free FITC-Tris was negligibly taken up by HeLa and COS-7 cells because of its impermeability across the cell membranes. When treated with guanidine-containing polypeptides, FITC-Tris uptake level was dramatically increased by 2 orders of magnitude. Polypeptides with longer hydrophobic side chains triggered higher FITC-Tris uptake level, which substantiated that the guanidine-rich, helical polypeptides triggered strong pore formation on cell membranes to facilitate the direct diffusion of FITC-Tris into the cytoplasm. The increment of hydrophobicity further strengthened their pore formation capabilities. In comparison, the random-coiled PLR and other short CPPs including HIV-TAT and Arg9, showed remarkably lower pore formation properties. Such pore formation mechanism of polypeptides thus allowed direct permeation of the gene cargos into cells, and accordingly internalized YOYO-1-DNA was distributed to the whole cytoplasm in a permeated manner without being entrapped in endosomal/lysosomal compartments as shown in CLSM images (Fig. 7E).
3.5. Cytotoxicity
Cytotoxicity of the polypeptide/DNA polyplexes at various DNA concentrations or N/P ratios was assessed in both HeLa and COS-7 cells by the MTT assay. As shown in Fig. 8 and Supplementary Fig. S14, the cytotoxicity was both dose- and cell line-dependent. At various DNA concentrations or N/P ratios, guanidine-containing polypeptide showed higher cytotoxicity than the amine-containing analogues, and polypeptide with longer side chains also exhibited higher cytotoxicity, which correlated well with their pore formation capacities. It was thus assumed that at high concentrations, polypeptides with excessive membrane activities would lead to irreversible membrane damage. For each individual polypeptide, their toxicity to HeLa cells was higher than to COS-7 cells, which was attributed to the different tolerability of each cell type towards polymer-induced cytotoxicity.
Fig. 8.

Cytotoxicity of polypeptide/DNA polyplexes towards HeLa (A, C) and COS-7 (B, D) cells as determined by the MTT assay. The N/P ratio was maintained constant at 10 (A and B) while the DNA amount was maintained constant at 0.1 μg/well (C and D).
4. Discussion
Low transfection efficiency is the key drawback of non-viral gene delivery, stemming from multiple extra- and intracellular barriers associated with delivery process. Above all, vectors should be able to effectively condense DNA into nano-scale complexes, maintain stable under physiological conditions, and then mediate efficient cellular uptake via either endocytic or non-endocytic pathway. Among the existing non-viral vectors, polycations containing cationic amine groups such as poly(ethylenimine) (PEI), PLL and poly(amidoamine) dendrimer (PAMAM) are most widely used, and upon protonation under physiological conditions, they can effectively condense DNA to form polyplexes. Further functionalization, exemplified by modification with targeting ligands or PEGylation, could further potentiate the cellular delivery efficiency while reducing the material toxicity induced by cationic charges [36]. Studies have also shown that such polyplexes are often internalized via endocytosis [37], which leads to the entrapment by endosomes and ultimate degradation of the gene cargo in late lysosomes filled with hydrolytic enzymes. Therefore, endosomal entrapment poses a critical barrier that impedes the efficacy of non-viral vectors. PEI, one of the most widely used non-viral vectors containing large amount of primary, secondary, and tertiary amine groups, is able to buffer the acidic environment in endosomes, cause influx of extra protons into the endosomes, and finally osmotically rupture the endosomes, commonly known as the “proton sponge” effect [37]. However, this hypothesis has been debated and recent studies have suggested that the incorporation of amine groups with buffering capacity does not necessarily result in effective endosomal escape [38, 39]. At later stage, DNA needs to be released from complexes and transported from cytoplasm to the nuclei in order to initiate successful transcription.
Compared with amine groups, guanidine groups can bind phosphate anions in the DNA molecule to form characteristic pairs of parallel hydrogen bonds, which provide additional binding affinity with DNA other than electrostatic interaction [40]. Similarly, such bidentate hydrogen bonding could also be formed between guanidine groups and negatively charged carboxylates, sulfates, and phosphates on cell surface. The resultant ion-pairs could then translocate across the cell membrane due to the influence of the membrane potential, which is a different pathway from conventional endocytosis [30, 41]. Although multiple mechanisms have been proposed and there is no consensus on a universal mechanism, such membrane activity of guanidine groups has been confirmed to enable efficient cellular uptake and endosomal escape of various cargoes by destabilizing the cellular as well as endosomal membranes [42]. Herein, we incorporated guanidine groups into our design of helical polypeptide-based non-viral vectors, and compared the gene delivery efficiencies with their amine-containing analogues. G3 possessed much higher gene transfection efficiency than P3 and T3, despite their comparable DNA binding strength and cellular uptake level (Supplementary Fig. S13). Given that guanidine-containing polypeptides (G3, G6 and G8) were mainly internalized via energy-independent permeation or caveolae-mediated endocytosis that did not experience endosomal entrapment, we speculated that such effective pathways of cell entry largely contributed to the excellent gene transfection efficiencies of the guanidine modified polypeptides.
In addition to the membrane-penetrating capabilities of guanidine groups, secondary conformation and hydrophobicity also affect the membrane activities of CPPs. We thus hypothesized that the incorporation of helical structures and hydrophobic content could alter the membrane activities of polypeptides and ultimately affect their gene delivery efficiencies. In comparison to PLR which lacks hydrophobicity and adopts random-coiled structure, G3 possessed long hydrophobic side chains and thus was able to adopt helical conformation because the electrostatic interactions among pendant charged groups was greatly diminished. Greatly enhanced pore formation and gene transfection efficiency were noted when compared to PLR, which substantiated the essential roles of the introduced helicity and hydrophobicity. Indeed, our previous studies on the comparison of cationic polypeptides with exactly the same chemical composition yet different secondary structure (helix vs. random coil) also revealed that helical structure remarkably contributed to the strong pore formation properties of polypeptides to mediate effective gene transfection [15, 32]. Further increasing the hydrophobicity by elongating the side chain length (G3, G6, G8) resulted in further elevated pore formation activities, and consistently contributed to enhanced transfection capabilities. As such, G8 was able to mediate effective transfection at the N/P ratio of 5 while comparable efficiencies were noted at the N/P ratio 10 for G3 and G6 (Fig. 5A and 5B). In consistence, G6 and G8 showed notably higher resistance against serum than G3, a desired property towards in vivo gene delivery.
The pore formation mechanism, although highly effective in triggering cellular internalization and endosomal escape, will at the same cause irreversible damage to the cell membranes at excessive levels. This holds true for guanidine-containing helical polypeptides, where longer hydrophobic side chain length rendered the polypeptides with higher membrane activities but also higher cytotoxicities. Therefore, in HeLa cells that are more vulnerable to material-induced cytotoxicity, G8 showed decreased rather than increased transfection efficiency compared to G6 because of greatly compromised cell viability. COS-7 cells were more robust in terms of material tolerance, and thus G6 and G8 showed comparable transfection efficiencies. Serum can shield the surface charge of the polyplexes to alleviate material cytotoxicity [43], and therefore at the same DNA amount, G8 with the highest membrane activity revealed the highest transfection efficiency. At higher DNA amount, excessive G8 exerted appreciable cytotoxicities and thus the transfection efficiency remained slightly lower than G6, the same trend as observed under serum-free condition. These results collectively suggest that an optimal balance between membrane activity and cytotoxicity is crucial towards the maximization of the gene delivery efficiencies of helical polypeptides and other polycation-based non-viral vectors.
5. Conclusion
A set of α-helical, cationic polypeptides with various side charged groups and hydrophobic side chain lengths were synthesized, and the effect of polypeptide structure on the gene delivery efficiencies was systematically explored. Incorporation of guanidine group, helical conformation, and hydrophobic content into the polypeptide design collectively led to materials with high membrane activities and transfection efficiencies, significantly outperforming commercial CPPs and transfection reagent LPF2000. Although hydrophobicity enhanced the membrane activities of polypeptides via the pore formation mechanism, excessive hydrophobicity will simultaneously potentiate the irreversible damage to cell membranes and ultimately impair the transfection efficiencies. Therefore, a proper balance between membrane activities and cytotoxicities is demanded during the material design in order to maximize the gene delivery efficiency. Such study on the structure-property relationship provides insights into the rational design of non-viral vectors and the top-performing materials identified (G6 and G8) are promising additions to existing vectors.
Supplementary Material
Scheme 1.

Synthetic routes of γ-(4-propargyloxybenzyl)-L-glutamic acid based N-carboxyanhydride (POB-L-Glu-NCA) (A), azido amines/guanidines (B), and amine/guanidine functionalized polypeptides (C).
Acknowledgments
J.C. acknowledges support from the NSF (CHE-1153122), the NIH (Director’s New Innovator Award 1DP2OD007246 and 1R21EB013379), and the CABPN (I/UCRC NSF A1422).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Candolfi M, Xiong W, Yagiz K, Liu C, Muhammad AK, Puntel M, et al. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc Natl Acad Sci USA. 2010;107:20021–6. doi: 10.1073/pnas.1008261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang JL, Tang GP, Shen J, Hu QL, Xu FJ, Wang QQ, et al. A gene nanocomplex conjugated with monoclonal antibodies for targeted therapy of hepatocellular carcinoma. Biomaterials. 2012;33:4597–607. doi: 10.1016/j.biomaterials.2012.02.045. [DOI] [PubMed] [Google Scholar]
- 3.Cho SK, Pedram A, Levin ER, Kwon YJ. Acid-degradable core–shell nanoparticles for reversed tamoxifen-resistance in breast cancer by silencing manganese superoxide dismutase (MnSOD) Biomaterials. 2013;34:10228–37. doi: 10.1016/j.biomaterials.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin L, Song Z, Qu Q, Kim KH, Zheng N, Yao C, et al. Supramolecular self-assembled nanoparticles mediate oral delivery of therapeutic TNF-α siRNA against systemic inflammation. Angew Chem Int Ed. 2013;52:5757–61. doi: 10.1002/anie.201209991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–58. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 6.Zhang QF, Yi WJ, Wang B, Zhang J, Ren LF, Chen QM, et al. Linear polycations by ring-opening polymerization as non-viral gene delivery vectors. Biomaterials. 2013;34:5391–401. doi: 10.1016/j.biomaterials.2013.03.083. [DOI] [PubMed] [Google Scholar]
- 7.Kolusheva S, Shahal T, Jelinek R. Peptide-membrane interactions studied by a new phospholipid/polydiacetylene colorimetric vesicle assay. Biochemistry-Us. 2000;39:15851–9. doi: 10.1021/bi000570b. [DOI] [PubMed] [Google Scholar]
- 8.Smith BA, Daniels DS, Coplin AE, Jordan GE, McGregor LM, Schepartz A. Minimally cationic cell-permeable miniature proteins via alpha-helical arginine display. J Am Chem Soc. 2008;130:2948–9. doi: 10.1021/ja800074v. [DOI] [PubMed] [Google Scholar]
- 9.Saw PE, Ko YT, Jon S. Efficient Liposomal Nanocarrier-mediated oligodeoxynucleotide delivery involving dual sse of a cell-penetrating peptide as a packaging and intracellular delivery agent. Macromol Rapid Comm. 2010;31:1155–62. doi: 10.1002/marc.200900861. [DOI] [PubMed] [Google Scholar]
- 10.Chu DSH, Schellinger JG, Bocek MJ, Johnson RN, Pun SH. Optimization of Tet1 ligand density in HPMA-co-oligolysine copolymers for targeted neuronal gene delivery. Biomaterials. 2013;34:9632–7. doi: 10.1016/j.biomaterials.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei H, Volpatti LR, Sellers DL, Maris DO, Andrews IW, Hemphill AS, et al. Dual responsive, stabilized nanoparticles for efficient in vivo plasmid delivery. Angew Chem Int Ed. 2013;52:5377–81. doi: 10.1002/anie.201301896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okuda T, Sugiyama A, Niidome T, Aoyagi H. Characters of dendritic poly((L)-lysine) analogues with the terminal lysines replaced with arginines and histidines as gene carriers in vitro. Biomaterials. 2004;25:537–44. doi: 10.1016/s0142-9612(03)00542-8. [DOI] [PubMed] [Google Scholar]
- 13.Munoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 14.Lu H, Wang J, Bai YG, Lang JW, Liu SY, Lin Y, et al. Ionic polypeptides with unusual helical stability. Nat Commun. 2011:2. doi: 10.1038/ncomms1209. [DOI] [PubMed] [Google Scholar]
- 15.Gabrielson NP, Lu H, Yin LC, Kim KH, Cheng JJ. A cell-penetrating helical polymer for siRNA delivery to mammalian cells. Mol Ther. 2012;20:1599–609. doi: 10.1038/mt.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop CJ, Ketola TM, Tzeng SY, Sunshine JC, Urtti A, Lemmetyinen H, et al. The effect and role of carbon atoms in poly(beta-amino ester)s for DNA binding and gene delivery. J Am Chem Soc. 2013;135:6951–7. doi: 10.1021/ja4002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parelkar SS, Chan-Seng D, Emrick T. Reconfiguring polylysine architectures for controlling polyplex binding and non-viral transfection. Biomaterials. 2011;32:2432–44. doi: 10.1016/j.biomaterials.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Venkataraman S, Ong WL, Ong ZY, Loo SCJ, Ee PLR, Yang YY. The role of PEG architecture and molecular weight in the gene transfection performance of PEGylated poly(dimethylaminoethyl methacrylate) based cationic polymers. Biomaterials. 2011;32:2369–78. doi: 10.1016/j.biomaterials.2010.11.070. [DOI] [PubMed] [Google Scholar]
- 19.Hinton TM, Guerrero-Sanchez C, Graham JE, Le T, Muir BW, Shi SN, et al. The effect of RAFT-derived cationic block copolymer structure on gene silencing efficiency. Biomaterials. 2012;33:7631–42. doi: 10.1016/j.biomaterials.2012.06.090. [DOI] [PubMed] [Google Scholar]
- 20.Eltoukhy AA, Siegwart DJ, Alabi CA, Rajan JS, Langer R, Anderson DG. Effect of molecular weight of amine end-modified poly(beta-amino ester)s on gene delivery efficiency and toxicity. Biomaterials. 2012;33:3594–603. doi: 10.1016/j.biomaterials.2012.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed M, Jawanda M, Ishihara K, Narain R. Impact of the nature, size and chain topologies of carbohydrate-phosphorylcholine polymeric gene delivery systems. Biomaterials. 2012;33:7858–70. doi: 10.1016/j.biomaterials.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliver Rev. 2008;60:452–72. doi: 10.1016/j.addr.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Som A, Reuter A, Tew GN. Protein transduction domain mimics: the role of aromatic functionality. Angew Chem Int Ed. 2012;51:980–3. doi: 10.1002/anie.201104624. [DOI] [PubMed] [Google Scholar]
- 24.Som A, Tezgel AO, Gabriel GJ, Tew GN. Self-activation in de novo designed mimics of cell-penetrating peptides. Angew Chem Int Ed. 2011;50:6147–50. doi: 10.1002/anie.201101535. [DOI] [PubMed] [Google Scholar]
- 25.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, et al. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry-Us. 2000;39:11657–66. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X, Yin LC, Ding JY, Tang C, Gu SH, Yin CH, et al. Thiolated trimethyl chitosan nanocomplexes as gene carriers with high in vitro and in vivo transfection efficiency. J Control Release. 2010;144:46–54. doi: 10.1016/j.jconrel.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 27.Ter-Avetisyan G, Tuennemann G, Nowak D, Nitschke M, Herrmann A, Drab M, et al. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284:3370–8. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin L, Song Z, Kim KH, Zheng N, Tang H, Lu H, et al. Reconfiguring the architectures of cationic helical polypeptides to control non-viral gene delivery. Biomaterials. 2013;34:2340–9. doi: 10.1016/j.biomaterials.2012.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engler AC, Lee HI, Hammond PT. Highly efficient “grafting onto” a polypeptide backbone using click chemistry. Angew Chem Int Ed. 2009;48:9334–8. doi: 10.1002/anie.200904070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothbard JB, Jessop TC, Wender PA. Adaptive translocation: the role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliver Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Srinivasachari S, Liu YM, Zhang GD, Prevette L, Reineke TM. Trehalose click polymers inhibit nanoparticle aggregation and promote pDNA delivery in serum. J Am Chem Soc. 2006;128:8176–84. doi: 10.1021/ja0585580. [DOI] [PubMed] [Google Scholar]
- 32.Yin L, Song Z, Kim KH, Zheng N, Gabrielson NP, Cheng J. Non-viral gene delivery via membrane-penetrating, mannose-targeting supramolecular self-assembled nanocomplexes. Adv Mater. 2013;25:3062–70. doi: 10.1002/adma.201205088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 34.Kwon YJ. Before and after endosomal escape:roles of stimuli-converting siRNA/polymer interactions in determining gene silencing efficiency. Acc Chem Res. 2012;45:1077–88. doi: 10.1021/ar200241v. [DOI] [PubMed] [Google Scholar]
- 35.Tang H, Yin L, Kim KH, Cheng J. Helical poly(arginine) mimics with superior cell-penetrating and molecular transporting properties. Chem Sci. 2013;4:3839–44. doi: 10.1039/C3SC51328A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian HY, Deng C, Lin H, Sun J, Deng M, Chen X, et al. Biodegradable cationic PEG-PEI-PBLG hyperbranched block copolymer: synthesis and micelle characterization. Biomaterials. 2005;26:4209–17. doi: 10.1016/j.biomaterials.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Rejman J, Bragonzi A, Conese M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol Ther. 2005;12:468–74. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 38.Funhoff AM, van Nostrum CF, Koning GA, Schuurmans-Nieuwenbroek NME, Crommelin DJA, Hennink WE. Endosomal escape of polymeric gene delivery complexes is not always enhanced by polymers buffering at low pH. Biomacromolecules. 2004;5:32–9. doi: 10.1021/bm034041+. [DOI] [PubMed] [Google Scholar]
- 39.Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther. 2013;21:149–57. doi: 10.1038/mt.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aissaoui A, Oudrhiri N, Petit L, Hauchecorne M, Kan E, Sainlos M, et al. Progress in gene delivery by cationic lipids: guanidinium-cholesterol-based systems as an example. Curr Drug Targets. 2002;3:1–16. doi: 10.2174/1389450023348082. [DOI] [PubMed] [Google Scholar]
- 41.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–7. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 42.Stanzl EG, Trantow BM, Vargas JR, Wender PA. Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications. Acc Chem Res. 2013 doi: 10.1021/ar4000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Audouy S, Molema G, de Leij L, Hoekstra D. Serum as a modulator of lipoplex-mediated gene transfection: dependence of amphiphile, cell type and complex stability. J Gene Med. 2000;2:465–76. doi: 10.1002/1521-2254(200011/12)2:6<465::AID-JGM141>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.