

Abstract
Fluorescent proteins have become indispensable imaging tools for biomedical research. continuing progress in fluorescence imaging, however, requires probes with additional colors and properties optimized for emerging techniques. Here we summarize strategies for development of red-shifted fluorescent proteins. We discuss possibilities for knowledge-based rational design based on the photochemistry of fluorescent proteins and the position of the chromophore in protein structure. We consider advances in library design by mutagenesis, protein expression systems and instrumentation for high-throughput screening that should yield improved fluorescent proteins for advanced imaging applications.
The discovery of homologs of GFP, which emit not only green but also yellow, orange and red fluorescence, has provided a powerful boost for in vivo labeling1. Similarly to GFP, these fluorescent proteins have been developed into monomers suitable for protein tagging: this includes conventional fluorescent proteins, fluorescent proteins with a large Stokes shift of fluorescence emission (LSS-FPs), photoactivatable and photoswitchable fluorescent proteins (PA-FPs and PS-FPs, respectively) and fluorescent timers2. Several advances in the design of red-shifted—such as orange, red, and far-red—fluorescent proteins (here generally referred to as red fluorescent proteins (RFPs)), and RFP-based biosensors with new spectral and photochemical properties have also been achieved: reduced autofluorescence, low light scattering and minimal absorbance at the longer wavelengths make RFPs superior probes for super-resolution, deep-tissue and two-photon imaging. However, no existing RFP is perfect; each still has some suboptimal key characteristics such as brightness, pH stability, photostability, maturation rate, photoactivation, photoswitching contrast or monomeric state (Supplementary Table 1). Some RFPs undergo undesirable photochromism and photoconversion during imaging or complex photobehavior during switching, and few attempts have been made to optimize such properties as intracellular life span and cytotoxicity. Although many RFPs have properties that are optimal for specific applications, no single RFP combines several of them (Supplementary Table 2). Moreover, there is a large demand for RFPs with improved brightness. Also, monomeric LSS-FPs and PA-FPs are available in green and red colors only2,3. Finally, red-shifted fluorescent proteins also hold a great potential for the engineering of biosensors, but this has not been exploited in full as yet. Thus, we expect that new strategies for generating enhanced RFPs will have an impact on many fields.
Here we first describe chemical transformations of the RFP chromophores, the understanding of which gives the basis for a knowledge-guided design of new red-shifted fluorescent proteins. The chemistry of the RFP chromophores is more diverse and complex than that of the GFP-like chromophore and thus has substantially more potential for selection and fine-tuning. Nonetheless, although we focus on the development of new RFPs, the general themes we discuss apply to fluorescent proteins of all colors. Based on in depth analysis of the dependence of RFP properties on amino acids surrounding the chromophore, we describe approaches for the development of new red-shifted fluorescent proteins with desired phenotypes. Finally, we consider new methods to improve molecular evolution and discuss possible resulting RFPs for emerging imaging techniques.
Rational design of fluorescent proteins
A typical process for the development of fluorescent proteins with desired properties includes rational design followed by several steps of directed molecular evolution (Fig. 1 and Supplementary Note). Rational design relies on knowledge about chromophore transformations in RFPs.
Figure 1.
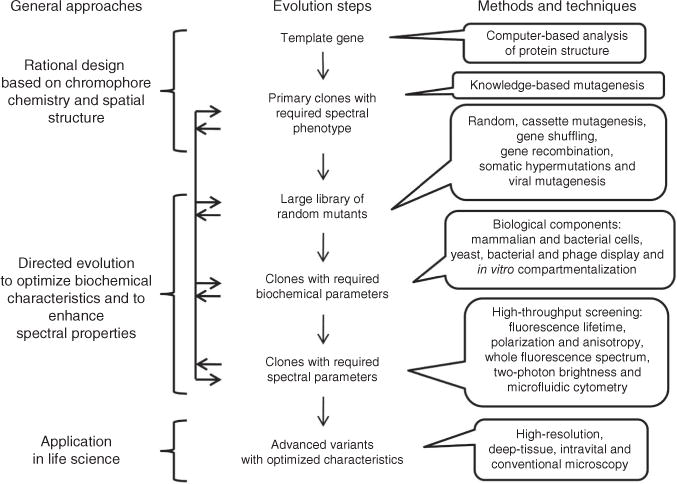
Steps in the directed molecular evolution of fluorescent probes. Vertical arrows indicate the typical order of steps. Horizontal arrows represent possible transitions between the steps of molecular evolution, which can be repeated several times in different order.
Chromophore transformations
The chemistry of RFPs is determined by the chromophore-forming tripeptide and its immediate environment. Although most chemical transformations occur in the chromophore, its amino acid microenvironment has a crucial role for catalysis. Currently known red-shifted fluorescent proteins have one of two major types of red chromophores, called the DsRed-like chromophore 5a, 6a (see Fig. 2 for chromophore structure and numbering) and the Kaede-like chromophore 14, after the first proteins in which they had been found1,2.
Figure 2.
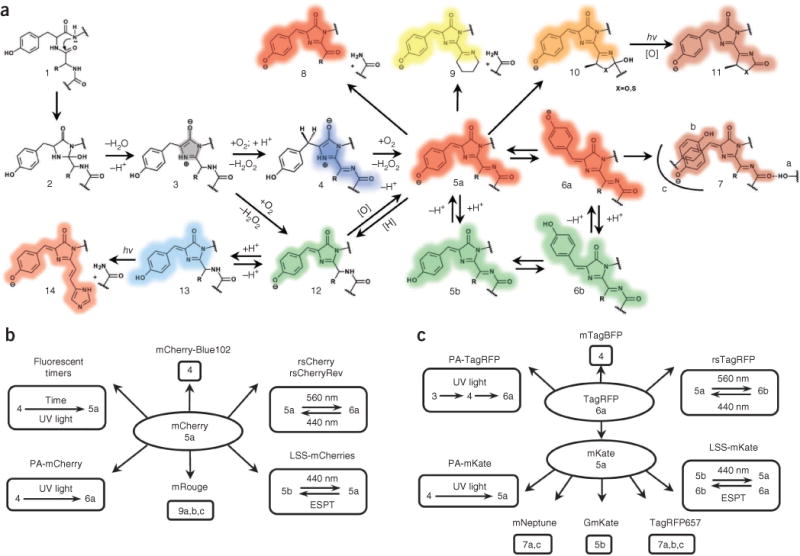
Major chemical transformations of the chromophores in red fluorescent proteins. (a–c) Transformations in fluorescent protein subfamilies derived from red fluorescent protein (a), mCherry (b) and TagRFP (c). The colored shading of the chemical structures (a) and chromophore numbers (b,c) correspond to the spectral range of the chromophore fluorescence emission. Gray shading denotes the nonfluorescent state; [H] denotes reduction; and [O] denotes oxidation. The chromo states (structures 5, 10 and 13) are not necessarily caused by a cis-trans chromophore isomerization but may result from modifications of the chromophore environment of the same isoform that decrease quantum yield. hv, photon.
Two chemical mechanisms for DsRed-like chromophore formation have been discovered2: it is formed either through autocatalytic post-translational modifications of a blue monomeric (m)TagBFP-like chromophore 4 (refs. 4,5) (found in mTagBFP6 and in the blue form of fluorescent timers7) or via induction by irradiation with violet light8, as in the case of PAmCherry9, PATagRFP10 and PAmKate11. The DsRed-like chromophore can exist in neutral 5b, 6b or anionic 5a, 6a states, which interconvert upon pH changes and excited-state proton transfer3,12. Cyclization of the N-acylimine group in the DsRed-like chromophore results in an mOrange-like or mKO-like 10 chromophore or a zFP538-like 9 chromophore, exhibiting orange emission1,2. Formation of the zFP538-like chromophore 9 is accompanied by backbone cleavage. The photoconversion of the DsRed-like chromophore 5a, 6a into the GFP-like chromophore 12 has been demonstrated for mKate, HcRed and DsRed using high-power irradiation13. The converse transition 12 to 5a, 6a, called ‘redding’, was autocatalytic14 or induced by oxidants for several different GFPs15. Unlike the GFP-like chromophore, the illumination of mOrange-like or mKO-like chromophores 10 resulted in an appearance of far-red 11 and red 5a species, respectively13,16; the absorbance (610 nm) and emission (650 nm) of these far-red species suggested formation of new type of chromophore (Supplementary Table 3). The far-red–shifted spectral properties of chromophore 7 observed in mRouge17, E2-Crimson18, mNeptune19 and TagRFP657 (ref. 20) result from the realization of hydrogen bonding, stacking interactions and/or hydrophobic residues surrounding the DsRed-like chromophore.
The Kaede-like chromophore 14 is characteristic for the red state of green-to-red PS-FPs including Kaede, EosFP, mEos2, mIrisFP, Dendra2, mKikGR and mClavGRs1,2,21. In the green state, these Kaede-like PS-FPs have the GFP-like chromophore 12. The formation of the Kaede-like chromophore requires a histidine residue at the first position of the chromophore-forming tripeptide Xnn65-Tyr66-Gly67 (where Xnn is any amino acid and numbering of amino acids follows that of GFP), limiting its possible chemical transformations.
Based on the current data on fluorescent proteins, three conclusions can be formulated: (i) most chemical transitions between chromophore structures occur autocatalytically, photochemically (by photoinduction) or are blocked; (ii) the same chromophore structure can be either in a fluorescent state (that absorbs and emits) or in a chromo state (that absorbs but does not emit); and (iii) autocatalytic versus photoinduced versus blocked states and fluorescent versus chromo states are mainly determined by amino acid residues in the chromophore and in its nearby environment. Some of these conclusions can be illustrated by two RFP subfamilies, derived from mCherry and TagRFP (Fig. 2b,c). These observations form the physicochemical basis for the development of RFPs with new photochemical behavior.
Choosing a template
Choosing an appropriate starting template for development of fluorescent proteins is an important step that strongly influences the final results of the process. A list of possible templates is made based on their spectral and biochemical properties, using as criteria both the required phenotype and the availability of three-dimensional structures. High amino acid homology between potential precursors and already known RFPs with the desired phenotype but having different spectral characteristics or other suboptimal properties can be used as an additional screening criterion. It is preferable to select templates with beneficial characteristics such as high brightness, pH stability, photostability, fast maturation and low cytotoxicity. For example, the brightness, maturation and pH stability of PATagRFP and PAmCherry1 proteins correlates with those characteristics of their precusors10.
Knowledge-based mutagenesis
The knowledge of chromophore chemistry and overall protein structure enables the directed engineering of variants with required phenotypes (Table 1 and Supplementary Table 4). As stated above, the diverse chemical transitions and spectral and photochemical properties of red-shifted fluorescent proteins are mainly due to the interactions between the chromophore and its immediate environment. The main targets for rational design are therefore the amino acid residues around the chromophore.
Table 1.
Residues responsible for RFP phenotypes and properties
| Phenotype or property | Structure or transition | Key positions | ||||
|---|---|---|---|---|---|---|
| mTagBFP-like | 4 | 84a Phe, Trp, Leu |
148a Phe, Ile |
203a Ile, Tyr, Phe |
||
| Fluorescent timer-like | 4 → 5a, 6a | 69a Arg |
84a Trp |
|||
| Photoactivatable-like | 4 → 5a,6a | 69a Asn, Ser, Lys, Gln |
203a Arg |
|||
| Photoswitchable-like | 5a ↔ 6a | 148a Cys, Ala, Thr, Ser |
165a Ser, Gly, Ile |
167a Met, Gln |
203a His, Ile |
|
| Far-red shifted–like | ||||||
| Hydrogen bonding to N-acylimine | 7a | 14b Thr, Phe |
16b Thr, Glu, Gln, Val |
44b Gly, Gln, Glu, Ala, Cys, Met |
||
| Hydrophobic packing | 7c | 84c Phe, Leu, Trp |
148c His, Ser, Cys, Asn |
165c Met,T,C, Asn, Ser |
167c Mer, Leu, Gln |
|
| π stacking | 7b | 65c Phe, Met, Gln |
69c His |
148c His, Ser, Cys, Asn |
203c Tyr, His, Arg, Thr, Ile |
|
| Large Stokes shift–like | 5b, 6b ↔ 5a, 6a | 148b Asp |
165b Asp, Glu |
167b Asp, Glu, Lys |
||
| High quantum yield | 70c Thr |
148c Ser, Asn |
167c Met |
|||
| High photostability | 64a His |
99a Tyr |
165c Thr, Ala, Ile |
|||
| High pH stability | 69b Arg |
167b Lys |
||||
| Fast maturation | 42c Gln |
69c Lys, Arg |
179c Val, Ala, Cys |
224c Ser, Ala |
||
| Monomeric state | 126c Arg |
162c Glu, Lys, Arg |
166c Asp, Lys, His |
168c Ala, Arg |
||
Residues at this position provide the respective phenotype in a concerted manner (residue numbering follows that for jellyfish GFP).
Residues at this position provide the respective phenotype independently.
Residues at this position provide the respective phenotype either in a concerted manner or independently.
There are currently ~80 red-shifted fluorescent proteins of different phenotypes. An analysis of the properties of these existing RFPs and of their chromophore’s immediate environment suggests key and supporting positions for each phenotype. Amino-acid residues at the key positions provide a principal RFP phenotype, whereas residues at the supporting positions tune the RFP properties (Supplementary Note). Both key and supporting residues are responsible for the different photoinduced or autocatalytic chromophore transitions, photophysical chromophore properties and oligomeric state of RFPs (Table 1, Supplementary Table 4 and Supplementary Fig. 1). For example, an alignment of the amino acid sequences of red-shifted PA-FPs with those of the parental RFPs and with each other reveals positions at which the residues were substituted. This allows us to identify positions such as 69 and 203, minimally required for the photoactivatable-like phenotype, as key positions (Table 1). Additional analysis of literature on available mutants and on X-ray structures of the related RFPs allowed us to determine ten supporting positions responsible for this phenotype (Supplementary Table 4). We note that there are many examples where a particular property (for example, red color in DsRed) is a synergistic effect of the large number of residues, including many that are remote from the chromophore. How these positions control fluorescence properties via coupling to each other is poorly understood.
Based on suggested residues at the key (Table 1) and supporting positions (Supplementary Table 4), the template can be subjected to multiple site-specific mutagenesis at the chosen positions, beginning with residues at key positions and, in subsequent rounds, targeting supporting positions to optimize properties of fluorescent proteins. The PAmCherry9, PATagRFP10, rsTagRFP22, mRouge17 and mTagBFP-like6 and LSS-FP–like3 fluorescent proteins have different colors and photochemical behavior, and all of them were developed using rational mutagenesis at amino acids around the chromophore to find a weak phenotype, followed by random mutagenesis for the improvement of mainly brightness, maturation and photostability. A computer-based approach using the PHOENIX protein design software and FASTER algorithm can be used to eliminate amino acids incompatible with the β-barrel protein fold and generate focused small-size combinatorial libraries of RFP mutants17.
Directed evolution of fluorescent proteins
Creation of large libraries of mutants
Once rational design has resulted in primary clones with a required spectral phenotype, researchers typically take advantage of directed evolution for optimization. The first step in each round of molecular evolution is the generation of a large number (>105) of mutant genes. In vitro random mutagenesis, coupled with bacterial expression, has been effectively used to develop new red-shifted fluorescent proteins. In eukaryotic systems, different approaches must be taken to generate large mutant libraries. A recombinant vesicular stomatitis virus was recently reported to generate randomly mutated fluorescent proteins amenable to screening in mammalian cells23. The mutation rate and the size of the library were controlled by regulating the number of infected cells and the number of rounds of viral replication. However, the likely presence and continuous replication of several viral genomes per cell may hamper efficient screening of clones for specific spectral phenotypes (Fig. 3). Gene-diversification processes, such as somatic hypermutation and gene conversion, occur naturally in B lymphocytes via an introduction of random point mutations in a certain gene and through random shuffling of complex genetic domains, respectively. These approaches have been introduced for high-throughput screening of non-antibody proteins and have been applied to develop RFPs24.
Figure 3.
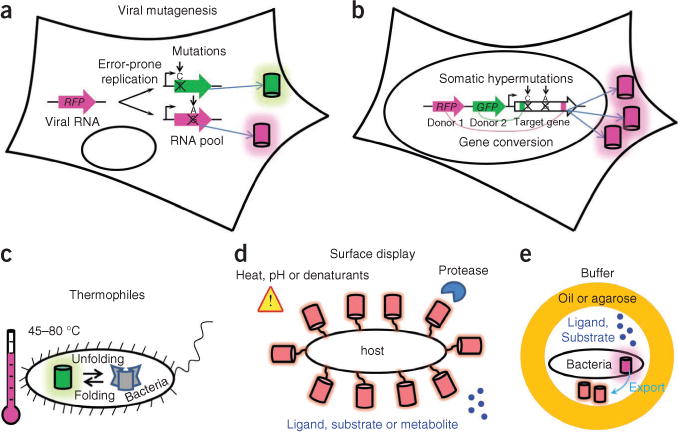
Methods that could improve molecular evolution of fluorescent proteins. (a–e) Schematics depict eukaryotic cell–based mutagenesis methods (a,b) and advanced protein expression systems (c–e). Cylinders denote fluorescent protein molecules. Error-prone replication of virus (a) causes point mutations in the viral genome containing a target fluorescent protein gene; after several rounds of replication, the cell expresses mutated fluorescent protein genes. Somatic hypermutations and gene conversion in eukaryotic cells (b) allow for creation of large random mutant gene libraries during cell proliferation (note that only one type of fluorescent protein mutant is produced per cell). Expression of fluorescent protein libraries (c) in thermophilic bacteria for selection of more stable fluorescent proteins. Surface display (d) of fluorescent protein libraries could facilitate screening for fluorescent protein stability under different environmental conditions or for fluorescent protein–based biosensors. In vitro compartmentalization (e) of bacteria in water-oil-water or water-agarose-water droplets should enable screening for fluorescent protein–based biosensors.
The main advantages of in vivo mutagenesis are expression of the protein library in the context of intracellular networks, substantially shorter time of sequence evolution and high diversity of mutants; viral mutagenesis is applicable to a wide spectrum of cell types and has advantage over somatic hypermutation and gene conversion approaches, which are limited to B lymphocytes. Furthermore, because these mutagenesis techniques use mammalian cells as hosts, they should result in the selection of mutants with low cytotoxicity and ones that are optimized for expression and stability in these cells.
Biological components of molecular evolution
New systems for the expression of gene libraries may help to improve directed evolution of fluorescent proteins and increase the number of biochemical parameters screened. With the exception of the above examples in eukaryotic cells, to date the molecular evolution of fluorescent proteins is mainly carried out via expression in Escherichia coli.
One property that could be substantially improved in new expression systems is intracellular half-life. Currently available fluorescent proteins typically have an intracellular half-life in mammalian cells of about 20–30 h (ref. 25). Short-lived fluorescent proteins variants with lifetimes of 0.5–10 h have been developed by fusing them with ubiquitinatable peptides25. However, fluorescent proteins with an extended intracellular lifespan are still in demand. Use of bacterial hosts other than E. coli, such as Thermus thermophilus that grow at 70 °C (ref. 26), could allow high-throughput screening for thermally stable fluorescent proteins, but this approach has not been implemented yet (Fig. 3). With some exceptions, a longer intracellular lifespan of proteins correlates with their higher thermostability in bacteria27 and mammalian cells, so that this approach could complement expression in mammalian cells as a way to screen for variants with longer half-lives and those that are more suitable for long-term monitoring of proteins in vivo.
Phage, bacterial and yeast surface display of a fluorescent protein library offers a spectrum of screening conditions (proteases, temperature and denaturants) for higher stability of the displayed protein28. In particular, surface display of fluorescent proteins could provide an important future tool for screening of biosensors. Surface display of biosensor variant libraries would enable screening for interactions with potential ligands, substrates or metabolites. Surface displays of large proteins are well developed in yeast eukaryotic cells29, but typical library sizes are limited to about 105 clones. Bacterial surface display is not well established for large proteins and requires improvement30. The Bacillus subtilis endospore system (proteins are targeted to the endospore surface) may be promising in this regard because the expressed proteins do not need to cross a cytoplasmic membrane, but the low transformation efficiency of B. subtilis is a substantial limitation. An approach that could in the future complement bacterial display for RFP-biosensor screening is in vivo compartmentalization31, based on compartmentalization of bacterial cells secreting RFP constructs in water-in-oil emulsions. But problems with ensuring the presence of single constructs per ‘cell’, with limiting the number of empty ‘cells’ and with preventing exchange of small molecules between compartments limit this approach and will need to be solved.
Instrumentation for high-throughput screening
The major high-throughput screening approach for evolution of fluorescent proteins is sorting of bacterial libraries using a fluorescence-activated cell sorter. However, it is limited by the available excitation sources32 and to the detection of essentially three parameters: (i) wavelength of fluorescence emission, (ii) fluorescence intensity and (iii) intensity of light-scattering at two angles. The substantial improvements of the optical detection modes and fluidics formats discussed below have been implemented in proof-of-principle experiments, but these achievements have not been applied for RFP screening yet.
Fluorescence-activated cell sorter optical components can be developed by introducing new types of excitation and detection formats (Fig. 4). Recording the whole fluorescence spectrum33 during fluorescence-activated cell sorting (FACS) may enable efficient screening for far-red– and near-infrared–shifted fluorescent proteins. Detection of polarization or anisotropy would enable screening for monomeric RFPs lacking fluorescence resonance energy transfer (FRET) between similar chromophores (homo-FRET). Incorporating sequential irradiation with several synchronized lasers will make it possible to sort for high-speed photoswitching PA-FPs for photoactivated localization microscopy (PALM) of live cells.
Figure 4.
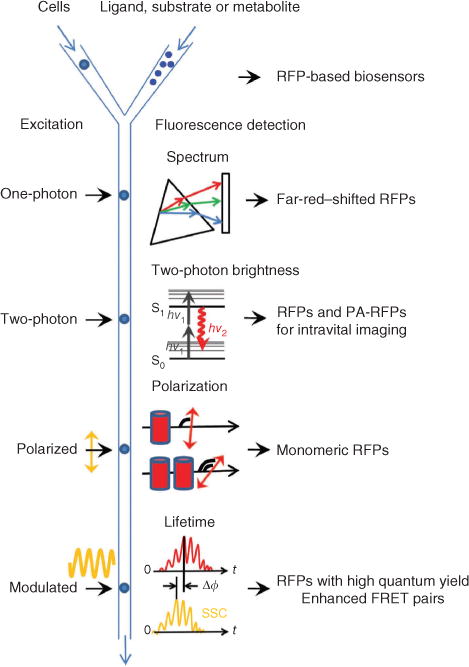
Possible FACS-based screening approaches for red-shifted fluorescent proteins. The respective red fluorescent proteins expected to result from each method are listed on the right. The schematic depicts cells or other hosts expressing fluorescent proteins being mixed with ligand, substrate or metabolite with different delays before fluorescence screening. A standard one-photon laser excites flowing cells, and the resultant fluorescent signal is dispersed with a diffraction grating (triangle) and projected onto an array detector (rectangle) for recording of a complete emission spectrum. A two-photon laser excites the cells with two low-energy photons (hv1) and the resulting fluorescence emission (hv2) is detected. Linearly polarized laser excitation and the emitted fluorescence signals have different degrees of polarization depending on the size of fluorescent molecules and FRET efficiency between them. Cylinders denote fluorescent proteins in monomeric and dimeric states. Modulated excitation (yellow sinusoid) results in a phase shift, Δϕ, between the fluorescence emission (red) and side-scattered excitation light (SSC; yellow), which is used to compute the average fluorescence lifetime of fluorescent proteins in a cell.
FACS with fluorescence lifetime detection34 will permit additional improvement of FRET pairs of fluorescent proteins. It will accelerate the development of RFPs with distinctive lifetimes, which would provide new possibilities for imaging proteins in vivo, using a single excitation source and emission channel35. The fluorescence lifetime is proportional to quantum yield, and fluorescence lifetime measurements are independent of changes in probe concentration, excitation intensity and other factors that limit intensity-based measurements36. Thus we believe that fluorescence lifetime screening either in a low-throughput format36, or using FACS34, holds great promise for the improvement of RFP brightness.
Finally, coupling a two-photon laser with a fluorescence-activated cell sorter37 provides a way to develop a new class of RFPs with greater two-photon excitation efficiency. Current RFPs have suboptimal two-photon brightness and photostability38, which are important properties for intravital imaging in animals.
Developments in the fluidics components of fluorescence-activated cell sorters could provide advanced platforms for screening of molecular fluorescent biosensors that include (i) rapid switching between buffers with distinctive properties or concentrations of substances to be ‘sensed’, (ii) automatic sampling from multiwell plates pretreated with such substances and (iii) use of biphasic unmixable suspensions of one liquid in another, such as aqueous drops in a nonpolar carrier for bead-based in vitro transcription-translation systems. Several recent approaches such as those using microfluidic cytometers39 and a combination of imaging scanning cytometers40 with cell-isolation technologies41 could provide additional options for screening. Compared to FACS, these techniques are slower, but they provide longer time periods for cell detection and manipulations such as buffer exchange.
Installing cell lysis42 and PCR microfluidic chips43 at the output of fluorescence-activated cell sorters or microfluidic cytometers could accelerate re-cloning of fluorescent protein genes from eukaryotic, viral and phage expression systems into bacterial systems, for high-throughput screening gene sequencing and protein production. Growing of cells in microdroplets44 would increase screening sensitivity, particularly for surface displays with a limited number of molecules of fluorescent proteins on the cell surface.
Advanced probes for emerging imaging approaches
Approaches such as super-resolution microscopy including stimulated emission depletion (STED) microscopy and PALM, deep-tissue imaging, intravital microscopy and two-photon microscopy demand fluorescent proteins with particular properties.
Commercial super-resolution STED microscopy requires far-red fluorescent proteins excitable with red lasers, such as 633 nm HeNe and 635–640 nm laser diodes18,20. Similarly, far-red or near-infrared fluorescent proteins are necessary for deep-tissue and whole-animal imaging, as oxyhemoglobin, deoxyhemoglobin and melanin do not absorb light at these wavelengths (650–900 nm), and light scattering from cellular components is decreased as well. To date the most far-red shifted fluorescent proteins, such as E2-Crimson18, mNeptune19 and TagRFP657 (ref. 20), have in common a DsRed-like chromophore, and have excitation and emission maxima limited to about 610 nm and 650 nm, respectively. The ‘cost’ of the far-red shift of the DsRed-like chromophore is a substantial drop in its quantum yield. Furthermore, quantum yield also decreases with the increasing Stokes shift of emission. As there is a correlation between the absorbance and emission wavelengths and the number of the conjugated double bonds (Supplementary Table 3), one possible way to develop far-red fluorescent proteins could be to design a chromophore with more double bonds than in the DsRed-like chromophore. The Kaede-like chromophore 14 is a good candidate; it has more double bonds than DsRed-like chromophore 5a, 6a and a far-red shoulder at 627 nm. Kaede-like far-red fluorescent proteins could be developed by causing the light-inducible transition from structure 12 to 14 to proceed autocatalytically. Another approach may be to exploit the fact that the mOrange-like chromophore 10 undergoes photoconversion to far-red species 11. Thus, far-red fluorescent proteins could be developed by causing the transition from structure 10 to 11 to occur autocatalytically, with no light. A third approach may exploit hydrogen bonding, stacking and hydrophobic interactions around the DsRed-like chromophore to obtain a far-red–shifted chromophore 7a,b,c.
STED microscopy is typically limited to fixed cells because of the extremely high light intensity (10 MW cm−2)—which is harmful to live cells—required for the depletion of the excited state of fluorescent proteins45. However, a STED microscopy modification, called reversible saturable optical transitions (RESOLFT), can be applied for live cell super-resolution microscopy. RESOLFT uses RS-FPs, which typically require relatively low photoswitching intensity of 1–10 W cm−2, but scanning is typically performed several tens of times. Presently available RS-FPs have low photostability, which does not allow their use for many switching cycles of RESOLFT. RS-FPs with substantially higher photostability need to be engineered. RS-FPs with photoswitchable absorbance spectra could be also used as acceptors in photochromic FRET approaches22. This should facilitate detection of protein-protein interactions, provide internal controls for FRET and could enable the determination of the distance between interacting protein pairs.
Finally, multicolor localization microscopy of single molecules, such as PALM and its variants, requires PA-FPs of different colors46. Currently, monomeric PA-FPs are limited to green and red1,9,10,46–49. Analysis of chemical transformations of the RFP chromophores provides putative rational strategies to develop orange and far-red PA-FPs photoactivatable with violet light, for instance via making light-inducible the autocatalytic steps from structure 4 to 10 and from structure 4 to 7a,b,c (Fig. 2). PA-FPs with photoactivation bands in the near-UV or green wavelength ranges could expand the palette of single-molecule super-resolution approaches even further16. For example, a blue PA-FP, photoactivatable with near-UV light and excitable with violet light, could be engineered by blocking the photoinducible transition from structure 4 to 5a,b (Fig. 2).
PALM with PA-FPs is limited to a localization precision of 15–20 nm9 that allows for estimation of co-localization of two proteins but makes it difficult to determine whether the proteins physically interact with each other. Combining FRET with two-color PALM could solve this problem, but it is difficult because simultaneous photoactivation of the PA-FP donor and PA-FP acceptor in the interacting protein pair is highly unlikely. However, an acceptor PA-FP photoactivatable via FRET from a conventional fluorescent protein donor could allow super-resolution imaging of protein-protein interactions. A PA-FP acceptor with the photoactivation band in a green-orange wavelength range would be required for this purpose. Such PA-FPs could be also useful for intravital photoactivation experiments because green-orange light is substantially less phototoxic than violet50.
Multicolor fluorescence imaging using a single excitation wavelength could be possible with LSS-FPs of different emission wavelengths. The excitation of such LSS-FPs with the same one-photon laser would permit researchers to image several probes simultaneously. Determination of chromophore environments in green and red LSS-FPs3 resulted in the elucidation of several excited-state proton transfer pathways that cause the LSS emission. Engineering of these pathways into existing conventional RFPs could yield LSS probes with orange and far-red fluorescence. Moreover, multicolor LSS-FPs with close excitation maxima could be applied for intravital imaging using two-photon excitation with a single wavelength of the two-photon laser.
Conclusions
Despite the availability of increasingly sophisticated rational design software, it is unlikely that the structure-based approaches alone will be sufficient to solve all future demands for engineering of fluorescent proteins. Analysis of chromophore transformation mechanisms suggests that fluorescent probes with novel features can be designed based on existing monomeric fluorescent proteins, using a combination of rational engineering and high-throughput screening techniques. Improvements both in the biological and instrumental components of directed molecular evolution should increase the palette of red-shifted fluorescent proteins with advanced biochemical and photochemical properties. Advanced fluorescent proteins will not only solve some of the limitations of current microscopy methods but will in turn stimulate the development of novel fluorescence-detection technologies, in vivo imaging approaches and image-processing techniques.
Supplementary Material
Acknowledgments
This work was supported by US National Institutes of Health grants GM073913 and CA164468 to V.V.V.
Footnotes
Note: Supplementary information is available on the Nature Methods website.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- 2.Piatkevich KD, Verkhusha VV. Advances in engineering of fluorescent proteins and photoactivatable proteins with red emission. Curr Opin Chem Biol. 2010;14:23–29. doi: 10.1016/j.cbpa.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piatkevich KD, Malashkevich VN, Almo SC, Verkhusha VV. Engineering ESPT pathways based on structural analysis of LSSmKate red fluorescent proteins with large Stokes shift. J Am Chem Soc. 2010;132:10762–10770. doi: 10.1021/ja101974k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pletnev S, Subach FV, Dauter Z, Wlodawer A, Verkhusha VV. Understanding blue-to-red conversion in monomeric fluorescent timers and hydrolytic degradation of their chromophores. J Am Chem Soc. 2010;132:2243–2253. doi: 10.1021/ja908418r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subach OM, et al. Structural characterization of acylimine-containing blue and red chromophores in mTagBFP and TagRFP fluorescent proteins. Chem Biol. 2010;17:333–341. doi: 10.1016/j.chembiol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subach OM, et al. Conversion of red fluorescent protein into a bright blue probe. Chem Biol. 2008;15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subach FV, et al. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat Chem Biol. 2009;5:118–126. doi: 10.1038/nchembio.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subach FV, et al. Photoactivation mechanism of PAmCherry based on crystal structures of the protein in the dark and fluorescent states. Proc Natl Acad Sci USA. 2009;106:21097–21102. doi: 10.1073/pnas.0909204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subach FV, et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subach FV, Patterson GH, Renz M, Lippincott-Schwartz J, Verkhusha VV. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132:6481–6491. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunewardene MS, et al. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101:1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, et al. Molecular mechanism of a green-shifted, pH-dependent red fluorescent protein mKate variant. PLoS ONE. 2011;6:e23513. doi: 10.1371/journal.pone.0023513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kremers GJ, Hazelwood KL, Murphy CS, Davidson MW, Piston DW. Photoconversion in orange and red fluorescent proteins. Nat Methods. 2009;6:355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mishin AS, et al. The first mutant of the Aequorea victoria green fluorescent protein that forms a red chromophore. Biochemistry. 2008;47:4666–4673. doi: 10.1021/bi702130s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogdanov AM, et al. Green fluorescent proteins are light-induced electron donors. Nat Chem Biol. 2009;5:459–461. doi: 10.1038/nchembio.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subach OM, et al. A photoswitchable orange-to-far-red fluorescent protein, PSmOrange. Nat Methods. 2010;8:771–777. doi: 10.1038/nmeth.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chica RA, Moore MM, Allen BD, Mayo SL. Generation of longer emission wavelength red fluorescent proteins using computationally designed libraries. Proc Natl Acad Sci USA. 2010;107:20257–20262. doi: 10.1073/pnas.1013910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strack RL, et al. A rapidly maturing far-red derivative of DsRed-Expres2 for whole-cell labeling. Biochemistry. 2009;48:8279–8281. doi: 10.1021/bi900870u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin MZ, et al. Autofluorescent proteins with excitation in the optical window for intravital imaging in mammals. Chem Biol. 2009;16:1169–1179. doi: 10.1016/j.chembiol.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morozova KS, et al. Far-red fluorescent protein excitable with red lasers for flow cytometry and superresolution STED nanoscopy. Biophys J. 2010;99:L13–L15. doi: 10.1016/j.bpj.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoi H, et al. A monomeric photoconvertible fluorescent protein for imaging of dynamic protein localization. J Mol Biol. 2010;401:776–791. doi: 10.1016/j.jmb.2010.06.056. [DOI] [PubMed] [Google Scholar]
- 22.Subach FV, et al. Red fluorescent protein with reversibly photoswitchable absorbance for photochromic FRET. Chem Biol. 2010;17:745–755. doi: 10.1016/j.chembiol.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis JN, van den Pol AN. Viral mutagenesis as a means for generating novel proteins. J Virol. 2010;84:1625–1630. doi: 10.1128/JVI.01747-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arakawa H, et al. Protein evolution by hypermutation and selection in the B cell line DT40. Nucleic Acids Res. 2008;36:e1. doi: 10.1093/nar/gkm616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corish P, Tyler-Smith C. Attenuation of green fluorescent protein halflife in mammalian cells. Protein Eng. 1999;12:1035–1040. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- 26.Cava F, Hidalgo A, Berenguer J. Thermus thermophilus as biological model. Extremophiles. 2009;13:213–231. doi: 10.1007/s00792-009-0226-6. [DOI] [PubMed] [Google Scholar]
- 27.Kwon WS, Da Silva NA, Kellis JT., Jr Relationship between thermal stability, degradation rate and expression yield of barnase variants in the periplasm of Escherichia coli. Protein Eng. 1996;9:1197–1202. doi: 10.1093/protein/9.12.1197. [DOI] [PubMed] [Google Scholar]
- 28.Martin A, Schmid FX, Sieber V. Proside: a phage-based method for selecting thermostable proteins. Methods Mol Biol. 2003;230:57–70. doi: 10.1385/1-59259-396-8:57. [DOI] [PubMed] [Google Scholar]
- 29.Pavoor TV, Cho YK, Shusta EV. Development of GFP-based biosensors possessing the binding properties of antibodies. Proc Natl Acad Sci USA. 2009;106:11895–11900. doi: 10.1073/pnas.0902828106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daugherty PS. Protein engineering with bacterial display. Curr Opin Struct Biol. 2007;17:474–480. doi: 10.1016/j.sbi.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Bergquist PL, Hardiman EM, Ferrari BC, Winsley T. Applications of flow cytometry in environmental microbiology and biotechnology. Extremophiles. 2009;13:389–401. doi: 10.1007/s00792-009-0236-4. [DOI] [PubMed] [Google Scholar]
- 32.Telford WG, Subach FV, Verkhusha VV. Supercontinuum white light lasers for flow cytometry. Cytometry A. 2009;75:450–459. doi: 10.1002/cyto.a.20687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goddard G, et al. Single particle high resolution spectral analysis flow cytometry. Cytometry A. 2006;69:842–851. doi: 10.1002/cyto.a.20320. [DOI] [PubMed] [Google Scholar]
- 34.Houston JP, Naivar MA, Freyer JP. Digital analysis and sorting of fluorescence lifetime by flow cytometry. Cytometry A. 2010;77:861–872. doi: 10.1002/cyto.a.20930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Kwon D, Lee J, Pasquier H, Grailhe R. The use of Cyan Fluorescent Protein variants with a distinctive lifetime signature. Mol Biosyst. 2009;5:151–153. doi: 10.1039/b815445g. [DOI] [PubMed] [Google Scholar]
- 36.Goedhart J, et al. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat Methods. 2010;7:137–139. doi: 10.1038/nmeth.1415. [DOI] [PubMed] [Google Scholar]
- 37.Buschke DG, et al. Multiphoton flow cytometry to assess intrinsic and extrinsic Fluorescence in cellular aggregates: applications to stem cells. Microsc Microanal. 2010;17:540–554. doi: 10.1017/S1431927610000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Two-photon absorption properties of fluorescent proteins. Nat Methods. 2011;8:393–399. doi: 10.1038/nmeth.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Godin J, et al. Microfluidics and photonics for Bio-System-on-a-Chip: a review of advancements in technology towards a microfluidic flow cytometry chip. J Biophotonics. 2008;1:355–376. doi: 10.1002/jbio.200810018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heng X, Hsiung F, Sadri A, Patt P. Serial line scan encoding imaging cytometer for both adherent and suspended cells. Anal Chem. 2011;83:1587–1593. doi: 10.1021/ac102408g. [DOI] [PubMed] [Google Scholar]
- 41.Pai JH, Xu W, Sims CE, Allbritton NL. Microtable arrays for culture and isolation of cell colonies. Anal Bioanal Chem. 2010;398:2595–2604. doi: 10.1007/s00216-010-3984-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J, et al. Cell lysis on a microfluidic CD (compact disc) Lab Chip. 2004;4:516–522. doi: 10.1039/b401106f. [DOI] [PubMed] [Google Scholar]
- 43.Zhang C, Xing D, Li Y. Micropumps, microvalves, and micromixers within PCR microfluidic chips: advances and trends. Biotechnol Adv. 2007;25:483–514. doi: 10.1016/j.biotechadv.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Barbulovic-Nad I, Au SH, Wheeler AR. A microfluidic platform for complete mammalian cell culture. Lab Chip. 2010;10:1536–1542. doi: 10.1039/c002147d. [DOI] [PubMed] [Google Scholar]
- 45.Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci USA. 2005;102:17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippincott-Schwartz J, Patterson GH. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu B, Piatkevich KD, Lionnet T, Singer RH, Verkhusha VV. Modern fluorescent proteins and imaging technologies to study gene expression, nuclear localization, and dynamics. Curr Opin Cell Biol. 2011;23:310–317. doi: 10.1016/j.ceb.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davidson MW, Campbell RE. Engineered fluorescent proteins: innovations and applications. Nat Methods. 2009;6:713–717. doi: 10.1038/nmeth1009-713. [DOI] [PubMed] [Google Scholar]
- 49.Nienhaus GU, Wiedenmann J. Structure, dynamics and optical properties of fluorescent proteins: perspectives for marker development. ChemPhysChem. 2009;10:1369–1379. doi: 10.1002/cphc.200800839. [DOI] [PubMed] [Google Scholar]
- 50.Post JN, Lidke KA, Rieger B, Arndt-Jovin DJ. One- and two-photon photoactivation of a paGFP-fusion protein in live Drosophila embryos. FEBS Lett. 2005;579:325–330. doi: 10.1016/j.febslet.2004.11.092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.