

Abstract
Long noncoding RNAs (lncRNAs) have been identified as oncogenes or tumor suppressors that are involved in tumorigenesis and chemotherapy drug resistance. Maternally expressed gene 3 (MEG3) is an imprinted gene located at 14q32 that encodes an lncRNA, and decreased MEG3 expression plays an important role in multiple cancers. However, its biological role in the development of the chemoresistance phenotype of human lung adenocarcinoma (LAD) is unknown. This study aimed to observe the expression of MEG3 in LAD and to evaluate its biological role and clinical significance in the resistance of LAD cells to cisplatin. MEG3 expression was markedly decreased in cisplatin-resistant A549/DDP cells compared with parental A549 cells as shown by an lncRNA microarray. MEG3 overexpression in A549/DDP cells increased their chemosensitivity to cisplatin both in vitro and in vivo by inhibiting cell proliferation and inducing apoptosis. By contrast, MEG3 knockdown in A549 cells decreased the chemosensitivity. Moreover, MEG3 was decreased in cisplatin-insensitive LAD tissues while p53 protein levels were decreased and Bcl-xl protein levels increased. Furthermore, patients with lower levels of MEG3 expression showed worse responses to cisplatin-based chemotherapy. These findings demonstrate that MEG3 is significantly downregulated in LAD and partially regulates the cisplatin resistance of LAD cells through the control of p53 and Bcl-xl expression. Thus, MEG3 may represent a new marker of poor response to cisplatin and could be a potential therapeutic target for LAD chemotherapy.
Introduction
Lung cancer is one of the most prevalent neoplasms worldwide, ranking as the first and second leading causes of cancer-related deaths in males and females, respectively. Non-small cell lung cancer (NSCLC) currently accounts for about 80% of all lung cancer cases [1, 2], and more than 65% of NSCLC patients present with locally advanced or metastatic disease [3]. Lung adenocarcinoma (LAD) is the most common histological subtype of NSCLC [4], but affected patients have limited access to early detection and timely treatment. Platinum-containing drugs such as cisplatin and carboplatin are typically used in LAD chemotherapy [5], although cisplatin resistance is the greatest obstacle of clinical LAD treatment.
Recent improvements in high-throughput gene expression analysis have led to the discovery that transcription from <2% of the human genome yields many short or long noncoding RNAs (lncRNAs) with limited or no protein-coding capacity because of a lack of open reading frames [6–9]. These show dysregulated expression in several human diseases but their exact biological functions are unclear. In our previous study, we reported that miR-224 could promote cisplatin resistance in A549/DDP cells via regulating G1/S transition and apoptosis by targeting p21WAF1/CIP1 [10]. The dysregulation of lncRNAs has also been shown to participate in tumorigenesis through promoting cellular proliferation and inducing apoptosis in multiple tumors. For example, lncRNA uc002mbe.2 expression was significantly downregulated in hepatocellular cancer (HCC), and its overexpression contributed to the trichostatin-induced apoptosis of HCC cells [11]. Similarly, lncRNA AK126698 was thought to regulate the chemoresistance of NSCLC cells through the Wnt signaling pathway [12], and our previous study found that lncRNA HOTAIR overexpression contributed to LAD cell cisplatin resistance via the regulation of p21 expression [13]. Moreover, in breast cancer patients with the single nucleotide polymorphism rs6983267 genotype, the novel lncRNA CCAT2 was demonstrated to reduce chemosensitivity to fluorouracil [14]. However, although some studies have elucidated the lncRNA functions in tumor chemoresistance, the underlying mechanisms are less well documented. An understanding of these mechanisms would improve LAD treatment and enable new targets for tumor chemotherapy to be identified.
LncRNA MEG3 is a maternally expressed imprinted gene that is part of the DLK1–MEG3 locus located on human chromosome 14q32 [15]. It is expressed in normal human tissues, especially in brain and the pituitary, and is thought to be a tumor suppressor [16]. Recent studies showed that MEG3 expression is disrupted in various human cancers, such as bladder cancer, glioma, and HCC [17–19]. Moreover, Zhang et al. demonstrated that MEG3 expression was reduced in meningioma compared with normal controls, and that this loss was associated with tumor grade [20]. Conversely, increased MEG3 expression was reported to regulate NSCLC cell proliferation and apoptosis through the activation of p53 [21]. However, the functions and detailed mechanisms of MEG3 in the cisplatin resistance of LAD remain elusive.
In this study, therefore, we investigated the role of MEG3 in the chemosensitivity of LAD cells to cisplatin by analyzing its function both in vitro and in vivo. We demonstrated that MEG3 expression was significantly decreased in cisplatin-resistant A549/DDP cells compared with that in parental cells, using lncRNA microarray and quantitative reverse transcriptase qRT-PCR. Ectopic expression of MEG3 reduced A549/DDP cell cisplatin resistance, while MEG3 knockdown increased this through regulation of cell proliferation and apoptosis. We further verified that overexpression of MEG3 induced the mitochondrial apoptosis pathway via p53 and Bcl-xl activation. Our research confirms for the first time that, as a tumor suppressor, MEG3 improves LAD chemosensitivity, and shows that it has potential to be used as a therapeutic target to reverse the cisplatin resistance of LAD patients.
Materials and Methods
Cell lines and culture
The human LAD cell line A549 was purchased from the Cancer Institute, Chinese Academy of Sciences. The cisplatin-resistant cell line A549/DDP was selected by continuous exposure to increasing concentrations of cisplatin followed by culturing in medium containing 1.0 μg/ml cisplatin to maintain the cisplatin resistance. All cell lines were cultured in RPMI 1640 medium (GIBCO-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin. They were grown under an atmosphere of 5% CO2 with humidity at 37°C. In all experiments, exponentially growing cells were used.
Patients and tissue samples
Forty-one tumor tissues were obtained from advanced LAD patients who underwent cisplatin-based chemotherapy at the First or Second Affiliated Hospital of Nanjing Medical University between April 2007 and November 2009. All patients were histopathologically diagnosed with LAD and accepted first-line chemotherapy with cisplatin in combination with gemcitabine or paclitaxel for a maximum of four cycles. Tissue samples were divided into “sensitive” (complete or partial response, CR+PR; n = 20) and “insensitive” (stable or progressive disease, SD+PD; n = 21) groups according to the patients’ responses, as assessed by medical image analysis and detection of serum tumor markers after four cycles of cisplatin-based chemotherapy. All tissues were immediately frozen in liquid nitrogen and stored at −80°C until required for total RNA extraction. Tumor staging was determined according to the tumor–node–metastasis (TNM) classification of the International Union against Cancer. Written informed consent was provided by patients or their guardians. This study was approved by the Chinese Medical Association Society of Medicine’s Ethics Committee.
RNA extraction and qRT-PCR
Total RNA was extracted from tissues or transfected cells using TRIzol reagent (Invitrogen, Carlsbad, CA). RT reactions were performed using MEG3-specific primers and a Takara Reverse Transcriptase kit (Takara, Dalian, China) according to the manufacturer’s instructions. The MEG3 expression level was quantified using SYBR Green-based PCR (Takara) using MEG3-specific primers: forward, 5′-CTGCCCATCTACACCTCACG-3′; and reverse, 5′-CTCTCCGCCGTCTGCGCTAGGGGCT-3′, and normalized to the expression of GAPDH, which was amplified using specific primers: forward, 5′-GTCAACGGATTTGGTCTGTATT-3′; and reverse, 5′-AGTCTTCTGGGTGGCAGTGAT-3′.
LncRNA microarray
Total RNA was isolated from cisplatin-resistant A549/DDP cells and parental A549 cells as described above, and the quality and quantity of the RNA samples were assessed. LncRNA microarray V2.0 analysis supported by CapitalBio Corporation (Beijing, China) was performed to detect lncRNAs and protein-coding mRNAs in the genome. LncRNAs were purified from total RNA after repeat sequences and ncRNAs shorter than 200 bp were deleted. In total, information about 30,756 lncRNAs was obtained from authoritative data sources including UCSC, RefSeq, H-Invitational Database, and other related literature, and bioinformatic analysis of microarray data was performed by CapitalBio Corporation.
Transfection of LAD cells
MEG3 was subcloned into the pCDNA 3.1 vector (Invitrogen, Shanghai, China), and this was purified using DNA Midiprep or Midiprep Kits (QIAGEN, Hilden, Germany). Exponentially growing A549/DDP cells were seeded into 6-well plates and transfected with pCDNA-MEG3 vector or empty vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. MEG3 overexpression was confirmed by qRT-PCR and western blot analyses 48 h after transfection.
Three small interfering (si)RNAs targeting MEG3 (si-MEG3 I, II, and III) were also transfected into A549 cells, and the most stable was chosen (si-MEG3 II). A549 cells were then transfected with this si-MEG3 or scrambled negative control siRNA (si-NC), and MEG3 expression was detected by qRT-PCR 48 h after transfection.
Flow cytometric analysis of cell cycle or apoptosis
LAD cells for apoptotic analysis were double stained with Annexin V–FITC and propidium iodide 48 h after transfection and analyzed using a flow cytometer (FACScan; BD Biosciences, Shanghai, China) equipped with CellQuest software (BD Biosciences). Cells were classified as viable, dead, early apoptotic, or apoptotic. The percentage of early apoptotic cells was counted and compared between cells receiving different treatment. Cells for cell cycle analysis were stained with propidium iodide using the BD Cycletest Plus DNA Reagent Kit (BD Biosciences). The relative ratio of cells in G0/G1, S, or G2/M phase was counted and respectively compared with control groups. Each experiment was performed in triplicate.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and colon formation assay
The IC50 was determined by an MTT assay. Transfected cells (3.5 × 103/well) were seeded in 96-well plates and maintained in standard growth medium. Overnight, cells were exposed to different concentrations (0, 1, 5, 10, 15, 20, 25, 30, 35, and 40 ìg/ml) of cisplatin. After incubation for 48 h, 0.5 mg/ml MTT was added and incubated for another 4 h. The medium was then replaced with 150 μl dimethyl sulfoxide (Sigma–Aldrich, St. Louis, MO) and vortexed for 10 min. Absorbance at 490 nm was then surveyed by an automatic microplate reader. Cell viability was assessed at 0, 24, 48, 72, and 96 h with and without cisplatin treatment. For the colony formation assay, transfected cells (0.5 × 103/well) were seeded in 6-well plates. After 14 d, cells were fixed with methanol and stained with 0.1% crystal violet (Sigma–Aldrich) and the number of colonies was counted.
Hoechst staining assay
Cells were cultured in 6-well plates and Hoechst 33342 (Sigma–Aldrich) was added to the culture medium for 10 min. Cells were then washed with PBS, and changes in nuclear morphology were detected by fluorescence microscopy using a 365 nm filter for Hoechst 33342. The percentage of Hoechst-positive nuclei per optical field (at least 50 fields) was counted to quantify Hoechst 33342 staining.
Western blot assay
Cells were lysed using the mammalian protein extraction reagent RIPA (Beyotime, Shangahi, China), together with a protease inhibitor cocktail (Roche, Pleasanton, CA), and phenylmethylsulfonyl fluoride (Roche). Protein lysates were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to 0.22 μm nitrocellulose membranes (Sigma–Aldrich), and incubated with specific antibodies against p53, mouse double minute 2 homolog (MDM2), Bcl-2, and Bcl-xl (Cell Signaling Technology, Danvers, MA). An anti-GAPDH antibody was used as a control. Signals were visualized using the ECL chromogenic substrate and quantified by densitometry using Quantity One software (Bio-Rad, Berkeley, CA).
In vivo chemosensitivity assay
Male athymic BALB/c nude mice aged 4 weeks were housed under specific pathogen-free conditions and manipulated according to protocols approved by the Shanghai Medical Experimental Animal Care Commission. Transfected A549/DDP cells (pCDNA-MEG3 and empty vector) were harvested and resuspended at a concentration of 2.0 × 107 cells/ml. Suspended cells (0.1 ml) were subcutaneously injected into a single side of the posterior flank of each mouse. Tumor growth was examined every 3 days, and tumor volume was calculated using the equation V = 0.5 × D × d2 (V, volume; D, longitudinal diameter; d, latitudinal diameter). When the average tumor size reached approximately 50 mm3, cisplatin was administered by intraperitoneal injection at a dose of 3 mg/kg, once every other day, for a total of three doses. Four weeks after the injection, mice were killed and the subcutaneous growth of each tumor was examined. Primary tumors underwent hematoxylin and eosin staining and immunostaining analysis for proliferating cell nuclear antigen, p53, and Bcl-XL protein expression. All procedures were performed in accordance with the Nanjing University Guide for the Care and Use of Laboratory Animals formulated by the National Society for Medical Research.
Statistical analysis
All data are shown as means ± SE and were analyzed using Prism 5.0 software. Statistical analysis in the form of a Student’s t-test (two-tailed), one-way analysis of variance, and Mann–Whitney U test was performed using SPSS 16.0 software. P values less than 0.05 were considered significant.
Results
Involvement of MEG3 expression in cisplatin-resistant LAD cells (A549/DDP) and parental LAD cells (A549)
To identify the lncRNAs differentially expressed between cisplatin-resistant A549/DDP and parental A549 cells, we performed lncRNA microarray analysis. Of the 30,756 lncRNAs identified from databases, 664 showed differential expression between the cell types by more than 5-fold. Of these, 304 were downregulated and 360 were upregulated (Table 1), and MEG3 showed the most downregulation in A549/DDP cells (by 61-fold). To validate the lncRNA microarray data, MEG3 expression was determined in cisplatin-resistant A549/DDP and parental A549 cells by qRT-PCR and normalized to GAPDH levels. This found MEG3 expression to be downregulated in A549/DDP cells by 4.5-fold compared with A549 cells (Fig 1A).
Table 1. Differentially expressed lncRNAs in A549/DDP cells compared with A549 cells as determined by microarray.
| lncRNAs | Regulation | Ratio | lncRNAs | Regulation | Ratio |
|---|---|---|---|---|---|
| HIT000322758.11 | Up | 914.2004 | ENST00000443252 | Down | 192.118 |
| uc002llc.1 | Up | 250.9595 | MEG3 | Down | 61.61361 |
| uc002bbp.2 | Up | 141.9445 | BX537613 | Down | 51.442579 |
| uc003hsl.1 | Up | 135.3685 | uc001yia.2 | Down | 48.626953 |
| HIT000087455.12 | Up | 92.1891 | 14q(II-3) | Down | 46.406529 |
| ASO1629 | Up | 86.4334 | NR_002766 | Down | 45.985177 |
| ASO3765 | Up | 80.1581 | LIT3318 | Down | 45.487044 |
| NONE | Up | 79.4499 | NONE | Down | 45.117236 |
| uc003wyd.2 | Up | 72.9465 | NR_033360 | Down | 42.646661 |
| HIT000326960.4 | Up | 68.7971 | uc010txc.1 | Down | 38.913981 |
| uc001luz.1 | Up | 61.1428 | AB074169 | Down | 35.9206 |
| uc001uib.2 | Up | 53.4206 | 14q(II-2) | Down | 35.540013 |
| uc001lvd.2 | Up | 47.9377 | uc010txe.1 | Down | 35.344072 |
| NONE | Up | 43.1377 | NR_033358 | Down | 35.038002 |
| BC032761 | Up | 27.0711 | NONE | Down | 33.132646 |
| NR_003503 | Up | 26.0497 | uc002sag.2 | Down | 31.316989 |
| uc003kzj.2 | Up | 22.2284 | AF070581 | Down | 31.148076 |
| HIT000076908.10 | Up | 21.1488 | NONE | Down | 29.847659 |
| ENST00000410370 | Up | 21.0256 | NONE | Down | 27.103947 |
| uc002pec.2 | Up | 20.3612 | NR_033359 | Down | 25.112085 |
| uc004edk.1 | Up | 18.354 | uc010txh.1 | Down | 24.008096 |
| ASO3473 | Up | 17.2485 | uc010txd.1 | Down | 22.722551 |
| NR_036555 | Up | 17.1851 | 14q(II-21) | Down | 21.993253 |
| uc001uie.2 | Up | 16.2843 | uc010txf.1 | Down | 21.907636 |
Fig 1. The level of MEG3 expression level in LAD cells.

(Fig 1A) qRT-PCR analysis of MEG3 expression levels in A549 and A549/DDP cells. (Fig 1B) MTT assay of the IC50 values of A549 and A549/DDP cells to cisplatin. (Fig 1C) qRT-PCR analysis of MEG3 expression levels following the transfection of A549/DDP cells with empty vector or pCDNA-MEG3. (Fig 1D) MTT assay of the IC50 values of empty vector- and pCDNA-MEG3-transfected A549/DDP cells to cisplatin. *P < 0.05, ** P < 0.01.
We previously reported differences in morphological characteristics and biological functions of A549/DDP and parental A549 cells [13]. In the present study, the IC50 of A549/DDP cells to cisplatin appeared to be higher than that of A549 cells (Fig 1B). We therefore explored the potential role of MEG3 in the chemoresistance of LAD cells. MEG3 was overexpressed in A549/DDP cells by transfecting them with pCDNA-MEG3 for 48 h. qRT-PCR analysis revealed that MEG3 expression was significantly increased by 487-fold in pCDNA-MEG3-transfected A549/DDP cells compared with A549/DDP cells transfected with empty vector (Fig 1C). Furthermore, an MTT assay showed that the IC50 of pCDNA-MEG3-transfected A549/DDP cells to cisplatin was significantly decreased compared with respective control cells (Fig 1D), indicating that the MEG3 expression level of LAD cells affects their chemoresistance to cisplatin.
MEG3 overexpression inhibits A549/DDP cell apoptosis in vitro
Because high MEG3 expression appeared to increase the chemosensitivity of A549/DDP cells to cisplatin, we further investigated the biological role and mechanism of MEG3 in LAD cell chemoresistance. We used Hoechst staining and flow cytometric analysis to determine whether apoptosis was a contributing factor in chemoresistance. A549/DDP cells transfected with pCDNA-MEG3 combined with cisplatin treatment showed a significantly increased rate of apoptosis with increasing doses of cisplatin (0.0, 1.0, and 2.0 ug/ml) compared with respective controls (Fig 2A). Thus, MEG3 appeared to significantly increase cisplatin-induced apoptosis in cisplatin-resistant LAD cells. Flow cytometric analysis was also consistent with these findings. When treated with increasing doses of cisplatin (0.0, 1.0, and 2.0 ug/ml), the apoptotic rate of A549/DDP cells transfected with pCDNA-MEG3 increased gradually compared with control cells transfected with empty vector (Fig 2B). Thus, MEG3 reversed the cisplatin resistance of A549/DDP cells through the enhancement of apoptosis.
Fig 2. MEG3 overexpression promotes A549/DDP cell apoptosis in vitro.

(Fig 2A) Hoechst staining assay for cell apoptosis; the percentage of Hoechst-positive nuclei per optical field (at least 50 fields) was counted. (Fig 2B) Flow cytometry analysis of apoptosis in transfected A549/DDP cells in combination with increasing concentrations of cisplatin (0.0, 1.0, and 2.0 μg/ml). *P < 0.05.
MEG3 overexpression impairs A549/DDP cell proliferation in vitro
We next performed MTT and colony formation analysis to examine whether MEG3 could affect cell proliferation in vitro. The MTT assay revealed that cell proliferation was impaired in A549/DDP cells transfected with pCDNA-MEG3 combined with cisplatin treatment compared with control cells (Fig 3A). Similarly, colony formation analysis revealed that MEG3 combined with increasing doses of cisplatin (0.0, 1.0, and 2.0 ug/ml) gradually decreased the number of colonies formed (Fig 3B).
Fig 3. MEG3 overexpression impairs A549/DDP cell proliferation in vitro.
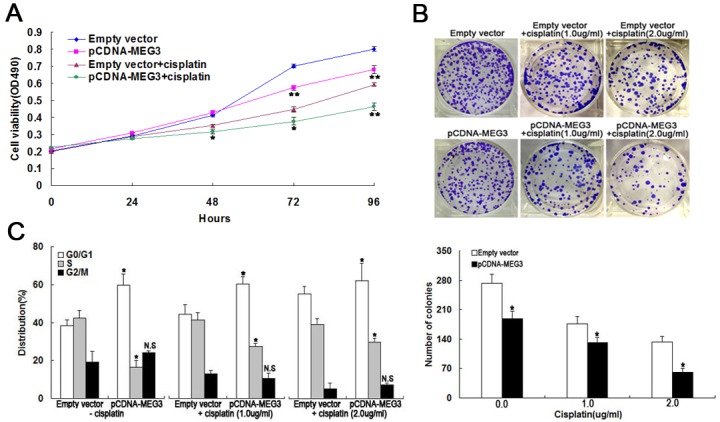
(Fig 3A) MTT assay of A549/DDP cell proliferation with or without cisplatin. (Fig 3B) Colony formation analysis of cell proliferation in combination with increasing concentrations of cisplatin (0.0, 1.0, and 2.0 μg/ml). (Fig 3C) Flow cytometry analysis of cell cycle distribution in combination with increasing concentrations of cisplatin. *P < 0.05, ** P < 0.01.
Flow cytometric analysis showed that the percentage of pCDNA-MEG3-transfected A549/DDP cells in G1/G0 phase increased gradually and those in S phase decreased gradually with increasing doses of cisplatin compared with control cells (Fig 3C). Thus, the overexpression of MEG3 also seemed to reverse the cisplatin resistance of LAD cells through the inhibition of proliferation.
MEG3 knockdown impairs A549 cell proliferation and apoptosis in vitro
To further investigate the effect of MEG3 on the cisplatin resistance of LAD cells, A549 cells were transfected with si-NC or siRNA-MEG3 (si-MEG3 I, si-MEG3 II, and si-MEG3 III). qRT-PCR showed that MEG3 expression was significantly decreased in si-MEG3 II-transfected cells compared with controls (Fig 4A). Moreover, MEG3 knockdown significantly increased the IC50 of A549 cells to cisplatin, as demonstrated by the MTT assay (Fig 4B). Flow cytometry revealed that MEG3 knockdown by si-MEG3 gradually decreased the cisplatin-induced apoptosis of A549 cells treated with increasing doses of cisplatin compared with respective controls (Fig 4C).
Fig 4. Knockdown of MEG3 inhibits A549 cell apoptosis in vitro.

(Fig 4A) qRT-PCR analysis of MEG3 expression levels following the transfection of A549 cells with si-NC and si-MEG3. (Fig 4B) IC50 values of si-NC- and si-MEG3-transfected A549 cells to cisplatin. (Fig 4C) Flow cytometry analysis of apoptosis in si-NC- and si-MEG3-transfected A549 cells treated with increasing concentrations of cisplatin (0.0, 1.0, and 1.5 μg/ml). *P < 0.05, ** P < 0.01.
Cell proliferation was also found to be promoted in si-MEG3-transfected A549 cells with or without cisplatin treatment compared with si-NC-transfected controls (Fig 5A). Flow cytometric analysis revealed that the percentage of cells in G1/G0 phase decreased gradually and those in S phase increased in si-MEG3-transfected A549 cells with increasing doses of cisplatin (0.0, 1.0, and 1.5 ug/ml) (Fig 5B). These results therefore suggest that MEG3 knockdown increases the cisplatin resistance of A549 cells through apoptosis inhibition and the promotion of cell proliferation.
Fig 5. Knockdown of MEG3 promotes A549 cell proliferation in vitro.

(Fig 5A) MTT assay of the proliferation of si-NC- and si-MEG3-transfected A549 cells. (Fig 5B) Flow cytometry analysis of cell cycle distribution in combination with increasing concentrations of cisplatin (0.0, 1.0, and 1.5 μg/ml). *P < 0.05, ** P < 0.01.
MEG3 induces the activation of p53 and Bcl-xl in LAD cells
To highlight the impact of MEG3 in reversing the cisplatin resistance of A549/DDP cells through the enhancement of apoptosis, we performed western blotting to detect apoptotic signals in the form of MDM2 and Bcl-XL proteins 48 h after MEG3 transfection combined with cisplatin treatment. This was because MEG3 is thought to function as a tumor suppressor by inducing the activation of p53, which can translocate to the mitochondria during apoptosis, and MDM2 is responsive to p53 activation [22,23]. As shown in Fig 6, A549/DDP cells transfected with pCDNA-MEG3 showed increased p53 protein and decreased MDM2 protein expression compared with controls. We next determined whether MEG3 could increase apoptosis by regulating the mitochondrial apoptosis pathway. When MEG3 was overexpressed, western blotting revealed decreased Bcl-xl expression, but no significant difference in Bcl-2 expression. These data further suggested that MEG3-induced apoptosis in LAD cell cisplatin resistance is induced by the mitochondrial apoptosis pathway involving p53 and Bcl-xl.
Fig 6. MEG3 induces the activation of p53 and Bcl-xl proteins.
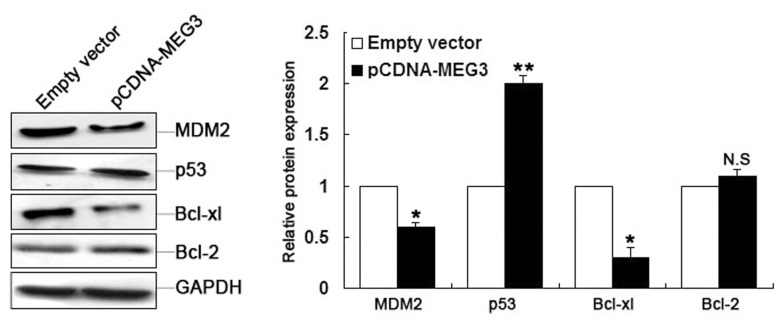
Western blot analysis of MDM2, p53, Bcl-xl, and Bcl-2 expression levels after pCDNA-MEG3 or empty vector transfection. *P < 0.05, ** P < 0.01.
MEG3 improves the in vivo sensitivity of LAD cells to cisplatin
To further investigate the underlying roles of MEG3 in enhancing the chemosensitivity of LAD cells to cisplatin, we used a nude mouse xenograft model. A549/DDP cells transfected with pCDNA-MEG3 or empty vector were subcutaneously injected into mice, followed by treatment with cisplatin. Four weeks after the initial cisplatin administration, the volume and average weight of tumor xenografts was recorded (Fig 7A and 7B). As shown in Fig 7C, the tumors formed from pCDNA-MEG3-transfected A549/DDP cells grew significantly more slowly than those from controls following cisplatin treatment. It was also observed that the upregulation of MEG3 led to the inhibition of tumor growth. qRT-PCR analysis of MEG3 expression found it to be significantly higher in tumor tissues formed from pCDNA-MEG3-transfected A549/DDP cells than those from controls (Fig 7D). Additionally, immunostaining indicated that PCNA levels were lower in tumors formed from pCDNA-MEG3-transfected A549/DDP cells compared with tumors from control cells, while p53 protein levels were increased and Bcl-xl protein decreased, respectively, in these tumor types (Fig 7E). These results suggested that MEG3 overexpression increased the in vivo chemosensitivity of LAD cells to cisplatin.
Fig 7. MEG3 improves the in vivo sensitivity of A549/DDP cells to cisplatin.

(Fig 7A, 7B) Four weeks after initial cisplatin administration, the tumor volume and average weight of mice receiving pCDNA-MEG3- or empty vector-transfected A549/DDP cells were recorded. (Fig 7C) Tumor volume was calculated twice weekly after cisplatin treatment. (Fig 7D) qRT-PCR analysis of MEG3 expression levels in tumor tissues formed from pCDNA-MEG3- or empty vector-transfected A549/DDP cells. (Fig 7E) Tumors developed from pCDNA-MEG3-transfected A549/DDP cells showed lower PCNA protein levels, higher p53 protein expression, and lower Bcl-xl protein expression than those developed from control cells. Upper: H&E staining; second, third row and lower: immunostaining. Original magnification, ×100 or ×200. *P < 0.05, ** P < 0.01.
MEG3 correlates with the chemotherapy of LAD patients
We next used qRT-PCR to investigate the MEG3 expression of tumor tissues from 41 patients with advanced LAD treated with cisplatin-based chemotherapy. As shown in Fig 8A, the average MEG3 expression level of ‘‘insensitive” tissues was significantly lower than ‘‘sensitive” tissues. Thus, the level of MEG3 expression was strongly positively correlated with the response of patients to cisplatin-based chemotherapy. However, correlation analysis of MEG3 expression level of two groups with their clinical pathological features revealed no significant association (S1 Table). Kaplan–Meier survival analysis was performed to establish the association between MEG3 expression and LAD patient progression-free survival (PFS). Those LAD patients with low MEG3 levels (n = 17) had a shorter PFS than those with high MEG3 levels (n = 24) (Fig 8B). Finally, we observed that p53 protein expression was increased and Bcl-xl protein expression decreased in ‘‘sensitive” tissues compared with ‘‘insensitive” tissues (Fig 8C). Together, these data strongly indicated that MEG3 expression in advanced LAD patients is correlated with the response of patients to cisplatin-based chemotherapy.
Fig 8. MEG3 expression levels are positively correlated with the responses of patients to cisplatin-based chemotherapy.

(Fig 8A) qRT-PCR analysis of MEG3 expression levels in cisplatin-sensitive (n = 20) and cisplatin-insensitive (n = 21) LAD tissues. (Fig 8B) Kaplan–Meier survival analysis of the association between PFS of LAD patients and MEG3 expression (log rank P < 0.001). (Fig 8C) Cisplatin-sensitive tissues exhibited higher p53 and lower Bcl-xl protein expression than cisplatin-insensitive tissues. Original magnification, ×200 or ×400. ** P < 0.01.
Discussion
As the most common subtype of NSCLC, LAD is attracting considerable attention particularly because its acquired resistance to drugs makes treatment difficult. Although the dysregulation of some lncRNAs is thought to affect LAD chemoresistance, the underlying molecular mechanisms of this have not been fully elucidated. Cisplatin is a commonly used chemotherapy drug and is the first chemotherapeutic choice for NSCLC. However, acquired drug resistance is a major obstacle to clinical chemotherapy.
Some mechanisms of cisplatin resistance have been reported, for instance, the Nrf2/ARE pathway in NSCLC and the calcium signaling pathway in ovarian cancer [24, 25]. Studies across a variety of cancer types demonstrate that misregulation of lncRNAs contributes to human disease or tumorigenesis, such as pituitary adenomas and gastric cancer [26–28]. Moreover, lncRNAs have been found to regulate a series of biological functions including genomic imprinting and transcriptional regulation. In this study, we found that the MEG3 expression level was significantly decreased in cisplatin-resistant A549/DDP cells compared with parental A549 cells. This increased MEG3 expression appeared to reverse the chemoresistance of A549/DDP cells to cisplatin. We found that lncRNA MEG3 appeared to function as a tumor suppressor by regulating the p53 and Bcl-xl-induced mitochondrial apoptosis pathway, and showed that its increased expression was positively correlated with the sensitivity of LAD patients to cisplatin treatment.
Cell apoptosis is involved in the response of cancers to chemotherapy, radiation, or targeted therapy, and its induction may offer potential benefits to cancer therapeutics [29]. However, apoptosis is just one of the responses to cisplatin treatment. Wang et al. demonstrated that cisplatin exerted its cytotoxic effect partially through regulation of the p53 signaling pathway [30]. p53 is a DNA-binding tumor suppressor that plays a key role in many physiological processes, including DNA repair and apoptosis [31]. Furthermore, it can be induced by cisplatin to regulate cell cycle arrest [30]. MDM2 may inhibit the p53 transactivation function by engaging its amino-terminal transactivation domain [32], while the reactivation of p53 is a viable strategy for cancer treatment [33]. p53 is thought to act at multiple levels to induce mitochondrially-directed apoptosis [34], and triptolide was shown to be a potential chemotherapeutic agent for endometrial cancer via its actions on the p53-independent mitochondrial pathway [35].
Bcl-xl is a member of the Bcl-2 family of proteins that suppresses tumor apoptosis. This family is also reported to play an important role in mitochondrial permeabilization-induced apoptosis particularly through the p53–Bcl-xl interaction [36, 37]. In the present study, we showed that p53 expression was increased and that of Bcl-xl was decreased in MEG3-overexpressing LAD cells, while apoptosis was increased and the chemoresistance was partially reversed. We speculate that these observations reflect changes to the mitochondrial apoptosis pathway via p53 and Bcl-xl, but further work is needed to confirm this.
Taken together, our data demonstrated that the dysregulation of lncRNA MEG3 underlies the cisplatin resistance of LAD, indicating that its overexpression may contribute to increased cisplatin chemosensitivity via the p53 and Bcl-xl induced mitochondria apoptosis pathway. This study enables us to better understand the functions of MEG3 in LAD chemotherapy and suggest that MEG3 could be used as a novel marker of poor response to cisplatin and a potential therapeutic target for LAD chemotherapy. Future study should investigate the molecular mechanisms underlying the functions of MEG3 in the chemoresistance of LAD.
Supporting Information
There was no significant association between pathological features and MEG3 expression level of LAD patient tissues.
(DOC)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (No.81272601,81372397,81472198), the Key Clinical Medicine Technology Foundation of Jiangsu Province (No.BL2014096), the Medical Key Talented Person Foundation of the Jiangsu Provincial Developing Health Project (No.RC2011080), Innovation Team Project of the Second Affiliated Hospital of Nanjing Medical University, the Scientific Research Foundation of Jiangsu Province Health Department(No.H201310),“333 high class Talented Man Project” (No.2011-III-2630) and the School key Fund of Nanjing Medical University (2013NJMU054). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. (2011) Global cancer statistics. CA Cancer J Clin 61: 69–90. 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 2. Devesa SS, Bray F, Vizcaino AP, Parkin DM (2005) International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer 117: 294–299. [DOI] [PubMed] [Google Scholar]
- 3. Sculier JP (2013) Nonsmall cell lung cancer. Eur Respir Rev 22: 33–36. 10.1183/09059180.00007012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morgensztern D, Ng SH, Gao F, Govindan R (2010) Trends in stage distribution for patients with non-small cell lung cancer: a National Cancer Database survey. J Thorac Oncol 5: 29–33. 10.1097/JTO.0b013e3181c5920c [DOI] [PubMed] [Google Scholar]
- 5. Boni C, Tiseo M, Boni L, Baldini E, Recchia F, et al. (2012) Triplets versus doublets, with or without cisplatin, in the first-line treatment of stage IIIB-IV non-small cell lung cancer (NSCLC) patients: a multicenter randomised factorial trial (FAST). Br J Cancer 106: 658–665. 10.1038/bjc.2011.606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gibb EA, Brown CJ, Lam WL (2011) The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10: 38 10.1186/1476-4598-10-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kapranov P, Willingham AT, Gingeras TR (2007) Genome-wide transcription and the implications for genomic organization. Nat Rev Genet 8: 413–423. [DOI] [PubMed] [Google Scholar]
- 8. Mattick JS, Makunin IV (2006) Non-coding RNA. Hum Mol Genet 15 Spec No 1: R17–R29. [DOI] [PubMed] [Google Scholar]
- 9. Brosnan CA, Voinnet O (2009) The long and the short of noncoding RNAs. Curr Opin Cell Biol 21: 416–425. 10.1016/j.ceb.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 10. Wang H, Zhu LJ, Yang YC, Wang ZX, Wang R (2014) MiR- 224 promotes the chemoresistance of human lung adenocarcinoma cells to cisplatin via regulating G1/S transition and apoptosis by targeting p21(WAF1/CIP1). Br J Cancer 111:339–354 10.1038/bjc.2014.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang H, Zhong Y, Xie H, Lai X, Xu M, et al. (2013) Induction of the liver cancer-down-regulated long noncoding RNA uc002mbe.2 mediates trichostatin-induced apoptosis of liver cancer cells. Biochem Pharmacol 85: 1761–1769. 10.1016/j.bcp.2013.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y, Li H, Hou S, Hu B, Liu J, et al. (2013) The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PLoS One 8: e65309 10.1371/journal.pone.0065309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Z, Sun M, Lu K, Liu J, Zhang M, et al. (2013) The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregualtion of p21(WAF1/CIP1) expression. PLoS One 8: e77293 10.1371/journal.pone.0077293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Redis RS, Sieuwerts AM, Look MP, Tudoran O, Ivan C, et al. (2013) CCAT2, a novel long non-coding RNA in breast cancer: expression study and clinical correlations. Oncotarget 4: 1748–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyoshi N, Wagatsuma H, Wakana S, Shiroishi T, Nomura M, et al. (2000) Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 5: 211–220. [DOI] [PubMed] [Google Scholar]
- 16. Zhang X, Zhou Y, Mehta KR, Danila DC, Scolavino S, et al. (2003) A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J Clin Endocrinol Metab 88: 5119–5126. [DOI] [PubMed] [Google Scholar]
- 17. Ying L, Huang Y, Chen H, Wang Y, Xia L, et al. (2013) Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst 9: 407–411. 10.1039/c2mb25386k [DOI] [PubMed] [Google Scholar]
- 18. Wang P, Ren Z, Sun P (2012) Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem 113: 1868–1874. 10.1002/jcb.24055 [DOI] [PubMed] [Google Scholar]
- 19. Anwar SL, Krech T, Hasemeier B, Schipper E, Schweitzer N, et al. (2012) Loss of imprinting and allelic switching at the DLK1-MEG3 locus in human hepatocellular carcinoma. PLoS One 7: e49462 10.1371/journal.pone.0049462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X, Gejman R, Mahta A, Zhong Y, Rice KA, et al. (2010) Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res 70: 2350–2358. 10.1158/0008-5472.CAN-09-3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu KH, Li W, Liu XH, Sun M, Zhang ML, et al. (2013) Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer 13: 461 10.1186/1471-2407-13-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pei D, Zhang Y, Zheng J (2012) Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget 3: 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, et al. (2007) Activation of p53 by MEG3 non-coding RNA. J Biol Chem 282: 24731–24742. [DOI] [PubMed] [Google Scholar]
- 24. Lim J, Lee SH, Cho S, Lee IS, Kang BY, et al. (2013) 4-methoxychalcone enhances cisplatin-induced oxidative stress and cytotoxicity by inhibiting the Nrf2/ARE-mediated defense mechanism in A549 lung cancer cells. Mol Cells 36: 340–346. 10.1007/s10059-013-0123-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Al-Bahlani S, Fraser M, Wong AY, Sayan BS, Bergeron R, et al. (2011) P73 regulates cisplatin-induced apoptosis in ovarian cancer cells via a calcium/calpain-dependent mechanism. Oncogene 30: 4219–4230. 10.1038/onc.2011.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huarte M, Rinn JL (2010) Large non-coding RNAs: missing links in cancer? Hum Mol Genet 19: 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gejman R, Batista DL, Zhong Y, Zhou Y, Zhang X, et al. (2008) Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. Journal of Clinical Endocrinology and Metabolism: 934119–934125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun M, Xia R, Jin F, Xu T, Liu Z, et al. (2014) Downregulated long noncoding RNA MEG3 is associated with poor prognosis and promotes cell proliferation in gastric cancer. Tumour Biol 35:1065–1073. 10.1007/s13277-013-1142-z [DOI] [PubMed] [Google Scholar]
- 29. Cotter TG (2009) Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 9: 501–507. 10.1038/nrc2663 [DOI] [PubMed] [Google Scholar]
- 30. Wang S, Li W, Xue Z, Lu Y, Narsinh K, et al. (2013) Molecular imaging of p53 signal pathway in lung cancer cell cycle arrest induced by cisplatin. Mol Carcinog 52: 900–907. 10.1002/mc.21930 [DOI] [PubMed] [Google Scholar]
- 31. Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9: 402–412. 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- 32. Wang X, Jiang X (2012) Mdm2 and MdmX partner to regulate p53. FEBS Lett 586: 1390–1396. 10.1016/j.febslet.2012.02.049 [DOI] [PubMed] [Google Scholar]
- 33. Wade M, Li YC, Wahl GM (2013) MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13: 83–96. 10.1038/nrc3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walia V, Kakar S, Elble R (2011) Micromanagement of the mitochondrial apoptotic pathway by p53. Front Biosci (Landmark Ed) 16: 749–758. [DOI] [PubMed] [Google Scholar]
- 35. Wang XF, Zhao YB, Wu Q, Sun ZH, Li HJ (2014) Triptolide induces apoptosis in endometrial cancer via a p53independent mitochondrial pathway. Mol Med Rep 9: 39–44. 10.3892/mmr.2013.1783 [DOI] [PubMed] [Google Scholar]
- 36. Tomita Y, Marchenko N, Erster S, Nemajerova A, Dehner A, et al. (2006) WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem 281: 8600–8606. [DOI] [PubMed] [Google Scholar]
- 37. Orrenius S (2004) Mitochondrial regulation of apoptotic cell death. Toxicol Lett 149: 19–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
There was no significant association between pathological features and MEG3 expression level of LAD patient tissues.
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.