

Abstract
We report a new method for fast and sensitive analyses of biologically relevant fatty acids (FAs) in red blood cells (RBC) by liquid chromatography mass spectrometry (LC–MS). A new chemical derivatization approach was developed forming picolylamides from FAs in a quantitative reaction. Fourteen derivatized FA standards, including saturated and unsaturated FAs from C14 to C22, were efficiently separated within 15 min. In addition, the use of a recently introduced benchtop orbitrap mass spectrometer under positive electrospray ionization (ESI) full scan mode showed a 2–10-fold improvement in sensitivity compared with a conventional tandem MS method, with a limit of detection in the low femtomole range for saturated and unsaturated FAs. The developed method was applied to determine FA concentrations in RBC with intra- and interday coefficients of variation below 10%.
Fatty acids (FAs), particularly long chain polyunsaturated fatty acids (PUFAs), are present in living cells and body fluids in the form of free acids, esters, triglycerides, or phospholipids, play a key role in metabolic pathways, regulating human cardiovascular and immune systems,1 and are crucial in brain development. Accurate measurement of FAs, therefore, has important physiological and clinical implications. The most common quantification of FAs was previously performed by gas chromatography/mass spectrometry (GC/MS) using electron impact (EI) ionization after methyl ester derivatization.1,2 Recently, liquid chromatography–mass spectrometry (LC–MS) has been widely used for FA analysis.3 Electrospray ionization (ESI) in combination with tandem mass spectrometry or the FTICR (Fourier transform-ion cyclotron resonance) technique4,5 have offered an alternative way to ionize and detect nonvolatile and heat-sensitive FAs. However, the drawbacks of low specificity in the negative MS detection mode and requirement of postcolumn alkalization after acidic chromatographic separation have hindered a simple way of fatty acid analysis.
Several chemical derivatization methods6,7 have since been developed to improve the ESI-LC–MS detection responses in the positive detection mode. Methyl ester derivatives for measuring fatty acids have been hampered by the lack of specificity due to the difficulty in fragmentation in tandem mass analysis. Johnson et al.8 have developed a sensitive ESI-LC–MS method for the measurement of free fatty acids using dimethylaminoethyl ester (DMAE) derivatives which contain a nitrogen tag that was easy to protonate. This FA-DMAE derivative showed fast and complete ionization under positive ESI mode and rapid fragmentation by collision induced dissociation (CID) which provided useful information when measured by tandem MS. Furthermore, a 10-fold increase in sensitivity has been achieved by the precharged quaternary ammonium salt of the trimethylaminoethyl ester (TMAE).9,10 However, this derivatization requires harmful reagents, and the products lack good chromatographic resolutions.
In this study, we established a simple, fast, accurate, and sensitive ESI-LC–MS method for measuring biologically and clinically important FAs, by comparing different derivatizing reagents, including nitrogen-containing amines and alcohols, for positive ESI-LC–MS detection. Moreover, we applied a new benchtop orbitrap MS using exact full mass scan, as an alternative method for tandem MS that allowed exact mass measurements for unambiguous analyte identification without collision induced fragmentation. Finally, we applied the developed method in red blood cells (RBC), since FA composition in RBC provide a useful marker for oxidative stress, cardiovascular disease, and other chronic diseases.11
EXPERIMENTAL SECTION
Fatty Acids Derivatization
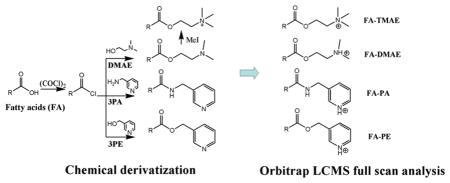
Chemicals, reagents, standards preparation, and fatty acids extraction from red blood cells (RBC) information is provided in supplemental notes in the Supporting Information. Both the FA standards and RBC samples underwent the same extraction, hydrolysis, and derivatization steps before LC–MS analysis. The derivatization procedure was modified from Johnson’s method.9 In brief, 150 μL of standard (0.1–100 μg/mL) and 20 μL of internal standard mixture were mixed and dried under nitrogen. To the dried residue was added 200 μL of oxalyl chloride (2 M in dichloromethane), and the mixture was incubated at 65 °C on a heating block for 5 min and then dried under nitrogen. To the residue was added 150 μL of dimethylaminoethanol, 3-picolylamine, or 3-pyridylcarbinol, respectively (1% in acetonitrile, v/v) to form the dimethylaminoethyl ester (FA-DMAE), 3-picolylamide (FA-PA), and 3-picolinyl ester (FA-PE) derivatives (Figure 1), respectively. The mixture was incubated at room temperature for 5 min, followed by drying under nitrogen to give the derivatized FAs. The FA-DMAE product was further converted to trimethylaminoethyl ester (FA-TMAE) by incubating with 150 μL of methyl iodide (50% in methanol, v/v) at room temperature for 5 min, followed by drying under nitrogen. The dried FAs derivatives were dissolved in 1000 μL of ethanol and further diluted up to 10-fold with ethanol prior to LC–MS analysis.
Figure 1.
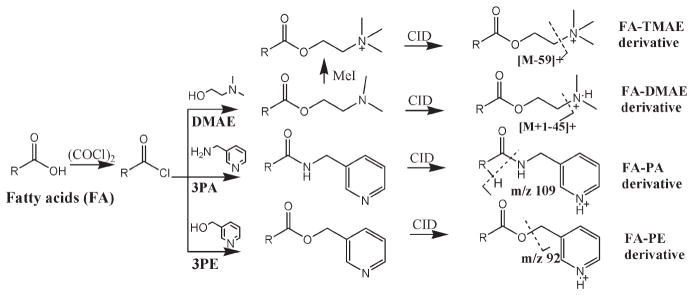
Fatty acid derivatization reaction scheme and their adducts fragmentation by MS/MS analysis.
LC–MS Analysis
The analysis was performed using a model HTC Pal autosampler (Leap Technologies, Carrboro, NC) connected to a model Accela ultra-HPLC system in combination with a model Exactive orbitrap mass spectrometer (both from Thermo Electron, Waltham, MA). Ten μL of the diluted FA derivative was injected onto a Agilent Zorbax SC-C18 column (3.0 × 50 mm, 1.8 μm, Agilent, Santa Clara, CA) using a mobile phase consisting of 0.1% formic acid in water (A) and 0.1% formic acid in MeCN (B) at a flow rate of 500 μL/min, with the following linear gradient A/B (v/v): 0–7 min 35/65, 7.1–11 min 10/90, and 11.1–15 min 35/65 for the separation of FA-PA and FA-DMAE derivatives or 0–5 min 20/80, 5.1–16 min 10/90, and 16.1–20 min 20/80 for FA-PE derivatives separation.
Mass detection was carried out after electrospray ionization (ESI) in positive-ion full scan mode. The settings of the mass spectrometer were as follows: spray voltage, 4.5 kV; capillary temperature, 250 °C; maxium injection time, 250 ms; and scan rage, 100–650. Nitrogen was used as sheath gas (pressure 30 units) and auxiliary gas (pressure 10 units). The in-source collision induced dissociation energy (CID) was set at 5 eV to dissociate dimers or sodium adducts, and the automatic gain control (AGC) was set at balanced. Data acquisition and analysis were performed using Thermo’s Xcalibur software. Detection of the analyte was set within 10 ppm of the calculated mass. Table 1 showed the detailed formula and exact molecular weight of each analyte for LC–MS full mass scan analysis.
Table 1.
Parameters of Fatty Acid Standards and 3-Picolylamide (PA), 3-Picolinyl Ester (PE), and Dimethylaminoethyl Ester (DMAE) Derivatives for Orbitrap Mass Spectrometry Quantification
| name | lipid name | FA-PA derivative
|
FA-PE derivative
|
FA-DMAE derivative
|
|||
|---|---|---|---|---|---|---|---|
| formula | [M + H]+ | formula | [M + H]+ | formula | [M + H]+ | ||
| Monounsaturated | |||||||
| oleic acid (OA) | C18:1 n-9 | C24H40N2O | 373.32134 | C24H39NO2 | 374.30536 | C22H43NO2 | 354.33666 |
| palmitoleic acid (PLA) | C16:1 n-7 | C22H36N2O | 345.29004 | C22H35NO2 | 346.27406 | C20H39NO2 | 326.30536 |
| ω-6 | Polyunsaturated | ||||||
| linoleic acid (LA) | C18:2 n-6 | C24H38N2O | 371.30569 | C24H37NO2 | 372.28971 | C22H41NO2 | 352.32101 |
| eicosadienoic acid (EDA) | C20:2 n-6 | C26H42N2O | 399.33699 | C26H41NO2 | 400.32101 | C24H45NO2 | 380.35231 |
| arachidonic acid (AA) | C20:4 n-6 | C26H38N2O | 395.30569 | C26H37NO2 | 396.28971 | C24H41NO2 | 376.32101 |
| ω-3 | |||||||
| α-linolenic acid (ALA) | C18:3 n-3 | C24H36N2O | 369.29004 | C24H35NO2 | 370.27406 | C22H39NO2 | 350.30536 |
| eicosapentaenoic acid (EPA) | C20:5 n-3 | C26H36N2O | 393.29004 | C26H35NO2 | 394.27406 | C24H39NO2 | 374.30536 |
| docosapentaenoic acid (DPA) | C22:5 n-3 | C28H40N2O | 421.32134 | C28H39NO2 | 422.30536 | C26H43NO2 | 402.33666 |
| docosahexaenoic acid (DHA) | C22:6 n-3 | C28H38N2O | 419.30569 | C28H37NO2 | 420.28971 | C26H41NO2 | 400.32101 |
| Saturated | |||||||
| stearic acid (SA) | C18:0 | C24H42N2O | 375.33699 | C24H41NO2 | 376.32101 | C22H45NO2 | 356.35231 |
| palmitic acid (PA) | C16:0 | C22H38N2O | 347.30569 | C22H37NO2 | 348.28971 | C20H41NO2 | 328.32101 |
| myristic acid (MA) | C14:0 | C20H34N2O | 319.27439 | C20H33NO2 | 320.25841 | C18H37NO2 | 300.28971 |
| Conjugated | |||||||
| α-eleostearic acid (ESA) | C18:3 n-5 | C24H36N2O | 369.29004 | C24H35NO2 | 370.27406 | C22H39NO2 | 350.30536 |
| α-parinaric acid (α-PA) | C18:4 n-3 | C24H34N2O | 367.27439 | C24H33NO2 | 368.25841 | C22H37NO2 | 348.28971 |
| Deuterated Internal Standards | |||||||
| docosahexaenoic acid-d5 (DHA-d5) | C28H33D5N2O | 424.33707 | C28H32NO2D5 | 425.32109 | C26H36NO2D5 | 405.35239 | |
| eicosapentaenoic acid-d5 (EPA-d5) | C26H31N2OD5 | 398.32142 | C26H30NO2D5 | 399.30544 | C24H34NO2D5 | 379.33674 | |
| linoleic acid-d4 (LA-d4) | C24H34N2OD4 | 375.33080 | C24H33NO2D4 | 376.31481 | C22H37NO2D4 | 356.34611 | |
| arachidonic acid-d8 (AA-d8) | C26H30N2OD8 | 403.35590 | C26H29NO2D8 | 404.33992 | C24H33NO2D8 | 384.37122 | |
| palmitoleic acid-d14 (PLA-d14) | C22H22N2OD14 | 359.37791 | C22H21NO2D14 | 360.36193 | C20H25NO2D14 | 340.39323 | |
| stearic acid-d3 (SA-d3) | C24H39N2OD3 | 378.35582 | C24H38NO2D3 | 379.33984 | C22H42NO2D3 | 359.37114 | |
LC–MS data were also obtained on an Accela ultra-HPLC system connected to a TSQ Ultra Quantum triple quadrupole mass spectrometer (Thermo Electron, Waltham, MA). The ion source was the same with the orbitrap, and the capillary temperature was 250 °C. CID was performed with the collision gas at 1.0 Torr. Scanning was performed in selected reaction monitoring (SRM) mode where the precursor ion transition to a diagnostically valuable and abundant fragment was recorded. For FA-DMAE derivatives, SRM was performed from the precursor [M + H]+ (FA plus 72 Da from the dimethylaminoehtyl group) to the corresponding [M + H − 45]+ ion (derived from loss of the dimethylamino group). The collision energy (CE) was set at 16 eV for polyunsaturated FAs and 20 eV for all other FAs. For FA-PA derivatives, SRM was monitored from the precursor [M + H]+ ion (FAs plus 91 from the picolylamine group) to the common fragment at m/z 109 generated by the picolylamine moiety. The CE was set at 24 eV for polyunsaturated FAs, 30 eV for all other FAs. The fragmentation of FA-PE derivatives requires higher collision energy; we chose to monitor the common fragment at m/z 92 (mass of picolyl moiety, Figure 1) generated from the precursor [M + H]+ (FAs plus 92 from the picolyl ester group) that requires less collision energy. The CE was set at 34 eV for all FAs. Figure 1 showed the detailed derivatization reaction schemes and their fragmentation in MS/MS analysis.
RESULTS AND DISCUSSION
Several derivatization methods have been previously investigated for the improvement of LC–MS analyses of FAs, including precharged quaternary amines or an electron-capture pentafluorobenzyl moiety.7,9,12 Those modifications were found to be valuable as to improve ionization and sensitivity, but they also have some limitations, for example, the use of harmful reagents and multiple derivatization steps,9 long analysis run,7 lack of complete separation of the physiological significant fatty acids,10 and not being applicable to saturated fatty acids.12 To overcome these problems, we intended to find a chemical tag with the following properties: (1) high proton-affinity (for example, an amine or a pyridine group which are readily ionized to be sensitively detected by positive ESI); (2) simple, mild, and quantitative derivatization reaction; (3) commercial availability, low cost, low toxicity, environmental friendliness, and high stability of derivatization reagents and products; (4) good chromatographic properties. On the basis of these requirements, 3-picolylamine (PA) and 3-pyridylcarbinol (PE) were selected as the derivatization reagents mainly because they possess a readily ionizable pyridine group and can be easily attached to the carboxylic acid moiety via an amide or an ester linkage, respectively. Furthermore, to compare the chromatographic resolution and limit of detection of the pyridine tag, we modified FAs to previously reported precharged quaternary ammonium FA-TMAE derivatives and FA-DMAE products. All products synthesized were highly soluble in ethanol which caused us to use ethanol for all dissolution and dilution steps.
The derivatization reactions were carried out using a modification from Johnson’s method9 via a straightforward one-pot two-step reaction procedure (Figure 1). A total of 14 FAs including saturated FAs (14–18 carbons), ω-3 and ω-6 polyunsaturated FAs (PUFAs) (18–22 carbons), some mono- and conjugated-FAs (16–18 carbon), and 6 internal standards were derivatized to the corresponding 3 types of products as shown in Table 1 (FA-TMAE data not shown). Free fatty acids were first converted to the acyl chloride intermediate by the treatment of oxalyl chloride, followed by coupling with an amine or alcohol to form the corresponding amide or ester. The FA-DMAE derivative was further converted to the precharged quaternary amine FA-TMAE derivative by reacting with methyl iodide. This de-rivatizing method is fast and quantitative, without a complicated handling procedure. Higashi et al.13 have investigated the coupling reaction of 2-picolylamine with carboxylic acid using triphenylphosphine (TPP) and 2,2′-dipyridyl disulfide (DPDS) via a one-pot synthesis step. We found, however, that the TPP/DPDS catalyzed reaction products interfere with late eluting FAs (e.g., SA and PA) under our LC conditions, and the reaction time is longer (30 min) compared to the 10 min total reaction time via acyl chloride.
Currently, most, if not all reported LC–MS methods for FA analysis use tandem MS in SRM mode. This technique provides good sensitivity for FAs in biological samples but has several restrictions. First, some FA derivatives, such as methyl ester derivatives, are hard to fragment after ionization and, therefore, cannot be detected by SRM MS; second, increasing the number of analytes may lead to compromised sensitivity. This is based on the scan events on a particular time period: the more scan events, the less scan time there is for a particular SRM transition. Third, our experiment showed that tandem MS lacks selectivity due to the SRM selection of noncharacteristic product ions. Last but not least, unlike full scan mode monitoring, tandem MS performs only with high sensitivity and selectivity for known targets but cannot be applied successfully to nontargeted analytes. In contrast, the orbitrap mass spectrometers including the recently introduced benchtop models have high resolving power, fast scan speed, high in-scan dynamic range, and accurate mass analysis. They overcome the problems associated with tandem MS and provide a sensitive and selective way for analyzing all analytes including those that were not targeted within a recorded HPLC run. Also, reinterrogation of recorded analyses is a great advantage, especially for precious clinical, epidemiologic, and other biological samples with limited specimen amounts. In our study, we intended to take advantage of the orbitrap technology to analyze a wide range of biological and clinical relevant fatty acids based on accurate mass measurements and evaluated whether a recently introduced benchtop model would fulfill the required needs.
Among many columns tested, good separation of the FA derivatives were achieved on an Agilent Zorbax SC-C18 reverse phase column using a mobile phase consisting of 0.1% formic acid in water and acetonitrile that exhibits a better chromatographic resolution than using methanol. A mixture of 20 fatty acids, including 14 FA standards and 6 deuterated internal standards, were derivatized and successfully separated with a retention time between 2 and 14 min (Figure 2). ESI orbitrap MS was performed in positive full scan mode with a mass scan range from m/z 100 to 650 and 5 eV in-source CID to avoid adduct or dimer formation without using higher energy collision induced dissociation (HCD) in the collision cell. Quantitation was carried out using exact masses of the protonated [M + H]+ ions for FA-PA (FA plus 91 from the picolylamine group), FA-PE (FA plus 92 from the pyridinyl methanol group), and FA-DMAE (FA plus 72 from the dimethylaminoethyl group) or the M+ ion (FA plus 86 from the trimethylaminoethyl group) for FA-TMAE derivatives. The calibration curves were linear (R2 > 0.99) in the experimental concentration range from 1.5 to 1500 ng/mL. Among the four different FA derivatives, FA-PA derivatives provided intense protonated molecules in the positive ESI and exhibited the best sensitivity and chromatographic property. The limit of detection (LOD) was in the low femtomole range for the saturated and unsaturated FAs, 2–4-fold more sensitive than the DMAE method, largely caused by the high proton-affinity pyridyl group (Table 2). The conjugated FAs, including cisPA and αESA, however, showed a LOD in the nanomole range, possibly due to the labile nature of the conjugated system during sample preparation. The FA-PE derivatives also gave intense protonated [M + H]+ signals as base peaks in the positive ESI-MS mode, but the FA-PA derivatives exhibited better results in terms of chromatography and LOD, with the lipophilic FA-PE derivatives more retained on the column that required a high portion of organic solvent for elution; in addition, the LOD was 2–3-fold higher than the FA-PA derivatives. Interestingly, contrary to the 10-fold increase in sensitivity using precharged quaternary amine FA-TMAE derivatives via methyl iodide treatment under a tandem mass method as reported previously,9 we found FA-DMAEs and FA-TMAEs to possess similar LODs in the femtomol range using orbitrap MS in the full scan method. DMAE products are preferable to TMAE, because the harmful reagent methyl iodide is not needed and an additional derivatization step can be avoided. In summary, we found that FA-PA derivatives are superior to PEs, DMAEs, and TMAEs when measured by orbitrap MS, due to 2–4-fold lower LODs, requiring short chromatographic runs (2–12 min) with good resolutions.
Figure 2.

LC–MS chromatogram of FA-PA derivatives from standards (75 ng/mL for all analytes). Profiles were collected using orbitrap LC–MS described in the Experimental Section. Quantitation was performed by the exact masses of [M + H]+ ions (protonated molecular weight of FA plus 91 Da from the picolylamine group). The abbreviations of each analyte are shown in Table 1. Mass spectra of each peak are included in the Supporting Information.
Table 2.
Comparison between the On-Column Limit of Detection (LOD) of 3-Picolylamide (PA), 3-Picolinyl Ester (PE), and Dimethylaminoethyl Ester (DMAE) Derivatives of FAs Analyzed by Orbitrap (Exactive) and Tandem (TSQ) Mass Spectrometersa
| name | FA-PA derivatives
|
FA-PE derivatives
|
FA-DMAE derivatives
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| tR (min) | orbitrap LOD (fmol) | TSQ LOD (fmol) | tR (min) | orbitrap LOD (fmol) | TSQ LOD (fmol) | tR (min) | orbitrap LOD (fmol) | TSQ LOD (fmol) | |
| OA | 8.6 | 11 | 280 | 9.0 | 44 | 84 | 5.8 | 30 | 180 |
| PLA | 3.6 | 7 | 30 | 6.0 | 43 | 63 | 2.8 | 22 | 30 |
| LA | 4.7 | 6 | 96 | 6.0 | 20 | 81 | 3.5 | 22 | 70 |
| EDA | 9.4 | 8 | 98 | 9.6 | 11 | 34 | 6.8 | 37 | 65 |
| AA | 4.6 | 9 | 101 | 5.4 | 20 | 176 | 3.4 | 63 | 167 |
| ALA | 2.9 | 25 | 55 | 4.4 | 30 | 165 | 2.3 | 13 | 39 |
| EPA | 2.9 | 20 | 38 | 3.6 | 21 | 106 | 2.2 | 24 | 190 |
| DPA | 5.5 | 15 | 89 | 6.1 | 64 | 70 | 3.8 | 135 | 62 |
| DHA | 4.3 | 4 | 40 | 4.4 | 42 | 140 | 3.2 | 13 | 23 |
| SA | 11.3 | 5 | 130 | 14.0 | 112 | 135 | 9.7 | 26 | 148 |
| PA | 7.0 | 20 | 138 | 8.9 | 35 | 59 | 4.8 | 34 | 97 |
| MA | 2.9 | 13 | 28 | 5.7 | 12 | 45 | 2.4 | 28 | 23 |
| α-ESA | 3.9 | 1.3 nmol | NDb | 5.2 | ND | ND | 2.9 | ND | ND |
| cisPA | 2.6 | 8.4 nmol | ND | 3.3 | ND | ND | 2.1 | ND | ND |
The abbreviation of each analyte is shown in Table 1.
ND: Not determined.
For comparison purposes, the FA derivatives were also analyzed on a triple quadrupole MS. The LC and ion source conditions were the same as those used with the orbitrap. The quantification was performed under SRM mode, and collision energy (CE) was operated at different levels for the saturated and unsaturated FAs as well as for FA-DMAE, PA, and PE derivatives to obtain the optimal sensitivity for each analyte (see Experimental Section part for detailed description). Compared to FA-PAs, the FA-PEs required higher collision energies to form the 3-pyridylcarbinol ion (m/z 110) at above 45 eV; we, therefore, chose to monitor the transition to the next abundant 3-pyridyl methyl ion (m/z 92) which required less energy (CE below 34 eV). For FA-PA derivatives, the needed CE was relatively mild, but saturated FAs needed higher energy than unsaturated FAs. Protonated 3-picolylamine (m/z 109) was selected for SRM transitions. The FA-DMAE derivatives required less CE (16 eV) for fragmentation; SRM was selected in this case following the transition from the adduct to the loss of the dimethylamine moiety (Figure 1). The calibration curves were linear (R2 > 0.99) in the experimental concentration range from 0.75 to 150 nM. Overall, TSQ triple quadruple MS/MS also provided a sensitive detection for FAs with a LOD (signal-to-noise = 3) in the midfemtomole range, but the orbitrap assay was found to be more sensitive than the tandem mass assay with detection limits being 2–10-fold lower. Table 2 shows a detailed comparison between different LC–MS methods.
Finally, we applied the FA-PA orbitrap MS method to the detection of FAs in RBCs (Figure 3). Due to the highly sensitive nature of FA-PAs, the sample size could be significantly reduced to around 20 μL of liquid or 5 mg of lyophilized RBCs. The consistency of the assay was evaluated by repeated analysis of quality control (QC) RBC samples for 5 days. The intraday CVs for the lyophilized and liquid RBC ranged from 4 to 19% (mean of 7%) and 5 to 18% (mean of 7%), respectively, on the basis of 10 analytes. Dried RBC showed better interday CV ranges (4–26% with a mean of 15%) than liquid RBC (11–22% with a mean of 20%) on the basis of 10 analytes, possibly due to lack of homogeneity in the liquid RBC. It is important to note that high CV values were only obtained by the low concentrated analytes in RBC. The mean intra- and interday CV of lyophilized RBC was improved to 5 and 8%, respectively, on the basis of the 6 most concentrated FA analytes (i.e., conc >0.1 mg/g such as PA, SA, LA, AA, OA, and DHA). More RBC material would be needed for the analysis of low level FAs.
Figure 3.

Typical orbitrap MS chromatogram of fatty acids extracted from 20 μL of red blood cells (diluted in 10 mL of AcN) after 3-picolylamide (PA) derivatization. The abbreviation of each analyte is shown in Table 1.
The accuracy of the FA-PA method was validated using NIST standard reference material SRM3274 (FAs in botanical oils). Four different botanical oils were hydrolyzed, extracted, and derivatized to FA-PAs (see Supporting Information), and the final products were analyzed by orbitrap MS. The concentrations of MA, SA, PA, OA, and LA were determined to be within the 95% confident level, except that the LA concentrations in evening primrose and perilla oils are slightly higher than that (Table 3).
Table 3.
FA-PA Method Validation with NIST SRM3274-FAs in Botanical Oilsa
| SRM oil | saturated FAs
|
unsaturated FAs
|
|||
|---|---|---|---|---|---|
| MA | PA | SA | OA | LA | |
| borage | 0.49 (0.62 ± 0.11) | 100(110 ± 12) | 33.6 (33.1 ± 4.0) | 152.4 (148.7 ± 8.7) | 396 (374 ± 35) |
| evening primrose | 0.344 (0.363 ± 0.03) | 54.7 (58.2 ± 6.1) | 17.8 (18.3 ± 0.838) | 69.9 (68.9 ± 3.7) | 787 (742 ± 24) |
| flax | 0.277 (0.271 ± 0.008) | 42.7 (44.8 ± 5.0) | 27.9 (30.4 ± 2.4) | 165.3 (165.7 ± 6.2) | 160 (171 ± 11) |
| perilla | 0.246 (0.206 ± 0.025) | 52.8 (56.4 ± 5.5) | 19.3 (20.9 ± 1.1) | 157.2 (166.8 ± 7.8) | 212 (160 ± 14) |
Certified concentration value was expressed as a mass fraction (mg/g) at 95% level of confidence and noted in bold in the parentheses. Reference values were also given in normal typeface in parentheses. The abbreviation of each analyte is shown in Table 1.
CONCLUSIONS
We developed a simple and fast fatty acid-picolylamine (FA-PA) derivatization method for the sensitive analysis of fatty acids using exact masses as measured by orbitrap mass spectrometry in positive ESI full scan mode. In comparison with previously reported methods using DMAE and TMAE derivatizations, the FA-PA method had superior sensitivity with a limit of detection in the low femtomole range, 2–4-fold lower than previous methods. Efficient separation for 14 calibrated saturated and unsaturated FAs was achieved within 15 min. In addition, the sensitivity and selectivity were further improved 2–10-fold by the use of orbitrap MS in full scan mode, which offers great advantages for trace amount analysis in addition to the potential of data reinterrogation of nontargeted FAs using full scan data. Finally, oribitrap also provides additional selectivity for nonreadily fragment molecules such as protonated FA esters.
Supplementary Material
Footnotes
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Moldovan Z, Jover E, Bayona JM. Anal Chim Acta. 2002;465:359–378. [Google Scholar]
- 2.Seppanen-Laakso T, Laakso I, Hiltunen R. Anal Chim Acta. 2002;465:39–62. [Google Scholar]
- 3.Rezanka T. J High Resolut Chromatogr. 2000;23:338–342. [Google Scholar]
- 4.Hughey CA, Rodgers RP, Marshall AG. Anal Chem. 2002;74:4145–4149. doi: 10.1021/ac020146b. [DOI] [PubMed] [Google Scholar]
- 5.Leavell MD, Leary JA. Anal Chem. 2006;78:5497–5503. doi: 10.1021/ac0604179. [DOI] [PubMed] [Google Scholar]
- 6.Johnson DW, Trinh MU. Rapid Commun Mass Spectrom. 2003;17:171–175. doi: 10.1002/rcm.889. [DOI] [PubMed] [Google Scholar]
- 7.Yang WC, Adamec J, Regnier FE. Anal Chem. 2007;79:5150–5157. doi: 10.1021/ac070311t. [DOI] [PubMed] [Google Scholar]
- 8.Johnson DW. Rapid Commun Mass Spectrom. 2000;14:2019–2024. doi: 10.1002/1097-0231(20001115)14:21<2019::AID-RCM121>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Johnson DW, Trinh MU, Oe T. J Chromatogr, B: Anal Technol Biomed Life Sci. 2003;798:159–162. doi: 10.1016/j.jchromb.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Pettinella C, Lee SH, Cipollone F, Blair IA. J Chromatogr, B: Anal Technol Biomed Life Sci. 2007;850:168–176. doi: 10.1016/j.jchromb.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 11.O’Brien DM, Kristal AR, Jeannet MA, Wilkinson MJ, Bersamin A, Luick B. Am J Clin Nutr. 2009;89:913–919. doi: 10.3945/ajcn.2008.27054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SH, Williams MV, DuBois RN, Blair IA. Rapid Commun Mass Spectrom. 2003;17:2168–2176. doi: 10.1002/rcm.1170. [DOI] [PubMed] [Google Scholar]
- 13.Higashi T, Ichikawa T, Inagaki S, Min JZ, Fukushima T, Toyo’oka T. J Pharm Biomed Anal. 2010;52:809–818. doi: 10.1016/j.jpba.2010.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.