

Abstract
We developed a multinomial probit model with singular value decomposition for testing a large number of single nucleotide polymorphisms (SNPs) simultaneously, using maximum likelihood estimation and permutation. The method was validated by simulation. We simulated 1000 SNPs, including 9 associated with disease states, and 8 of the 9 were successfully identified. Applying the method to study 32 genes in our Mexican-American samples for association with prediabetes through either impaired glucose tolerance (IGT) or impaired fasting glucose (IFG), we found 3 genes (SORCS1, AMPD1, PPAR) associated with both IGT and IFG, while 5 genes (AMPD2, PRKAA2, C5, TCF7L2, ITR) with the IGT mechanism only and 6 genes (CAPN10, IL4,NOS3, CD14, GCG, SORT1) with the IFG mechanism only. These data suggest that IGT and IFG may indicate different physiological mechanism to prediabetes, via different genetic determinants.
Introduction
Recent technology allows genome-wide association studies (GWAS) to involve a huge number of single nucleotide polymorphisms (SNPs). However, the study samples are often limited. This leads the situation that the number of SNPs (p) is much bigger than available sample (n), which makes the traditional statistical analysis methods unsuitable. Most of current practices analyze one SNP at a time, which create a huge multiple testing problem. In order to analyze all SNPs simultaneously when sample size is much smaller than number of SNPs, we introduced the Iterative Bayesian variable selection (IBVS) method (Kwon et al. 2007) and have successfully applied it to the simulated rheumatoid arthritis data provided by the Genetic Analysis Workshop 15 (GAW15).We later introduced a Bayesian classification with singular value decomposition (BCSVD) method (Kwon et al. 2009) to improve computer’s run-time. Both methods are still limited to dichotomous response variables. We introduce here a multinomial probit model with singular value decomposition (SVD) method for analyzing polychotomous responses in the situation of p ≫ n.We show the validity of the newly developed method by applying it to simulated data set as well as to a real study sample to identify genes contributing to two different mechanisms for prediabetes.
Method and Materials
Multinomial probit model with Singular Value Decomposition
Polychotomous ordinal responses are a type of data that is frequently encountered in common disease studies. A simple example could be found in a study of diabetes, in which subject can be categorized into three groups; normal, prediabetes, and diabetes. Multinomial probit model is commonly utilized to analyze polychotomous categorical responses. The multinomial probit model can be expressed by latent (unobserved) continuous variables associated with categorical responses.
In general, let us assume that the responses y1, y2, ⋯, yn are observed, where yi takes one of the J ordered categories and θ0, θ1, ⋯, θJ are real numbers of bin boundaries, which satisfy that −∞ = θ0 ≤ ⋯ ≤ θJ = ∞. As in Albert and Chib (1993), we denote that z1, z2, ⋯, zn are latent continuous random variables and assume that the latent variable, zi, associated with a categorical outcome, yi, can be explained in terms of a underlying linear model and that the observed response yi has the category j if and only if zi falls between θj−1 and θj. Therefore, we can notice that the ordinal probit model is equivalent to the following model.
| (1) |
where xi is a 1×p vector of the explanatory variables for the ith sample and β is a p×1 vector of parameters to be estimated. In vector-matrix notation, we can have the multinomial ordinal probit model
| (2) |
where z is the n×1 vector of latent variables, X is the n×p matrix of the explanatory variables, β is the p×1 vector of unknown regression coefficients, and ε follows an independent standard multivariate normal distribution, ε ~ N (0, In). By applying SVD to the matrix X in (2), we can notice that the matrix can be expressed as follows.
| (3) |
where A is the p×n singular value factor loading matrix with orthonormal columns so that A′A = In, D = diag(d1,⋯,dn), the diagonal matrix of positive singular values, ordered as d1 ≥ ⋯ ≥ dn > 0, and F is the n × n SVD orthogonal factor matrix with F′F = FF′ =In. Therefore, the model in (2) with the SVD of X can be written as follows.
| (4) |
where L = FD and . Therefore, the n×1 vector of latent variables, z in (4) has a multivariate normal distribution, i.e., z ~ N (Lγ, In).
As shown in (4), we can notice that γ is expressed by a linear combination of the original parameters, β, in (2). Hence, we call γ as super-factors. As mentioned in West et al. (2003), the model in (4) represents a possibly massive dimension reduction from p to n parameters. That is, the regression model with p parameters reduced to that with n parameters derived from the SVD of the design matrix X. Therefore, the statistical inference on the original parameter, β, in (2) turns into the super-factors, γ, in (4).
Model fitting with maximum likelihood estimation
The maximum likelihood estimates of the super-factors, γ, in (4) can be obtained by the iteratively reweighted least squares (IRLS) procedure (Jansen, 1991). The procedure can be briefly described as follows. Let η denote the vector of all model parameters (θ2, ⋯, θJ−1, γ1, ⋯, γn−2). Note that θ1 and θJ are not included in this vector because their values are assumed to be 0 and ∞, respectively. For example, let J = 4 and n = 15. We define a matrix
| (5) |
for i = 1, ⋯, 15. As we see that the vector of new covariates, , is preceded by a 3 × 3 identity matrix with the first column excluded corresponding to the imposed constant, θ1=0. Let Si be a 4 × 3 matrix,
| (6) |
and define Hi = diag(fi1, fi2, fi3), where fij denotes the derivative of a standard normal distribution at . Also, take Wi = diag(pi), where pi is the vector of probabilities that each individual falls in each category, and let N be a 15 × 4 matrix of counted responses. Accordingly, we can define the working dependent variable as ω=(ω′1, ⋯, ω′15)′, where ωi = Si HiTi + (Ni − pi), the regressor matrix, , and the weighted matrix . After initialization of all elements in the algorithm, the weighted least square estimate as a solution of a linear equation ω = Rη would be obtained for the (i+1) iteration as
| (7) |
The matrices ω, R, and W are all recalculated using the current values of η obtained at each least square iteration. This process is performed recursively until changes in the parameter vector η are negligible.
General Solution for the Original Parameters
Since a prior on β determines an prior on γ, the inferences of the original parameter β can be a result of transformation. Due to p ≫ n, the transformation from γ back to β indicates a one-to- many mapping, leading to infinite number of solutions for β. Since the regressor matrix, A, is not square, its inverse must be calculated using the generalized inverse to solve γ = A′β. Let G− denote a generalized inverse of A. As discussed in Graybill (1976, pp34–39), the most general solution to the linear equation can be expressed as
| (8) |
Where h is some p × 1 vector that can possibly be stochastic or deterministic. Therefore, a unique solution for β from the class of all solutions to the linear equation in (8) can be obtained by the choice of generalized inverse, G− and the characteristic of vector h. As discussed in Graybill (1976), we can achieve the unique solution for β by choosing the generalized inverse of A as G− = A′ since A′A = I.
Selection of the significant genes
Finding significant genes is similar to testing whether the regression coefficient of each SNP is statistically significant, i.e., testing a hypothesis: H0 : βi = 0 v.s. H1 : βi ≠ 0, i = 1, ⋯, p. However, when p ≫ n, we are unable to perform the test by comparing the test statistic, t = (β̂ − β)/se(β̂), to a critical value, t1−α/2(n − 2), because of singularity, where β̂ is an estimate of β, α is significant level of the test, se(β̂) is the standard error of β, and n is sample size. As an alternative, we utilized permutation to select significant SNPs. The rationale behind a permutation test is that, under the null hypothesis (H0 : βi = 0, i = 1, ⋯, p), the estimate of β obtained from the raw (unshuffled) data is similar to the estimate of β obtained from the shuffled data. Let β̂i (i = 1, ⋯, p) be the estimate of the ith SNP effect from the raw data and be the estimate of the ith SNP effect from the jth shuffled data. Let us define as the difference between β̂i and , i.e., . Then,
| (9) |
where is the sample mean of , j = 1, ⋯, k, i.e. and is the standard error of . Under the null hypothesis, the statistic Λi defined in (14) follows standard normal distribution when k is large. p-value for rejecting the null hypothesis and the rank of each Λi can be utilized to select significant SNPs.
Application of the multinomial probit model with SVD
Simulated multinomial data
To evaluate the validity of the multinomial probit model with SVD for polychotomous ordinal responses, we simulated 30 samples and 1000 SNPs, with 9 out of the 1000 SNPs (every 100th SNP, except the last one) contribute to disease status. We assumed that there are 3 disease development stages and a dominant genetic model for each of the 9 disease associated SNPs. Therefore, we would expect 9 strong signals corresponding to each of the 9 disease associated SNPs when applying the multinomial probit model with SVD to this data.
Mexican-American Coronary Artery Disease (MACAD) study
The Study population consists of probands who are Mexican American aged between 45 and 75 with coronary artery disease: Spouses of probands, adult offspring (≥ 18) and their spouses. For the offspring generation, we performed Oral Glucose Tolerance Test and genotyped 132 SNPs in 32 genes selected for a prior relationship to insulin physiology. Table 1 summarized the list of 132 SNPs in 32 genes. The goal of this study was to identify genes affecting the development of impaired glucose tolerance (IGT) and/or impaired fasting glucose(IFG). Impaired glucose tolerance (IGT) was defined as a 2hr glucose level between 140 and 199 mg/dl; Impaired fasting glucose (IFG) defined as a fasting glucose level between 100 and 125 mg/dl. In order to identify and compare genes affecting the development of IGT and/or IFG, we generated two study samples, D1 and D2 (Figure 1). Each has 3 disease stages: D1) both 2hr and fasting glucoses normal (N/N) (n1=60), IGT only (IGT/N) (n2=31) and IGT and IFG (IGT/IFG) (n3=15); D2) both 2hr and fasting glucoses normal (N/N) (n1=60), IFG only (N/IFG) and IGT and IFG (IGT/IFG) (n3=15).
Table 1.
List of 132 SNPs in the 32 candidate genes.
| Gene | SNP | Gene | SNP | Gene | SNP | Gene | SNP |
|---|---|---|---|---|---|---|---|
| LPL | m7315 | AMPD2 | rs12046107 | NOS3 | rs1800779 | rs1800874 | |
| m8292 | rs865774 | rs3918226 | MS4A2 | rs574700 | |||
| m8393 | rs568686 | rs1799983 | rs1441586 | ||||
| m8852 | rs523786 | NPPARS | rs5063 | rs2583471 | |||
| m9040 | PRKAG3 | rs650898 | PPARS | rs5065 | rs2070970 | ||
| m9712 | rs16859382 | SCNN1 | rs5742912 | FEM1B | rs10152450 | ||
| CAPN | n44 | rs6436094 | rs2228576 | rs11636081 | |||
| n43 | rs692243 | ADRB2 | rs1042713 | rs7172340 | |||
| n56 | PRKAA2 | rs11206887 | rs1042714 | GCG | rs13429709 | ||
| n63 | rs2051040 | CRP | rs3091244 | uw012629 | |||
| SORCS | rs2249022 | rs2143749 | TCF7L2 | m11196181 | rs6732914 | ||
| rs1537919 | rs2746349 | m17747324 | rs7581952 | ||||
| rs7897974 | rs857155 | m7901695 | rs5645 | ||||
| rs1530248 | CRP | rs1130864 | m11196187 | rs13001107 | |||
| rs11193190 | rs1800947 | m7077039 | ITR | rs6492722 | |||
| rs2243454 | rs2808630 | m11196199 | LPIN | rs11524 | |||
| rs4390282 | rs3093062 | m17685538 | PPARα | rs135549 | |||
| rs10736189 | C5 | rs25681m | m12255372 | rs135547 | |||
| rs10748924 | rs2416811 | AMPD1 | rs926938 | rs63382 | |||
| rs10509818 | rs2159776 | 1q12x | rs1800206 | ||||
| rs10748932 | IL4 | rs2070874 | rs2010899 | rs82789 | |||
| rs11193188 | rs2227284 | rs2268701 | PTPN1 | rs941798 | |||
| rs1251753 | rs2072130 | 1p48l | rs3787345 | ||||
| rs1269918 | rs3024622 | rs2268698 | rs754118 | ||||
| rs1322005 | IL6 | rs2069832 | rs2268697 | rs2282147 | |||
| rs1538417 | rs2069849 | rs3789627 | rs718050 | ||||
| rs2788677 | C5 | rs17611 | rs743041 | rs3787348 | |||
| rs607437 | IL4R | rs2243250 | rs761755 | SORCS3 | rs813756 | ||
| rs685316 | rs1805010 | rs6679869 | rs1670008 | ||||
| rs7067660 | rs1805015 | rs6701427 | SORT1 | rs4970843 | |||
| rs7086426 | rs1801275 | CD14 | rs4914 | rs1278664 | |||
| rs821994 | IL6 | rs1800796 | CCR5 | rs1799988 | rs11581665 | ||
| rs822000 | rs1800795 | rs2254089 | rs1149175 | ||||
Figure 1.
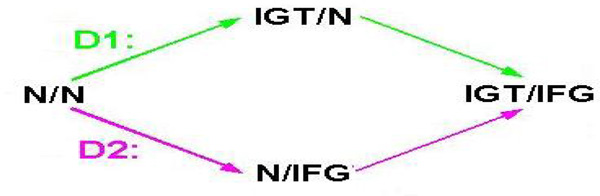
Two Study Samples: D1. Both 2hr and fasting glucoses normal (N/N)-IGT only (IGT/N)-IGT and IFG (IGT/IFG) and D2. Both 2hr and fasting glucoses normal (N/N), IFG only (N/IFG), and IGT and IFG (IGT/IFG)
Results
Simulated Multinomial Data
Figure 2 shows the result of estimate of parameters. There are 8 strong signals, showing that 8 out of the 9 disease associated SNPs were successfully identified by the method, even though some nosies are around each signals. This showed that multinomial probit model with SVD can be reliably used to analyze large scale association data when p ≫ n for polychotomous ordinal responses.
Figure 2.

Estimate of parameters in the simulated data
Mexican-American Coronary Artery Disease (MACAD) study
We analyzed two data sets generated from a subsample of subjects recruited through a coronary artery disease proband in the Mexican-American Coronary Artery Disease Project. Figure 3 shows the result of estimate of parameters, which are β′s in (1), using the MLE method and p-values of 132 SNPs in 32 genes to test each SNP effect for the data set D1. P-values are were calculated from (9). At significance level 0.05, which is 1.3 (= −log10(0.05)), we identified that 8 genes out of the 32 candidate genes that were associated with D1. Figure 4 gives the parameter estimates and −log10(p − value) of 132SNPs for data set D2. At significance level 0.05, 9 genes out of the 32 candidate genes were associated with D2. These results suggested that SNPs in 3 genes (SORCS1, AMPD, PPARα) were associated with both D1 and D2; SNPs in 5 genes (AMPD2, PRKAA2, C5, TCF7L2, ITR) were associated with D1 only; SNPs in 6 genes (CAPN, IL4, NOS3, CD14, GCG, SORT1) were associated with D2 only (Table 2). These results suggest that IGT and IFG may indicate different pathways to diabetes, with different genetic determinants. Multinomial Probit model with SVD can be utilized to identify associated markers with disease development when multi-disease stages are conside
Figure 3.

Genes for IGT/IFG through IGT pathway
Figure 4.

Genes for IGT/IFG through IFG pathway
Table 2.
Genes identified as significant for D1 and D2.
| Study sample | Genes selected |
|---|---|
| both IGT and IGF | SORCS1 (Sorcs receptor 1) AMPD1 (Adenosine monophosphate deaminase-1) PPARα (Peroxisome proliferators-activated receptor-α) |
 only only |
AMPD2 (Adenosine monophosphate deaminase-2) PRKAA2 (Protein kinase AMP-activated catalytic α2) C5 (Complement component 5) TCF7L2 (Transcription factor 7-like 2) |
 only only |
CAPN10 (Calpain 10) IL4 (Interleukin 4) NOS3 (Nitric oxide synthase 3) CD14 (Monocyte differentiation antigen cd14) GCG (Glucagon) SORT1 (Sortilin) |
References
- Albert J, Chib S. Bayesian analysis of binary and polychotomous response data. Journal of American Statistical Association. 1993;88:669–679. [Google Scholar]
- Anderson M, Legendre P. An empirical comparison of permutation methods for tests of partial regression coefficients in a linear model. Journal of Statistical Computation and Simulation. 1999;62:271–303. [Google Scholar]
- Croiseau P, Cordell H. Analysis of North American Rheumatoid Arthritis Consortium data using penalized logistic regression approaches. Genetic Analysis Work-shop. 2008;16 doi: 10.1186/1753-6561-3-s7-s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chib S, Greenberg E. Analysis of Multivariate Probit Models. Biometrika. 1998;85:347–361. [Google Scholar]
- Gelfand A. Model determination using sampling-based methods. In: Gilks, Richardson, Spiegelhalter, editors. Markov china Monte Carlo in Practice. London, England: Chapman & Hall; 1996. pp. 145–161. [Google Scholar]
- George E, McCulloch R. Variable selection via Gibbs sampling. Journal of American Statistical Association. 1993;88:881–889. [Google Scholar]
- George E, McCulloch R. Approached for Bayesian variable selection. Statistica Sinica. 1993;7:339–373. [Google Scholar]
- Graybill F. Theory and Application of the Linear Model. Belmont, California: Duxbury Press; 1976. [Google Scholar]
- Jansen J. Fitting regression models to ordinal data. Biometrical Journal. 1991;33:807–815. [Google Scholar]
- Johnson V, Albert J. Ordinal Data Models. New York: Springer-Verlag; 1999. [Google Scholar]
- Kennedy P. Randomization tests in econometrics. Journal of Business and Economic Statistics. 1995;13:85–94. [Google Scholar]
- Kwon S, Wang D, Guo X. Application of an iterative Bayesian variable selection method in a genome-wide association study of rheumatoid arthritis. BMC proceedings. 2007 doi: 10.1186/1753-6561-1-s1-s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P. Regression models for ordinal data (with discussion) Journal of the Royal Statistical Society. Series B. 1980;42:109–142. [Google Scholar]
- Meier L, van de Geer S, Buhlman P. The group lasso for logistic regression. Journal of the Royal Statistical Society. 2008;70:53–71. [Google Scholar]
- Sha N, Vannucci M, Tadesse M, Brown P, Dragoni I, Davies N, Roberts T, Contestabile A, Salmon M, Buckley C, Falciani F. Bayesian variable selection in multinomial probit models to identify molecular signatures of disease stage. Biometrics. 2004;60:812–819. doi: 10.1111/j.0006-341X.2004.00233.x. [DOI] [PubMed] [Google Scholar]
- West M. Bayesian factor regression models in the large p, small n paradigm. Bayesian Statistics. 2003;7:723–732. [Google Scholar]