

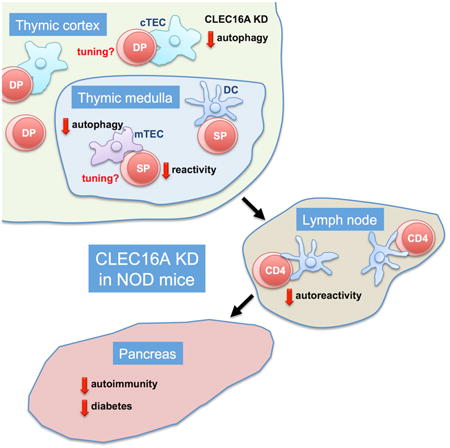
Summary
CLEC16A variation has been associated with multiple immune-mediated diseases, including type 1 diabetes, multiple sclerosis, systemic lupus erythematosus, celiac disease, Crohn's disease, Addison's disease, primary biliary cirrhosis, rheumatoid arthritis, juvenile idiopathic arthritis and alopecia areata. Despite strong genetic evidence implicating CLEC16A in autoimmunity, this gene's broad association with disease remains unexplained. We generated Clec16a knock-down (KD) mice in the nonobese diabetic (NOD) model for type 1 diabetes and found that Clec16a silencing protected against autoimmunity. Disease protection was attributable to T cell hyporeactivity which was secondary to changes in thymic epithelial cell (TEC) stimuli that drive thymocyte selection. Our data indicate that T cell selection and reactivity were impacted by Clec16a variation in thymic epithelium owing to Clec16a's role in TEC autophagy. These findings provide a functional link between human CLEC16A variation and the immune dysregulation that underlies the risk of autoimmunity.
Graphical abstract
Introduction
Genome-wide association studies (GWAS) have helped identify numerous gene variants that contribute to the risk of autoimmunity. Despite the vast catalog of causal candidate genes generated by GWAS, the functional contribution to disease of most autoimmunity-associated gene variations remains to be defined (Hu and Daly, 2012). Notably, several genetic loci stand out for having been very broadly associated with autoimmunity. Among these, variations within CLEC16A at chromosomal position 16p13 have been associated with no less than 10 diseases, including type 1 diabetes, multiple sclerosis, systemic lupus erythematosus, celiac disease, Crohn's disease, Addison's disease, primary biliary cirrhosis, rheumatoid arthritis, juvenile idiopathic arthritis and alopecia areata (Dubois et al., 2010; Gateva et al., 2009; Hakonarson et al., 2007; Hischfield et al., 2012; IMSGC, 2009; Jagielska et al., 2012; Marquez et al., 2009; Martinez et al., 2010; Skinningsrud et al., 2008; Skinningsrud et al., 2010; Todd et al., 2007; WTCCC, 2007). The association of CLEC16A variation with multiple autoimmune disorders thus implicates this gene in an as yet undefined but likely fundamental aspect of immune regulation.
CLEC16A encodes a large protein of 1053 amino acids that contains several putative functional domains, including a C-type lectin domain which led to its classification as C-type lectin domain family 16A (Berge et al., 2013). At the time CLEC16A was associated first with type 1 diabetes (Hakonarson et al., 2007; Todd et al., 2007; WTCCC, 2007) and then with multiple sclerosis (IMSGC, 2009), this gene formerly known as KIAA0350 had neither been classified nor was anything known of its function. The first data relating to CLEC16A's cellular function came from studies of its Drosophila ortholog, termed Ema, that was shown to participate in endosomal maturation (Kim et al., 2010) and autophagy (Kim et al., 2012). A role in autophagy was recently confirmed in mouse, where beta cell-specific deletion of Clec16a impaired mitophagy (Soleimanpour et al., 2014). In their study of mice with Clec16a-deficient beta cells, Soleimanpour and colleagues suggested that variation in CLEC16A function in the pancreas may be causal for this gene's association with type 1 diabetes. These investigators postulated that a defect in insulin secretion secondary to disrupted autophagy would predispose beta cells to the autoimmune destruction that causes type 1 diabetes. However, this hypothesis does not provide an explanation for CLEC16A's association with a broad array of immune-mediated diseases (Dubois et al., 2010; Gateva et al., 2009; Hakonarson et al., 2007; Hischfield et al., 2012; IMSGC, 2009; Jagielska et al., 2012; Marquez et al., 2009; Martinez et al., 2010; Skinningsrud et al., 2008; Skinningsrud et al., 2010; Todd et al., 2007; WTCCC, 2007). The functional link between CLEC16A variation and autoimmunity therefore remains to be convincingly explained.
The data presented herein indicate that Clec16a variation impacts thymic selection, owing to a role in thymic epithelial cell autophagy, thus implicating CLEC16A in a fundamental aspect of immune tolerance. Our findings thereby provide a functional link between CLEC16A variation and the immune dysregulation that broadly underlies the risk of autoimmune disease.
Results
Clec16a silencing diminishes the diabetogenicity of NOD T cells
To investigate CLEC16A function in autoimmunity, particularly in relation to autoimmune diabetes, we generated Clec16a KD mice in the NOD model for type 1 diabetes (Anderson and Bluestone, 2005) (Figure S1). Transgenic mice developed normally and were born with the expected Mendelian frequency. No changes in the gross distribution and number of immune cell populations were detected. Strikingly, Clec16a KD NOD mice were almost completely protected from spontaneous autoimmune diabetes (Figures 1A and 1B). Even when diabetes onset was accelerated using cyclophosphamide (Harada and Makino, 1984), Clec16a silencing afforded protection (Figure 1C). To test if this protection was conveyed by changes in lymphocyte function, we transferred splenocytes from Clec16a KD or WT animals into immunodeficient NOD.SCID mice. Recipients of Clec16a KD but not WT cells were largely resistant to cyclophosphamide-accelerated diabetes (Figure 1D). In contrast, transfer of WT splenocytes to Clec16a KD NOD.SCID mice restored full disease susceptibility, indicating that protection derived from changes in immune function (Figure 1E), and not from a pancreas-intrinsic resistance to autoimmune damage. Having established that loss of Clec16a renders NOD lymphocytes less diabetogenic, we next sought to localize this effect to a specific cell population. We purified T and B lymphocytes from WT and Clec16a KD mice and reconstituted NOD.SCID animals with all four possible combinations of cells. Disease protection was confined to groups that received transgenic T cells, irrespective of the genotype of co-transferred B cells (Figure 1F). We concluded that Clec16a KD reduces the pathogenicity of NOD T cells.
Figure 1. Clec16a KD prevents autoimmunity by reducing the pathogenicity of T cells.
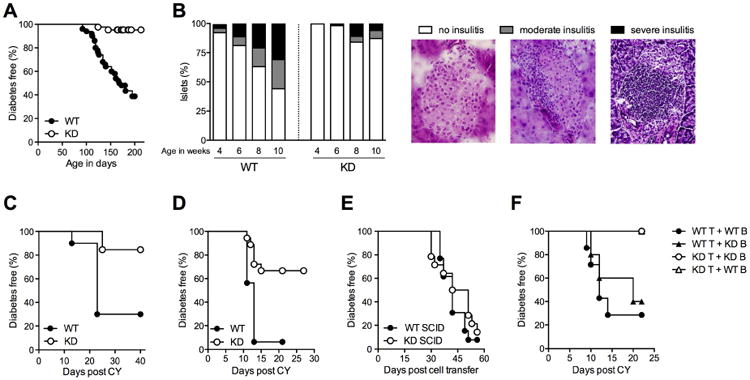
(A) Spontaneous diabetes frequency in cohorts of WT (n = 50) and Clec16a KD (KD, n = 42) NOD mice, P < 0.0001. (B) Insulitis in WT and KD mice at 4, 6, 8 and 10 weeks of age. n = 2-5 mice (150-500 islets) per group. Islets were scored as free of insulitis, or as having moderate or severe infiltration, as shown in representative images. (C) Cyclophosphamide (CY) diabetes in WT (n = 10) and KD (n = 13) mice, P = 0.0035. (D) CY diabetes in NOD.SCID mice reconstituted with WT (n = 16) or KD (n = 18) splenocytes, P = 0.0002. (E) Diabetes frequency in WT NOD.SCID (n = 13) or Clec16a KD NOD.SCID (n = 14) mice reconstituted with WT splenocytes, P = 0.36. (F) CY diabetes in NOD.SCID mice reconstituted with T and B cells, respectively, in the following combinations: WT/WT (n = 7), WT/KD (n = 5), KD/WT (n = 5), KD/KD (n = 4), P = 0.0046 for WT T cell groups vs. KD T cell groups using Fisher's exact test.
Clec16a silencing causes CD4+ T cell hyporeactivity
We proceeded to characterize T cell function in more detail in vitro. Stimulation of polyclonal CD4+ T cells from WT and Clec16a KD NOD mice revealed that Clec16a silencing caused T cells to be hyporesponsive to T cell receptor (TCR) stimulation, but not to PMA and ionomycin which provide mitogenic stimuli that circumvent TCR ligation (Figures 2A and 2B, and Figure S2). T cell hyporeactivity was confined to CD4+ cells, as CD8+ cells were not impaired in their response to TCR ligation (Figure S2). Because hyporesponsiveness could be overcome by mitogens that bypass proximal TCR signaling, we measured phosphorylation events immediately downstream of the TCR complex. We found that CD4+ T cells from Clec16a KD mice responded poorly to TCR ligation, as measured by diminished phosphorylation of the signaling molecules ZAP-70 and PLC-γ and ERK1/2 (Figure 2C). We then tested if antigen-specific stimulation was similarly impaired by Clec16a KD. The response to BDC2.5 mimotope peptide stimulation of T cells from Clec16a KD BDC2.5 TCR transgenic mice was reduced compared to WT BDC2.5 T cells. In this setting, hyporesponsiveness was again overcome by PMA and ionomycin stimulation, but not by CD3 antibody stimulation (Figure 2D). Of note, the suppressive capacity of Clec16a KD regulatory T (Treg) cells remained comparable to that of WT cells (Figure 2E), possibly because Treg cell function is less sensitive to changes in TCR signal strength (Plesa et al., 2012). Together, these results indicate that Clec16a KD affects TCR signaling, without however causing complete CD4+ T cell dysfunction.
Figure 2. Clec16a KD causes T cell hyporeactivity.
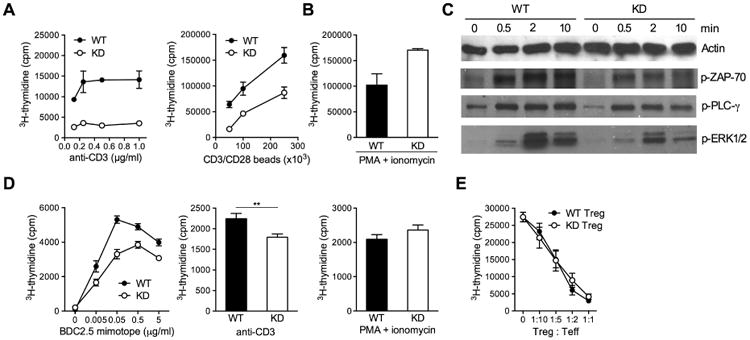
(A, B) Proliferation of CD4 T cells in response to CD3 antibody (A, left panel), CD3/CD28 antibody-coated beads (A, right panel) or PMA and ionomycin (B). (C) Western blot measurements of ZAP-70, PLC-γ and ERK1/2 phosphorylation in WT or KD CD4 T cells stimulated with cross-linked CD3 antibody for the indicated length of time (in min). (D) Proliferation of WT or KD TCR transgenic BDC2.5 CD4 T cells in response to BDC2.5 mimotope peptide (left panel), CD3 antibody (1 μg/ml, middle panel) or PMA and ionomycin (right panel). (E) Suppression of WT CD4 T cell proliferation by WT or KD CD4+CD25+ Treg cells. Results show mean ± SEM of triplicate measurements and are representative of 2 to 4 experiments. ** P < 0.01
See also Figure S2.
The effects of Clec16a silencing are not T cell-intrinsic
For initial comparisons, T cells had been purified by magnetic separation. Because expression of the lentiviral transgene is variegated, only 70-75% of T lymphocytes in Clec16a KD mice express the transgene which includes GFP. To more carefully characterize transgene-expressing cells, we sorted GFP+ and GFP- T cells from Clec16a KD mice by flow cytometry. Both T cell populations were impaired in their proliferative response (Figure 3A), suggesting that hyporeactivity was not caused by T cell-intrinsic effects of gene silencing. To test if the effect of Clec16a KD is instead conveyed to T cells during thymic selection, we generated bone-marrow chimeric mice. The reactivity of Clec16a KD T cells that developed in interaction with WT TECs in chimeric hosts was indistinguishable from that of WT T cells (Figure 3B). Critically, irradiated NOD mice reconstituted with Clec16a KD bone-marrow were not protected from diabetes (Figure 3C), while irradiated Clec16a KD mice reconstituted with WT bone-marrow remained diabetes resistant (Figure 3D), demonstrating that disease protection was not conveyed by Clec16a silencing in hematopoietic cells. Transplantation of fetal WT or Clec16a KD thymus into athymic WT NOD mice showed that protection from diabetes derived instead from Clec16a KD in thymic epithelium (Figure 3E). Clec16a KD has been reported to impair insulin release from beta cells (Soleimanpour et al., 2014). We therefore investigated whether limited availability of this self-antigen played a role disease protection. However, Clec16a KD mice secreted insulin and regulated their blood glucose in a manner indistinguishable from WT animals after intraperitoneal glucose challenge (Figure S3). The total pancreatic insulin content was also unaffected by Clec16a KD (Figure S3). Presentation of beta cell antigen in the pancreatic lymph node (PLN) is critical for the priming of diabetogenic T cells (Hoeglund et al., 1999). To test if antigen presentation was compromised in the PLN of Clec16a KD mice, we transferred BDC2.5 TCR transgenic T cells into WT or Clec16a KD animals. We measured robust BDC2.5 T cell proliferation in the PLNs of both Clec16a KD and WT recipients (Figure S3). We conclude that presentation of beta cell antigen in peripheral tissue is not affected by Clec16a KD, and that the protective effect of Clec16a silencing originates in the thymus.
Figure 3. The effect of Clec16a silencing is not T cell-intrinsic.
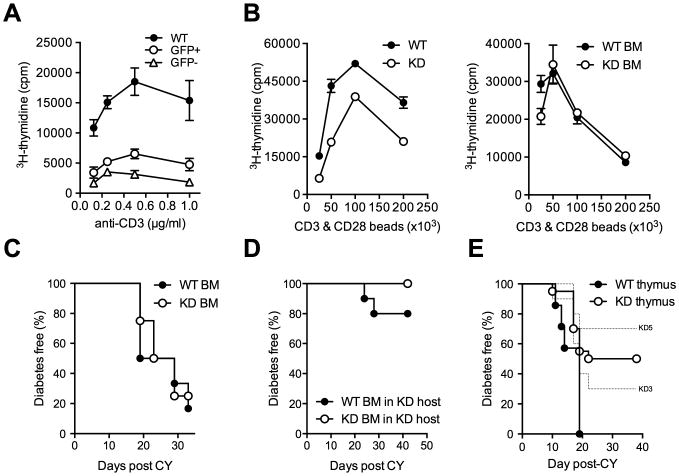
(A) Proliferation of GFP+ and GFP- Clec16a KD and WT CD4+ T cells stimulated with anti-CD3 antibody. (B) Proliferation of FACS-sorted CD4+ T cells from WT or KD mice (left panel) or from WT bone-marrow (BM) chimeric mice reconstituted with WT or KD BM (right panel) stimulated with anti-CD3/anti-CD28 coated beads. (C) CY diabetes in WT BM chimeric mice reconstituted with WT (n = 6) or KD (n = 4) BM. (D) CY diabetes in Clec16a KD BM chimeric mice reconstituted with WT (n = 10) or KD (n = 10) BM, P = 0.14. (E) Diabetes frequency in NOD mice thymectomized at 4 weeks of age and subsequently transplanted with fetal thymus (E14) from Clec16a KD (KD3 n = 10, KD5 n = 10) or WT (n= 7) embryos in conjunction with WT bone-marrow after irradiation, WT vs. KD - P = 0.0132. No difference was observed in either the size of thymuses post-transplantation or the frequency of T cells in transplanted mice at the time of diabetes onset (data not shown).
Clec16a silencing in thymic epithelium modifies T cell selection
To confirm that T cell hyporeactivity was imparted during thymic development, we tested the reactivity of Clec16a KD thymocytes. CD4 single-positive (SP) thymocytes from Clec16a KD mice were hyporeactive following in vitro stimulation (Figure 4A). In contrast, Clec16a KD did not diminish the reactivity of double-positive (DP) thymocytes. In vivo, CD3 antibody caused the deletion of a comparable proportion of DP cells in Clec16a KD and WT animals (Figure 4B). In vitro, differentiation of DP thymocytes into CD4SP cells using antibody stimulation (Cibotti et al., 1997) induced similar responses in WT and KD cells (Figure S4). These results suggest that hyporeactivity is acquired during thymic selection, in the transition from the DP to the CD4SP maturation stage.
Figure 4. Clec16a KD modifies the reactivity of CD4SP but not DP thymocytes.
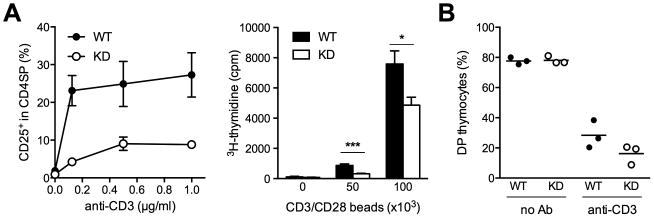
(A) CD25 up-regulation (left) and proliferation (right) of WT or KD CD4SP cells stimulated in vitro. (B) Frequency of DP cells in the thymus of WT or KD mice 48 h after injection with CD3 antibody. Results are representative of 3 experiments. * P < 0.05, *** P < 0.001.
See also Figure S4.
While we detected no evidence for altered TCR Vβ-chain selection (Figure 5A), we observed a striking change in the relative frequency of transgenic thymocytes during selection (Figures 5B and 5C, and Figure S5). When thymocytes were subdivided on the basis of their relative TCR and CD69 expression (Fu et al., 2009; Hare et al., 1999; Swat et al., 1993), preselection thymocytes (TCRlo/int CD69lo) were over-represented in Clec16a KD mice, whereas we measured a sharp drop in the frequency of Clec16a KD thymocytes undergoing selection (TCRint/high CD69hi). These changes were recapitulated in bone-marrow chimeric mice, and the phenotype observed in Clec16a KD mice segregated with the genotype of the irradiated host, and not with the genotype of transplanted hematopoietic cells (Figure 5D). These results indicate that Clec16a KD in thymic epithelium, and not in hematopoietic cells, impacts the selection of developing T cells.
Figure 5. Clec16a silencing impacts thymic selection.
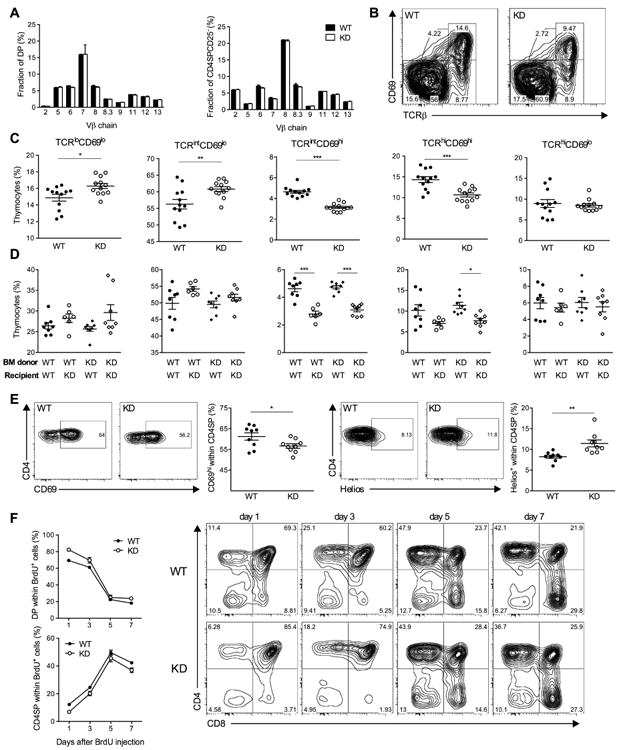
(A) Frequency of DP or CD4SPCD25- thymocytes that express different TCR Vβ chains in the thymus of WT (filled bars, n = 6) or KD3 (open bars, n = 6) mice. Vβ5 antibody recognizes both Vβ 5.1 and Vβ 5.2. Vβ8 antibody recognizes both Vβ8.1 and Vβ8.2. (B, C) Representative flow cytometry data (B) and frequency (C) of thymocyte populations distinguished by TCR and CD69 expression in WT (n = 12) and KD (n = 12) mice. (D) Frequency of thymocyte populations as in (C) measured in irradiated WT or KD mice reconstituted with WT or KD bone-marrow (BM) 6 weeks after reconstitution (n = 6-8 mice per group). (E) Representative flow cytometry data and frequency of CD69+ or Helios+ cells within the CD4SP population in WT (n = 9) or KD (n = 9) mice. (F) Representative flow cytometry data and frequency of BrdU-labeled thymocytes following a single injection of BrdU (n = 3 mice per group for each time point), P = 0.0422 for DP cells and P = 0.0004 for FoxP3-CD4SP cells, two-tailed paired t-test. All data are representative of 2 to 4 experiments. * P < 0.05, ** P < 0.01, ***P < 0.001.
See also Figure S5.
To evaluate the contribution of negative selection to the decreased frequency of CD69+ thymocytes in Clec16a KD mice, we measured the expression of Helios, a transcription factor reported to mark CD4SP cells undergoing deletion (Daley et al., 2013). We observed a significant increase in Helios+ cells within the FoxP3-CD4SP population (Figure 5E), suggesting that the loss of CD69+ thymocytes in Clec16a KD mice is caused, at least in part, by increased negative selection.
We further tracked the fate of developing thymocytes by BrdU pulse labeling (Figure 5F). The proportion of BrdU+ CD4SP cells was reduced in transgenic mice at all time points measured after the initial BrdU pulse. These results support the notion that Clec16a KD impairs the transition from DP to CD4SP cells. Subdivision of BrdU-labeled CD4SP cells into sequential maturation stages (Daley et al., 2013) defined by their relative expression of CD24 and CCR7 showed that the most immature (CD24+CCR7-) CD4SP cells were over-represented, while the most mature (CD24-CCR7+) cells were under-represented in transgenic mice (Figure S5), further indicating that Clec16a KD decreased the rate of CD4SP cell maturation.
Loss of Clec16a impairs thymic epithelial cell autophagy
Our experiments had shown that the effects of Clec16a KD originate in non-hematopoietic cells and impact thymic selection. T cell selection is largely mediated by TECs; we therefore speculated that Clec16a silencing alters the ability of TECs to stimulate thymocytes. The Drosophila ortholog of Clec16a was ascribed a function in autophagy (Kim et al., 2012), and this function was recently confirmed in mouse (Soleimanpour et al., 2014). Notably, constitutive autophagy is a distinguishing feature of thymic epithelium (Nedjic et al., 2008). Furthermore, TEC autophagy contributes to self-antigen presentation by feeding peptides derived from cellular proteins into the MHC class II presentation pathway (Kasai et al., 2009; Nedjic et al., 2008). By participating in antigen presentation, TEC autophagy was shown to be critical to thymic selection (Nedjic et al., 2008), providing a testable link between Clec16a silencing, changes in TEC autophagy and thymocyte stimulation. After confirming that Clec16a expression is diminished in TECs from Clec16a KD mice (Figure 6A), we measured autophagy in thymic epithelium by immunohistochemical staining of the autophagosome-associated protein LC3 (Kabeya et al., 2000). We observed a modest increase in the number of autophagosomes in Clec16a KD mice (Figure 6B), particularly in the cortical TEC (cTEC) compartment that displayed higher levels of autophagy than the thymic medulla, as reported previously (Kasai et al., 2009; Nedjic et al., 2008). However, autophagy was decreased overall, as measured by a reduction of the autophagosome-incorporated LC3-II isoform (Kabeya et al., 2004) (Figure 6C). These results are consistent with data reported for Ema mutant Drosophila showing a modest increase in the number of autophagosomes but an overall reduction in autophagic flux (Kim et al., 2012). Of note, decreased autophagy did not affect MHC II expression on Clec16a KD cTECs. To confirm these findings, we derived a new cTEC cell line, which we called MJC1. We proceeded to silence Clec16a or Atg5, a critical component of the autophagy machinery (Mizushima et al., 1998), in MJC1 cells. Both Clec16a and Atg5 silencing reduced the conversion of LC3-I into LC3-II (Figure 6D) and caused the accumulation of p62, a protein involved in and degraded by autophagy (Bjørkøy et al., 2005) (Figure 6E), confirming that Clec16a silencing impaired autophagic flux in TECs. To test if Clec16a's function in autophagy is conserved in human, we silenced CLEC16A in HEK293 cells and in HeLa cells. We found that CLEC16A KD disrupted autophagic flux to a similar extent as ATG5 KD, demonstrating the involvement of CLEC16A in human autophagy (Figure S6). Together, these results establish a role for Clec16a in mouse TEC autophagy and show that this function is conserved in human.
Figure 6. Clec16a KD disrupts TEC autophagy.
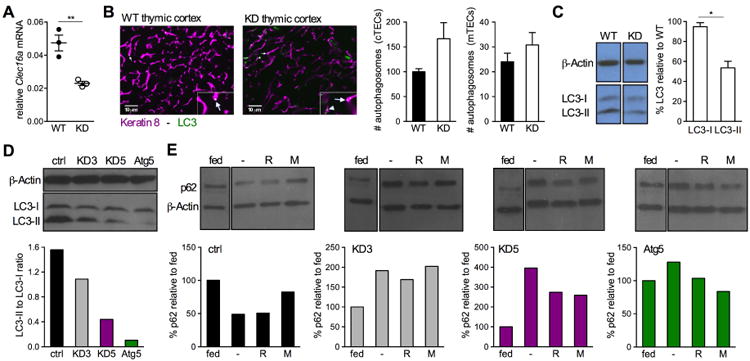
(A) Clec16a expression in WT or KD TECs (pooled from 4 mice/group, triplicate measurements); representative of 2 experiments. (B) Autophagosome quantification in WT or KD thymic cortex (shown) and medulla (not shown) (n = 5 mice/group, autophagosomes were enumerated per area of a fixed but arbitrary size and data were averaged from 5 images/mouse). Representative thymus sections used for quantification are shown. Insets show a magnified area marked by arrows that point to distinct autophagosomes within cells. (C) Quantification of LC3 protein in WT and KD TECs. Results show KD relative to WT and are representative of 2 experiments. (D, E) LC3 and p62 quantification in MJC1 cells transduced with control (ctrl), Clec16a (KD3 and KD5) or Atg5 shRNA. Cells were starved in the presence of the protease inhibitor E64d (D), or in the presence or absence (-) of rapamycin (R) or 3-MA (M) that induces and inhibits autophagy, respectively (E). Data shown in D and E are representative of 3 independent experiments.
Clec16a silencing modifies the stimulatory capacity of TECs
We next sought to determine whether Clec16a KD directly impacts the ability of TECs to stimulate thymocytes. We speculated that MJC1 cells, that were derived from cTECs, should have the capacity to stimulate DP thymocytes in vitro. While we found that MJC1 cells express very low amounts of MHC II, IFN-γ treatment caused MHC II upregulation on both control and Clec16a KD cells (data not shown). Therefore, MJC1 cells were pre-treated with IFN-g prior to being used for thymocyte stimulation. Taking into account the obvious caveat that a cell line could not fully recapitulate thymic selection in all respects, we sought to ask if Clec16a silencing in MJC1 cells would have a detectable effect thymocyte stimulation, as measured by co-receptor expression and activation marker upregulation. Co-culture of CD69lo DP cells with MJC1 cells did indeed cause a significant proportion of immature thymocytes to upregulate CD69 and to become CD4SP (Figure 7A and Figure S7). Using MJC1 cells stably transduced with two different Clec16a shRNA constructs, we found that Clec16a KD in TECs impaired the progression of thymocytes from the CD69lo to CD69hi stage (Figure 7A). Furthermore, Clec16a silencing in MJC1 cells decreased PD1 upregulation in CD4SP thymocytes that is dependent on TCR stimulation (Keir et al., 2005), and these results mirrored observations made in vivo (Figure S7). Together, these data suggest that Clec16a KD modifies the stimulatory capacity of cTECs.
Figure 7. Clec16a silencing affects the stimulatory capacity of TECs.
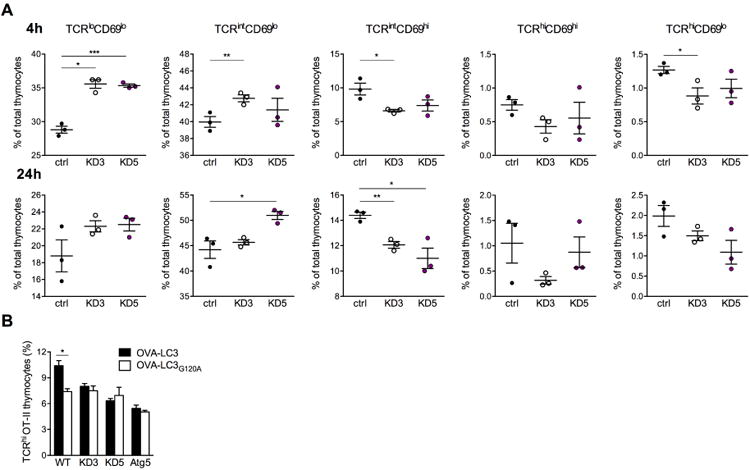
(A) Frequency of thymocyte subpopulations distinguished by their relative TCR and CD69 expression 4 h (top panels) and 24 h (bottom panels) after stimulation with control or Clec16a KD MJC1 cells. Input cells were WT immature (TCRloCD69lo) thymocytes; representative of 3 experiments. (B) Frequency of TCRhi OT-II cells 24 h after stimulation of immature OT-II thymocytes with control, Clec16a KD or Atg5 KD MJC1 TECs transduced with OVA323-339-LC3 or OVA323-339-LC3G120A; representative of 4 experiments. * P < 0.05, ** P < 0.01, ***P < 0.001.
See also Figure S7.
Clec16a silencing alters TEC stimulation of thymocytes owing to impaired autophagy
To test if a direct link exists between changes in TEC autophagy and thymocyte stimulation, we transduced MJC1 cells with an LC3 transgene that includes the OVA323-339 peptide and enables autophagy-dependent presentation of this antigen (Aichinger et al., 2013; Schmid et al., 2007). The OVA peptide was incorporated within the LC3 protein and is thereby shuttled to and processed in autophagic vesicles, from which the peptide gains access to the MHC class II presentation pathway. In this context, stimulation of OVA323-339 responsive OT-II thymocytes was impaired by both Atg5 KD and Clec16a KD (Figure 7B). Mutation of the OVA323-339-LC3 construct at a critical cleavage site (LC3G120A) to prevent its inclusion in autophagosomes (Aichinger et al., 2013; Schmid et al., 2007) diminished OT-II thymocyte stimulation by control MJC1 cells, but did not further impair stimulation by Atg5 KD or Clec16a KD TECs. The effect on thymocyte stimulation of inhibiting OVA323-339 presentation via autophagy by mutating the LC3 protein was thus redundant with the effect of Clec16a silencing that diminishes MJC1 cell autophagy (Figure S7). These results suggest that Clec16a impacts thymocyte stimulation owing to its role in autophagy.
Discussion
Collectively, our findings implicate Clec16a in a fundamental process of immune regulation. Through its role in autophagy, Clec16a modulates the antigen-presenting function of TECs. TEC autophagy was shown to contribute to the generation of MHC class II-peptide complexes that dictate the fate of developing thymocytes (Kasai et al., 2009; Nedjic et al., 2008). The role of autophagy in generating MHC class II ligands has been demonstrated by a number of groups (Brazil et al., 1997; Dengjel et al., 2005; Paludan et al., 2005; Schmid et al., 2007). Of interest, Münz and colleagues reported that LC3, a characteristic component of autophagosomes, co-localizes with MHC class II loading compartments not only in dendritic cells and B cells, but also in human epithelial cell lines (Schmid et al., 2007). This observation was extended by Mizuochi and colleagues who subsequently showed that autophagy intersects with the MHC class II pathway in TECs, indicating that autophagosomal cargo gains access to the MHC class II loading compartment in thymic epithelium. Analysis of the MHC class II ligandome of human B cell lymphoblastoid cells by mass spectrometry demonstrated that autophagy modifies antigen presentation both by shuttling proteins from different cellular sources towards the antigen-loading compartment and by modifying the proteolytic activitiy of these cells (Dengjel et al., 2005). How the repertoire of selecting peptides presented by TECs is affected either qualitatively or quantitatively by Clec16a silencing remains to be determined, but it is clear that an impairment of autophagy caused by loss of Clec16a should significantly alter the MHC class II ligandome.
Our data demonstrate that Clec16a silencing impacts thymic selection, and these results are consistent with prior experiments that tested the functional impact of autophagy in the thymus. Using Atg5-deficient thymic epithelium, Klein and colleagues showed that the loss of autophagy in TECs altered the selection of MHC class II restricted T cells (Nedjic et al., 2008). While autophagy was dispensable for the selection of a polyclonal T cell repertoire, its absence had a significant effect on individual TCR specificities. Depending on the model TCR examined, autophagy had a positive, neutral or deleterious effect on the selection of TCR transgenic thymocytes. Consequently, a change in autophagic flux, as caused by CLEC16A variation, can be predicted to broadly shape the T cell repertoire and modulate the risk of autoimmunity. Klein et al indeed reported that the complete absence of autophagy in thymic epithelium could lead to severe immune-mediated pathology when Atg5-deficient thymus was transplanted into athymic BALB/c mice. We speculate that the comparatively mild impairment in autophagy observed in Clec16a KD mice may have a qualitatively distinct effect on T cell selection, explaining why Clec16a silencing instead afforded protection against autoimmunity in the NOD model of type 1 diabetes. Of note, epithelial cell-specific deletion of Atg5 or Atg7 caused no obvious pathology in the C57BL/6 background (Sukseree et al., 2012; Sukseree et al., 2013), suggesting that the effect of impaired TEC autophagy on immune tolerance may differ between genetic backgrounds. Thus, a change in the contribution of autophagy to TEC antigen presentation does not merely have a quantitative impact on the MHC II ligandome, but rather modifies the quality of the selected T cell repertoire in a difficult-to-predict manner. How the human disease-associated CLEC16A variants affect T cell reactivity will therefore be a challenging question to address. Notwithstanding, a role for CLEC16A in TEC autophagy provides a compelling explanation for this gene's association with numerous immune-mediated diseases. CLEC16A variation can be anticipated to modify the reactivity of the T cell repertoire, and to fundamentally modulate the risk of autoimmunity. A role for CLEC16A in the thymus is supported by a study in human which reported that homozygocity for the disease-associated variant correlated with altered thymic expression (Mero et al., 2011).
A recent publication (Soleimanpour et al., 2014) described that Clec16a participates in autophagy in mouse fibroblasts and islet cells. These data confirmed earlier findings in Drosophila (Kim et al., 2012), and are consistent with our own data showing Clec16a to be involved in TEC autophagy. Our work further extends these findings by demonstrating that CLEC16A's role in autophagy is conserved in human cells. In their report, Soleimanpour and colleagues proposed that the involvement of CLEC16A in type 1 diabetes stems from effects within pancreatic beta cells, but had no disease data in support of this claim, which is not substantiated by our extensive characterization within the NOD model of autoimmune diabetes. These authors reported that loss of Clec16a in beta cells caused hyperglycemia as a consequence of defective beta cell function. In this respect, complete Clec16a deficiency would appear to differ significantly from a partial loss, as neither of the Clec16a KD mouse lines generated in our laboratory displayed a defective response to an intraperitoneal glucose tolerance test performed in young pre-diabetic animals. Our transplantation data instead suggest that the effects of Clec16a silencing on autoimmunity originate in the thymic epithelium. The hypothesis that CLEC16A's effect on disease stems from its role in beta cells is indeed difficult to reconcile with the fact that CLEC16A variation has been implicated in a wide range of autoimmune disorders unrelated to the pancreas.
In conclusion, our data highlight the critical function of TEC autophagy in establishing immune tolerance. TEC autophagy was previously shown to impact T cell selection in mouse (Nedjic et al., 2008). By linking CLEC16A, a gene associated with human autoimmunity, to this pathway of self-antigen presentation, our findings suggest that TEC autophagy plays a similarly pivotal role in human immune tolerance. CLEC16A's cellular function remained uncharted until very recently, and how this gene impinges on autoimmunity was therefore unclear. The functional link between CLEC16A variation and autoimmunity presented here now provides an explanation for this gene's broad involvement in human autoimmune disease.
Experimental Procedures
Mice
Clec16a KD mice were generated by lentiviral transgenesis in the NOD mouse strain as described previously (Kissler et al., 2006). Target sequences for Clec16a KD were CACCTTGTACGTCATTTCTATA (KD3) and GAGTGTCCACCTTGTACGTCAT (KD5). WT NOD mice used for comparison in all experiments were bred and housed in the same facility and in the same room as transgenic mice. NOD BDC2.5 TCR transgenic mice were purchased from Jackson Laboratories, and bred with Clec16a KD mice to generate the NOD BDC2.5/Clec16a KD line. OT-II TCRα-/- mice were kindly provided by T. Serwold. All experimental procedures in animals were approved by the Regional Government of Lower Franconia or by the Institutional Animal Care and Use Committee, for experiments performed at the University of Wurzburg and at the Joslin Diabetes Center, respectively.
Diabetes frequency studies
Onset of diabetes was monitored by weekly (for spontaneous disease) or thrice-weekly (for cyclophosphamide studies and cell transplantation studies) measurements of glycosuria using Diastix (Bayer).
Bone Marrow Chimeras
Irradiated NOD.SCID mice (250 rad) or NOD mice (900 rad) were injected intravenously with lineage-depleted bone-marrow cells (3-5 × 105/mouse).
Thymic transplantation
Thymectomized NOD WT mice were purchased from Jackson Laboratories and reconstituted with NOD WT bone marrow as described above, followed by transplantation of NOD WT or Clec16a KD E14 thymic lobes under the kidney capsule. Transplanted mice were challenged with cyclophosphamide to accelerate diabetes onset.
T cell in vitro assays
Purified T cells were stimulated with CD3 antibody or with the BDC2.5 mimotope peptide (Judkowski et al., 2001) in the presence of irradiated T cell-depleted splenocytes, with CD3/CD28 antibody-coated beads (Invitrogen/Life Technologies) or with PMA (25 ng/ml) and Ionomycin (1 μg/ml). Cell proliferation was measured by 3H-thymidine incorporation and analyzed with a Microbeta plate reader (Perkin-Elmer).
Reagents, Antibodies and Flow cytometry
Flow cytometry measurements were performed using a FACSCantoII or LSRII instrument (BD Biosciences). Cell sorting was performed with a FACSAriaIII instrument (BD Biosciences). Data were analysed with FlowJo software (TreeStar Inc.). Fluorescently conjugated antibodies were purchased from Biolegend, eBioscience and BD Biosciences. Intracellular staining was performed with a FoxP3-labelling kit (eBioscience) and BrdU staining using a flow kit (BD Biosciences).
Immunohistochemistry
Frozen sections were either stained with hematoxylin and eosin or with fluorescently conjugated antibodies. Slides were analysed on a light microscope (Olympus BX-60) or an Axioplan2 microscope (Zeiss). Image analysis was performed with ImageJ64 software.
Thymic epithelial cell isolation
Thymic epithelial cells were enriched using a Percoll (Fisher Scientific) density gradient as described previously (Nedjic et al., 2008) or purified by sequential digestion with Liberase TM (Roche) and DNase (Roche) followed by CD45 depletion using a MACS Kit (Miltenyi Biotec).
Immunoblotting
SDS-PAGE was performed followed by protein transfer onto nitrocellulose membrane and incubation with specific antibodies. Blots were analyzed using chemiluminescence. Band density was quantified using ImageJ64 software
Cell lines and in vitro cell assays
Generation of the MJC1 cell line and lentiviral cell transductions are described in Supplemental Experimental Procedures. Cell starvation experiments were performed in Earle's Balanced Salt Solution supplemented with Rapamycin or 3-methyladenine. Cells were treated with Bafilomycin A1 or E64d for autophagic flux measurements. Thymocyte differentiation assays were performed by stimulation of sorted DP thymocytes with CD3 and CD5 antibody or with MJC1 cells. Generation of LC3-OVA constructs is described in Supplemental Experimental Procedures.
Quantitative RT-PCR
RNA was isolated using the RNeasy Mini Kit (Qiagen) or the High Pure RNA isolation Kit (Roche) followed by cDNA synthesis using the Superscript III cDNA synthesis Kit (Invitrogen). Primers used for RT PCR are listed in Supplemental Experimental Procedures
Statistics
Data were analysed with the Prism software (Graphpad). Diabetes frequency comparisons were carried out using the Log-rank test, except for the experiment shown in Fig. 1F, where Fisher's exact test was used. All other comparisons were performed using a two-tailed t-test, with P < 0.05 considered significant.
Supplementary Material
Highlights.
Clec16a knockdown is protective in the NOD model for type 1 diabetes
Loss of Clec16a diminishes the pathogenicity of NOD CD4+ T cells
The effects of Clec16a are not T cell intrinsic, but imparted during thymic selection
Clec16a modifies T cell selection owing to its role in in thymic epithelial autophagy
Acknowledgments
The authors thank Nicole Hain for technical assistance, Daniela Blassfeld for help with the initial cloning and validation of lentiviral vectors, Jennifer Hollister-Lock for advice with islet purification and insulin measurements, and the Joslin DRC (NIH Award Number P30DK036836) Flow Cytometry Core facility for help with cell sorting. This work was supported in part by funding from the Deutsche Forschungsgemeinschaft to the Rudolf Virchow Center (DFG FZ82), by a Career Development Award from JDRF to S.K. (2-2010-383), and by a Mary K. Iacocca Fellowship provided by the Iacocca Foundation to C.S.
Footnotes
Author Contributions: C.S., K.D.G., K.S., L.P.,K.B. and A.R. designed and performed experiments and interpreted the data. M.-J.K. generated the MJC1 cell line. T.S. contributed to data interpretation. S.K. conceived and designed the study, performed experiments, interpreted the data and wrote the manuscript. C.S. and K.G. contributed equally to the study. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aichinger M, Wu C, Nedjic J, Klein L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J Exp Med. 2013;210:287–300. doi: 10.1084/jem.20122149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- Berge T, Leikfoss IS, Harbo HF. From Identification to characterization of the multiple sclerosis susceptibility gene CLEC16A. Int J Mol Sci. 2013;14:4476–4497. doi: 10.3390/ijms14034476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil MI, Weiss S, Stockinger B. Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur J Immunol. 1997;27:1506–1514. doi: 10.1002/eji.1830270629. [DOI] [PubMed] [Google Scholar]
- Cibotti R, Punt JA, Dash KS, Sharrow SO, Singer A. Surface molecules that drive T cell development in vitro in the absence of thymic epithelium and in the absence of lineage-specific signals. Immunity. 1997;3:245–255. doi: 10.1016/s1074-7613(00)80327-1. [DOI] [PubMed] [Google Scholar]
- Daley SR, Hu DY, Goodnow CC. Helios marks strongly autoreactive CD4+ T cells in two major waves of thymic deletion distinguished by induction of PD-1 and NF-kB. J Exp Med. 2013;210:269–285. doi: 10.1084/jem.20121458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GA, Adány R, Aromaa A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu G, Vallée S, Rybakin V, McGuire MV, Ampudia J, Brockmeyer C, Salek M, Fallen PR, Hoerter JA, Munshi A, et al. Themis controls thymocyte selection through regulation of T cell antigen receptor-mediated signaling. Nat Immunol. 2009;10:848–856. doi: 10.1038/ni.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H, Grant SF, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Casalunovo T, Taback SP, Frackelton EC, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- Harada M, Makino S. Promotion of spontaneous diabetes in non-obese diabetes-prone mice by cyclophosphamide. Diabetologia. 1984;27:604–606. doi: 10.1007/BF00276978. [DOI] [PubMed] [Google Scholar]
- Hare KJ, Jenkinson EJ, Anderson G. CD69 expression discriminates MHC-dependent and –independent stages of thymocyte positive selection. J Immunol. 1999;162:3978–3983. [PubMed] [Google Scholar]
- Hirschfield GM, Xie G, Lu E, Sun Y, Juran BD, Chellappa V, Coltescu C, Mason AL, Milkiewicz P, Myers RP, et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun. 2012;13:328–335. doi: 10.1038/gene.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Daly M. What have we learned from six years of GWAS in autoimmune diseases, and what is next? Curr Opin Immunol. 2012;24:571–575. doi: 10.1016/j.coi.2012.09.001. [DOI] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. The expanding genetic overlap between multiple sclerosis and type 1 diabetes. Genes Immun. 2009;10:11–14. doi: 10.1038/gene.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagielska D, Redler S, Brockschmidt FF, Herold C, Pasternack SM, Garcia Bartels N, Hanneken S, Eigelshoven S, Refke M, Barth S, et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J Invest Dermatol. 2012;132:2192–2197. doi: 10.1038/jid.2012.129. [DOI] [PubMed] [Google Scholar]
- Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kasai M, Tanida I, Ueno T, Kominami E, Seki S, Ikeda T, Mizuochi T. Autophagic compartments gain access to the MHC class II compartment in thymic epithelium. J Immunol. 2009;183:7278–7285. doi: 10.4049/jimmunol.0804087. [DOI] [PubMed] [Google Scholar]
- Keir ME, Latchman YE, Freeman GJ, Sharpe AH. Programmed death-1 (PD-1):PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. J Immunol. 2005;175:7372–7379. doi: 10.4049/jimmunol.175.11.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Wairkar YP, Daniels RW, DiAntonio A. The novel endosomal protein Ema interacts with the class C Vps-HOPS complex to promote endosomal maturation. J Cell Biol. 2010;188:717–734. doi: 10.1083/jcb.200911126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Naylor SA, DiAntonio A. Drosophila Golgi membrane protein Ema promotes autophagosomal growth and function. Proc Natl Acad Sci U S A. 2012;109:E1072–1081. doi: 10.1073/pnas.1120320109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet. 2006;38:479–483. doi: 10.1038/ng1766. [DOI] [PubMed] [Google Scholar]
- Márquez A, Varadé J, Robledo G, Martínez A, Mendoza JL, Taxonera C, Fernández-Arquero M, Díaz-Rubio M, Gómez-García M, López-Nevot MA, et al. Specific association of a CLEC16A/KIAA0350 polymorphism with NOD2/CARD15(-) Crohn's disease patients. Eur J Hum Genet. 2009;17:1304–1308. doi: 10.1038/ejhg.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez A, Perdigones N, Cénit MC, Espino L, Varadé J, Lamas JR, Santiago JL, Fernández-Arquero M, de la Calle H, Arroyo R, et al. Chromosomal region 16p13: further evidence of increased predisposition to immune diseases. Ann Rheum Dis. 2010;69:309–311. doi: 10.1136/ard.2008.098376. [DOI] [PubMed] [Google Scholar]
- Mero IL, Ban M, Lorentzen ÅR, Smestad C, Celius EG, Sæther H, Saeedi H, Viken MK, Skinningsrud B, Undlien DE, et al. Exploring the CLEC16A gene reveals a MS-associated variant with correlation to the relative expression of CLEC16A isoforms in thymus. Genes Immun. 2011;12:191–198. doi: 10.1038/gene.2010.59. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–598. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- Plesa G, Zheng L, Medvec A, Wilson CB, Robles-Oteiza C, Liddy N, Bennett AD, Gavarret J, Vuidepot A, Zhao Y, et al. TCR affinity and specificity requirements for human regulatory T-cell function. Blood. 2012;119:3420–3430. doi: 10.1182/blood-2011-09-377051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinningsrud B, Husebye ES, Pearce SH, McDonald DO, Brandal K, Wolff AB, Løvås K, Egeland T, Undlien DE. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab. 2008;93:3310–3317. doi: 10.1210/jc.2008-0821. [DOI] [PubMed] [Google Scholar]
- Skinningsrud B, Lie BA, Husebye ES, Kvien TK, Førre Ø, Flatø B, Stormyr A, Joner G, Njølstad PR, Egeland T, Undlien DE. A CLEC16A variant confers risk for juvenile idiopathic arthritis and anti-cyclic citrullinated peptide antibody negative rheumatoid arthritis. Ann Rheum Dis. 2010;69:1471–1474. doi: 10.1136/ard.2009.114934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, Spruce LA, Kushner JA, Groop L, Seeholzer SH, et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell. 2014;157:1577–1590. doi: 10.1016/j.cell.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukseree S, Mildner M, Rossiter H, Pammer J, Zhang CF, Watanapokasin R, Tschachler E, Eckhart L. Autophagy in the thymic epithelium is dispensable for the development of self-tolerance in a novel mouse model. PloS One. 2012;7:e38933. doi: 10.1371/journal.pone.0038933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukseree S, Rossiter H, Mildner M, Pammer J, Buchberger M, Gruber F, Watanapokasin R, Tschachler E, Eckhart L. Targeted deletion of Atg5 reveals differential roles of autophagy in keratin K5-expressing epithelia. Biochem Biophys Res Commun. 2013;430:689–694. doi: 10.1016/j.bbrc.2012.11.090. [DOI] [PubMed] [Google Scholar]
- Swat W, Dessing M, von Boehmer H, Kisielow P. CD69 expression during selection and maturation of CD4+CD8+ thymocytes. Eur J Immunol. 1993;23:739–746. doi: 10.1002/eji.1830230326. [DOI] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case-Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.