

SUMMARY
At presynaptic active zones, exocytosis of neurotransmitter vesicles (SVs) is driven by SNARE complexes that recruit Syb2 and SNAP25. However, it remains unknown which SNAREs promote the secretion of neuronal proteins, including those essential for circuit development and experience-dependent plasticity. Here, we demonstrate that Syb2 and SNAP25 mediate the vesicular release of BDNF in axons and dendrites of cortical neurons, suggesting these SNAREs act in multiple spatially-segregated secretory pathways. Remarkably, axonal secretion of BDNF is also strongly regulated by SNAP47 which interacts with SNAP25 but appears to be dispensable for exocytosis of SVs. Cell-autonomous ablation of SNAP47 disrupts the layer-specific branching of callosal axons of projection cortical neurons in vivo, and this phenotype is recapitulated by ablation of BDNF or its receptor, TrkB. Our results provide insights into the molecular mechanisms of protein secretion and define the functions of SNAREs in BDNF signaling and regulation of neuronal connectivity.
INTRODUCTION
Neurons activated during experience may nearly simultaneously release fast neurotransmitters and diffusible polypeptides from distinct membrane-trafficking organelles. Synaptic neurotransmitter vesicles (SVs) reside at axon terminals and undergo exocytosis at highly compartmentalized active zones, whereas peptidergic vesicles are broadly distributed in axons and dendrites (Dean et al., 2012; Sudhof, 2013). The core presynaptic release machinery includes SNAREs, synaptobrevin/VAMP2 (Syb2), SNAP25 and syntaxin1, whose assembly into ternary complexes primes SVs for rapid exocytosis and overcomes the energy barrier for their fusion with the plasma membrane (Schoch et al., 2001; Sudhof and Rothman, 2009; Wojcik and Brose, 2007). These SNAREs play similar roles in release of catecholamines from dense core vesicles (DCVs) in chromaffin cells and promote membrane fusion in vitro (Giraudo et al., 2009; Sakaba et al., 2005; Sorensen et al., 2003b), which supports the hypothesis that SNAREs universally control secretion. Surprisingly, the contribution of SNAREs to secretion of neuronal proteins is largely unexplored. Moreover, invertebrate and vertebrate genomes have relatively few VAMPs and SNAPs, which raises the question: as to what extent are individual SNARE isoforms shared in a given neuron between organelles with different content? These gaps in knowledge of basic aspects of membrane trafficking present a challenge for understanding the mechanisms of experience-dependent plasticity at molecular and circuit levels. For example, experimental evidence suggests that both neurotransmitters and peptides may influence axon branching and patterning of synaptic networks of various neuron classes (Bloodgood et al., 2013; Cao et al., 2007; Cheng et al., 2011; Kerschensteiner et al., 2009; Pieraut et al., 2014; Wang et al., 2007; Yu et al., 2004). Yet, virtually all contemporary methods for pharmacological, genetic or optogenetic control of network activity in live animals likely affect secretion in a non-selective manner, making it difficult to define the impacts of specific cues on neural circuit structure, function and, ultimately, animal behavior. Likewise, cleavage of Syb2 with genetically encoded tetanus toxin (TeNT) has become a popular approach for blocking exocytosis of SVs (Kerschensteiner et al., 2009; Pieraut et al., 2014; Wang et al., 2007; Yu et al., 2004), but it remains unclear how TeNT affects other vesicle types. Although these problems can be partially overcome by ablating neurotransmitter or peptide-specific receptors, such strategies have a limited use for identification of cellular sources of release.
The brain-derived neurotrophic factor (BDNF) has emerged as one of the key diffusible signals that is essential for axon growth, synaptogenesis, remodeling of mature synapses, learning, and memory (Lu et al., 2013; Park and Poo, 2013). While secretion of native neurotrophins is notoriously difficult to detect in real time, it is generally agreed that BDNF is transported by secretogranin2-positive vesicles that undergo exocytosis upon synaptic excitation and calcium influx (de Wit et al., 2009; Dean et al., 2012; Dieni et al., 2012; Kolarow et al., 2007; Matsuda et al., 2009; Sadakata et al., 2013; Sadakata et al., 2012). Structurally, the axonal pool of these organelles resembles DCVs, whereas dendritic vesicles do not have characteristic dense cores (Dieni et al., 2012; Miyazaki et al., 2011). Unlike SVs and chromaffin DCVs whose exocytosis is triggered by calcium binding to synaptotagmins 1, 2, and 9 (Fernandez-Chacon et al., 2001; Geppert et al., 1994; Maximov and Sudhof, 2005; Sorensen et al., 2003a; Xu et al., 2007), BDNF vesicles are believed to recruit calcium sensors CAPS and to be negatively regulated by Syt4, a synaptotagmin isoform that lacks calcium binding activity in vertebrates (Dai et al., 2004; Dean et al., 2009; Sadakata et al., 2012). We found that, in spite of their remarkable differences with SVs, BDNF vesicles employ Syb2 and SNAP25 for fusion in all subcellular domains of cortical neurons. Our results imply that Syb2 and SNAP25 broadly regulate neuronal secretion and offer an alternative interpretation of previously described phenomena associated with disruption of these SNAREs in the brain. Nevertheless, release of BDNF is also controlled by SNAP47, a SNAP isoform that associates with Syb2 and SNAP25 but does not contribute to exocytosis and recycling of SVs. Cell-autonomous loss of SNAP47 impairs the layer-specific branching of callosal axons of pyramidal neurons in the somato-sensory cortex in vivo, suggesting that differentiation of cortical axons depends upon autocrine BDNF/TrkB signaling rather than glutamatergic outputs onto postsynaptic targets.
RESULTS and DISCUSSION
Spatial and temporal dynamics of BDNF exocytosis
To investigate the molecular mechanisms of BDNF secretion, we infected cortical neurons in primary cultures with a letivirus encoding BDNF-pHluorin, a reporter comprised of full-length (pro) BDNF and a pH-sensitive form of GFP (Matsuda et al., 2009). The rationale for this approach and various controls that validate the appropriate post-translational modification, trafficking and biological activity of BDNF fusion proteins are described in accompanying supplementary materials. Confirming earlier studies (Dean et al., 2012; Matsuda et al., 2009; Miyazaki et al., 2011), immunofluorescence and live time-lapse imaging showed that BDNF vesicles were scattered in processes of mature neurons, contained another secreted peptide, secretogranin2, and were largely segregated from SVs. Unlike SVs, these organelles were not anchored at synapses, and underwent bi-directional transport in axonal and dendritic shafts (Figure 1A and Supplemental Figure 1).
Figure 1. Intracellular trafficking and exocytosis of BDNF vesicles.
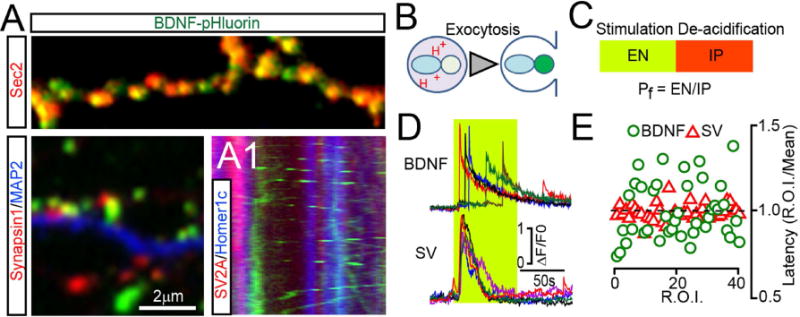
(A) DIV15 neurons expressing BDNF-pHluorin from a lentivirus were labeled with antibodies to GFP (as a probe for BDNF construct) and markers of either peptidergic (Sec2, top) or neurotransmitter vesicles (Synapsin1, bottom left). MAP2 labels dendrites. (A1) Kymograph from live neurons co-expressing BDNF-GFP and reporters of presynaptic terminals and dendritic spines, tdTomato-SV2A and EBFP2-Homer1c. See also Supplemental Figures 1 and 2.
(B) Schematic of reporter de-acidification during vesicle fusion with the plasma membrane.
(C) A protocol for quantitative analysis of exocytosis. EN = number of fusion events; IP = intracellular pool; Pf = probability of fusion.
(D and E) Exocytosis of BDNF and synaptic vesicles (SVs) was monitored at DIV15 by time-lapse TIRF imaging of BDNF-pHluorin and SyP-pHluorin, respectively. (D) Sample traces reflecting fusion in different sites of the same neuron during direct depolarization with KCl (50 mM). Green box marks the time-course of intracellular calcium rise, as determined in identical experimental settings in neurons expressing GCaMP3. (E) Latencies of isolated fusion events are plotted as ratios to averaged latency per cell. n = 4, 10 sites/neuron. See also Supplemental Figure 2, Movies SM1–4 and Supplemental methods.
We then monitored the release of BDNF-pHluorin in live neurons using total internal reflection fluorescent (TIRF) time-lapse microscopy. Under these conditions, the reporter is barely detectable in intracellular vesicles due to their low lumenal pH. During exocytosis, de-acidification leads to a rapid increase in fluorescence followed by a decay due to diffusion of cargo into the extracellular medium (Figure 1B). As shown previously (Balkowiec and Katz, 2000; Matsuda et al., 2009), secretion of BDNF could be reliably induced by direct membrane depolarization with KCl, high-frequency bursts of action potentials triggered by electrical stimulation, or pharmacological augmentation of network activity with the GABA receptor blocker, picrotoxin (PTX) (Supplemental Figure 2A–D and Movie SM1). To assess the efficacy of exocytosis, we measured three parameters: i) Number of isolated fusion events that occurred during fixed time intervals (EN); we confirmed that these events reflect release by applying a membrane-impermeable hydrophilic quencher, bromophenol blue (Harata et al., 2006); ii) Number of internal vesicles (IP, intracellular pool) in which BDNF-pHluorin could be detected following artificial de-acidification of lumens with ammonium chloride; and iii) Probability of fusion (Pf) = EN/IP ratio. This value was calculated to account for variability in the integrated area of plasma membrane and/or density of available vesicles across independent imaging sessions (Figure 1C and Supplemental Figure 2E–F). EN and Pf were diminished when stimulation was performed in the absence of extracellular calcium or in the presence of inhibitors of calcium channels known to be required for secretion of native BDNF (Balkowiec and Katz, 2000) (Supplemental Figure 2G).
SVs are clustered at presynaptic terminals where they form readily-releasable pools (RRP) adjacent to active zones. These pools are primed by SNARE complex assembly for rapid synchronous exocytosis in response to action potential-dependent calcium influx (Sudhof, 2013). By contrast, side-by-side comparison of neurons carrying BDNF-pHluorin or a reporter of SVs, synaptophysin (SyP)-pHluotrin (Peng et al., 2012), indicated that secretion of BDNF is asynchronous (Figure 1D–E). The kinetics of BDNF release also drastically differed from transferrin receptor-positive recycling endosomes (Supplemental Figure 2H–J). Moreover, only a fraction of BDNF vesicles fused at synapses regardless of stimulation type, as evidenced by simultaneous TIRF imaging of BDNF-pHluorin and genetically encoded pre- and postsynaptic markers, tdTomato-SV2A and EBFP2-Homer1c (Supplemental Figure 2K–N). These results indicate that no primed vesicle pools exist that supply a synaptic or extra synaptic RRP equivalent of BDNF in cortical neurons.
Secretion of BDNF is directly controlled by Tetanus toxin-sensitive SNAREs
To explore the possibility that release of neurotransmitters and BDNF is driven by distinct SNARE complexes, we first evaluated the effects of tetanus toxin (TeNT) – a protease that potently blocks the fusion of SVs by cleaving the vesicular SNARE, Syb2 (Schoch et al., 2001; Yamasaki et al., 1994). Although the extracellular application of recombinant TeNT polypeptide has been previously shown to interfere with release of BDNF in hippocampal cultures (Matsuda et al., 2009), it is unclear if this effect was direct. Indeed, TeNT-dependent loss of glutamatergic neurotransmission (Kerschensteiner et al., 2009; Pieraut et al., 2014; Yu et al., 2004; Zhang et al., 2008) may ultimately prevent the secretion of cargo from other membrane organelles whose exocytosis is coupled with synaptic excitation (Balkowiec and Katz, 2002; de Wit et al., 2009; Dean et al., 2012). We therefore investigated the effects of genetically-encoded TeNT that was expressed under the control of the synapsin promoter either globally or in a cell-autonomous manner, in only a small fraction of synaptically connected neurons. Similar approaches were used for all subsequent analyses of individual SNAREs described below.
Quantitative immunoblotting showed a strong increase in the levels of native BDNF and BDNF-pHluorin in cultured neurons that were uniformly infected with the TeNT lentivirus (Figure 2A–B). We also detected an accumulation of endogenous BDNF in membrane fractions isolated from the cortices of conditional mouse mutants that harbored TeNT in glutamatergic neurons in the postnatal forebrain (Pieraut et al., 2014) (Figure 2C–D). TIRF microscopy revealed that TeNT prevented the release of BDNF-pHluorin from cultured neurons resulting in its intra-vesicular retention during stimulation with KCl (Figure 2E–F). Conventional imaging of the entire vesicle pool suggested that TeNT interferes with exocytosis rather than transport from the endoplasmic reticulum to the Golgi, budding from Golgi, and endosomal trafficking (Supplemental Figure 3A–C).
Figure 2. Analysis of BDNF release in neurons carrying TeNT.
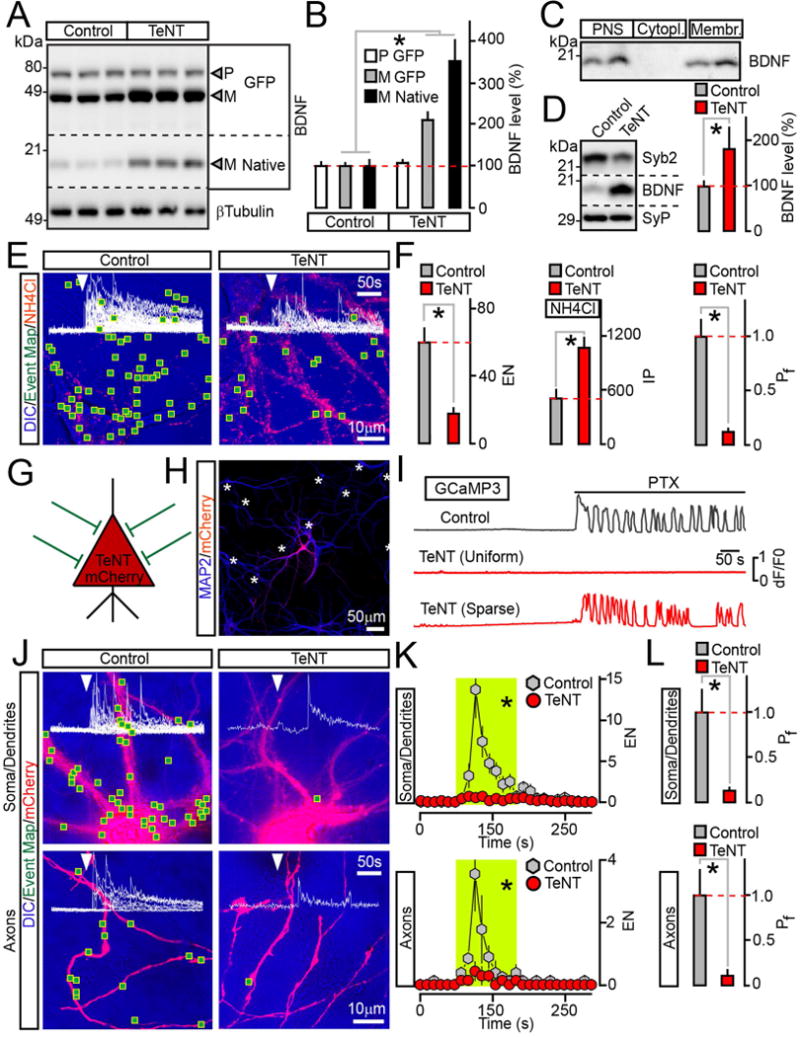
(A and B) Measurements of pro (P) and mature (M) BDNF levels in cultured neurons that uniformly expressed BDNF-pHluorin alone (Control) or with TeNT. Quantitative immunoblotting was performed with an antibody to BDNF which recognizes all native and recombinant forms. βTubulin was used as a loading standard. Representative blots and averaged values from 3 experiments are shown.
(C) Subcellular fractions of mouse forebrains were isolated by differential centrifugation and probed by immunoblotting for native BDNF. PNS=post nuclear supernatant.
(D) Quantitative immunoblot measurements of mature BDNF levels in membrane fractions prepared from cortices of wildtype P15 mice (Control) and their littermates carrying TeNT in glutamatergic neurons (CamKIIα:Cre/R26floxstopTeNT). n = 5 mice/genotype. The reduction of Syb2 band intensity is due to cleavage by TeNT, which makes Syb2 undetectable with antibodies.
(E and F) TIRF microscopy analysis of vesicle exocytosis in DIV15 cultured neurons that globally expressed BDNF-pHluorin without or with TeNT. (E) Pseudo-colored DIC images with superimposed maps of all sites where vesicle fusion was detected during transient depolarization with KCl (2 minutes). Red puncta are intracellular reporter-positive vesicles visualized by perfusing 50 mM of NH4Cl following stimulation. (F) Averaged EN (calculated for a 2 minute window of stimulation in entire fields of view), IP and Pf. n = 9/group.
(G to L) Cell-autonomous effects of TeNT on BDNF release. Neurons were sparsely infected (~5% of the total population) with lentiviruses encoding BDNF-pHluorin and mCherry alone or together with TeNT. These cells received synaptic inputs from wildtype neurons, as depicted in panel (G). (H) An example of infection pattern. Cultures were stained for MAP2. Asterisks label somas of reporter-negative cells. (I) PTX-induced (100 μM) calcium signals were recorded from neurons co-expressing TeNT, mCherry and GCaMP3 either globally or sparsely, as described above. (J to L) Exocytosis of BDNF vesicles was monitored by TIRF microscopy at DIV15. (J) DIC and mCherry images with mapped regions of somato-dendritic and axonal domains where vesicle fusion was observed during 2 minute depolarization with KCl. Superimposed traces of BDNF-pHluorin fluorescence sampled in time-lapse mode (inserts, arrows mark times of excitation) reflect exocytosis in each site. (K) Averaged EN in different subcellular compartments of individual neurons, plotted as a function of time (10 second bin). Green boxes mark the window of transient excitation. (L) Mean Pf of evoked fusion in somas, dendrites and axons. n = 7–8/group.
Quantifications are represented as Mean ± S.E.M. n corresponds to numbers of trials performed with at least 3 independent culture preparations. * p < 0.001 (t-test).
Because release of BDNF is regulated by synaptic network activity, we next tested if TeNT-sensitive SNAREs promote the secretion in a cell-autonomous manner. To accomplish this task, BDNF-pHluorin, TeNT and a red tracer, mCherry, were sparsely co-expressed in ~5% of cortical neurons in culture (Figure 2G–H and Supplemental Figure 3D–F). Infected neurons received intact glutamatergic inputs from surrounding wildtype cells, as demonstrated by GCaMP-based imaging of calcium signals elicited by application of PTX (Figure 2I and Supplemental Figure 3G–H). These experimental settings allowed us to bypass the effects of the protease on exocytosis of SVs and to monitor the fusion of BDNF vesicles in distinct subcellular domains of single neurons (Movies SM2 and SM3). TeNT abolished secretion in this case as well, even when cultures were depolarized continuously. Remarkably, quantifications of EN and Pf demonstrated that exocytosis was uniformly and nearly completely blocked in cell bodies, dendrites and axons (Figure 2J–L).
Exocytosis of BDNF is driven by the vesicular SNARE, Syb2
At least 9 genes encoding vesicular SNAREs are expressed in the mammalian brain, including 7 different Syb/VAMPs, YKT6 and Sec22b (Figure 3A, based on Allen Brain Atlas database). We systematically examined the sensitivity of these SNAREs to TeNT by immunoblotting following overexpression of cDNAs in HEK293 cells. Confirming earlier experiments with Sybs/VAMPs 1–3 (McMahon et al., 1993; Yamasaki et al., 1994), these proteins were cleaved in the presence of the protease whereas the remaining isoforms were resistant (Figure 3B). We then designed shRNAs that disrupt the expression of native TeNT-sensitive Syb/VAMPs and studied the outcomes of knockdowns (KD) on neuronal secretion of BDNF using a combination of biochemical and TIRF imaging readouts. We initially introduced pairs of lentiviruses driving hairpins under the control of H1 promoter to target two non-overlapping coding regions of each Syb/VAMP transcript. Single shRNAs from a mix that produces a phenotype were then tested in isolation or co-expressed with the rescue shRNA-resistant cDNA from a dual H1/Synapsin-promoter lentivirus vector (Supplemental Figure 4A and Table S1). The potencies and specificities of these hairpins were confirmed by immunohistochemistry and immunoblotting with relevant antibodies (Supplemental Figure 4D–F and data not shown).
Figure 3. Syb2 drives the exocytosis of BDNF vesicles in axons and dendrites independently from its presynaptic function.
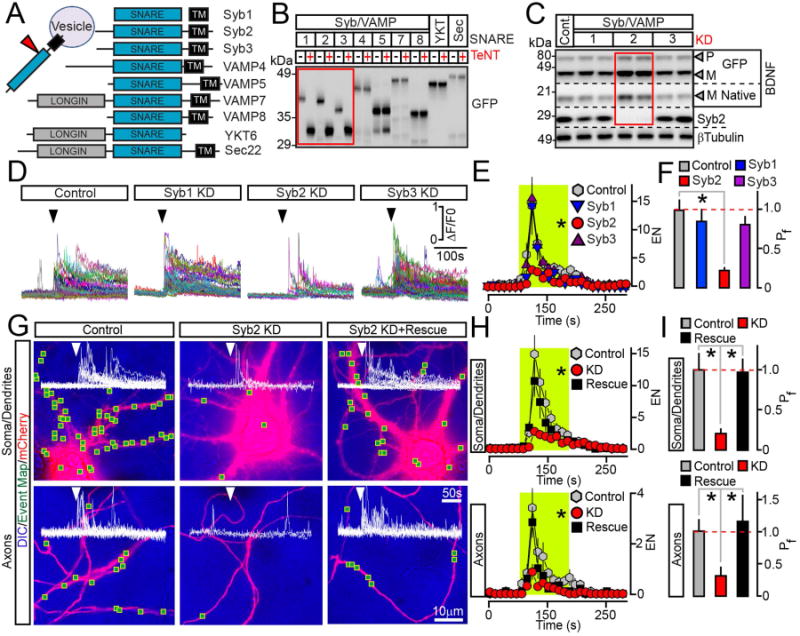
(A) Domain structures of vesicular SNAREs.
(B) Biochemical assessment of SNARE sensitivity to TeNT. Homogenates of HEK293 cells expressing different GFP-tagged SNAREs alone or together with TeNT were probed by immunoblotting for GFP.
(C to F) Cortical cultures were broadly co-infected at DIV5 with lentiviruses expressing BDNF-pHluorin and two shRNAs that target non-overlapping sequences of specific TeNT-sensitive Syb/VAMPs (KD). All experiments were performed at DIV15. (C) Immunoblot analysis of pro- (P) and mature (M) BDNF levels was performed using antibodies to BDNF and βTubulin (as a loading standard). n = 6 control and Syb/VAMP-deficient cultures, examined in duplicates. Quantifications are shown in Supplemental Figure 4B. (D) TIRF traces represent all events of depolarization-induced vesicle fusion detected in isolated sites of control and Syb1-3-deficient neurons. Arrows mark times of excitation. (E) Averaged EN, plotted as a function of time. Green box marks the window of transient excitation. (F) Mean Pf across the cell bodies and processes. Control, n = 7; Syb1 KD, n = 6; Syb2 KD, n = 7; Syb3 KD, n = 7. * p < 0.05 (ANOVA).
(G to I) Syb2 directly mediates BDNF release in all subcellular domains. Exocytosis was monitored by TIRF microscopy in somas, dendrites and axons of isolated neurons in DIV15 cultures that were sparsely co-infected with viruses encoding BDNF-pHluorin, mCherry and Syb2 shRNA without or with shRNA-resistant rescue cDNA. (G) DIC and mCherry images with mapped regions of somato-dendritic and axonal domains where fusion was observed during transient depolarization. Superimposed traces of BDNF-pHluorin fluorescence reflect exocytosis in each site. (H) Averaged EN in different subcellular compartments, plotted as a function of time. (I) Mean Pf in somas, dendrites and axons. n = 11–14/group. * p < 0.01 (ANOVA). See also Supplemental Figure 4.
Data are plotted as Mean ± S.E.M.
Loss of Syb2 alone fully recapitulated the effects of TeNT. KD of this SNARE in all neurons in culture resulted in an accumulation of mature BDNF forms and inhibited the depolarization-dependent exocytosis of BDNF-pHluorin without altering the subcellular localization of intracellular vesicles. In contrast, KDs of two other TeNT substrates, Syb1 and Syb3, failed to significantly affect the BDNF levels, EN and Pf (Figure 3C–F, Supplemental Figure 3C and 4B–C). TIRF imaging of BDNF-pHlorin in sparsely infected cultures demonstrated that, similar to TeNT, KD of Syb2 blocks the exocytosis in a cell-autonomous manner and in all subcellular domains. Single mCherry-tagged neurons carrying a Syb2-directed shRNA had a strong reduction of both the frequency of evoked fusion events and Pf across somas, dendrites and axons, despite receiving excitatory inputs from wildtype cells. These defects were rescued by the Syb2 cDNA containing silent mutations in the shRNA target site, hence ruling out off-target artifacts (Figure 3G–I and Supplemental Figure 4G). Also, native Syb2 could be detected in a significant fraction of BDNF vesicles by immunofluorescence microscopy, indicating that these organelles recruit Syb2 (Supplemental Figure 4H–J).
Secretion of BDNF is regulated by two membrane SNAPs
Our results imply that Syb2 directly promotes the release of BDNF independently of its presynaptic function and, surprisingly, does so in axons, dendrites and somas. To test if this previously unappreciated function of Syb2 involves interactions with unique vesicle-specific effectors on the plasma membrane, we focused on members of the membrane SNAP protein family with abundant expression in the brain, SNAPs 23, 25, 29 and 47 (Figure 4A, based on Allen Brain Atlas database and refs. (Holt et al., 2006; Johansson et al., 2008; Su et al., 2001; Suh et al., 2010)). Imaging of cortical cultures that broadly expressed SNAP-specific hairpins demonstrated that exocytosis of BDNF-pHluorin was suppressed after ablation of SNAP25 and SNAP47 (Figure 4B–D). However, only KD of SNAP25 resulted in an accumulation of bulk intracellular BDNF under these conditions, indirectly suggesting that SNAP47 has a modulatory and/or spatially-restricted function (Supplemental Figure 5A–B).
Figure 4. Exocytosis of BDNF vesicles is controlled by two different SNAPs.
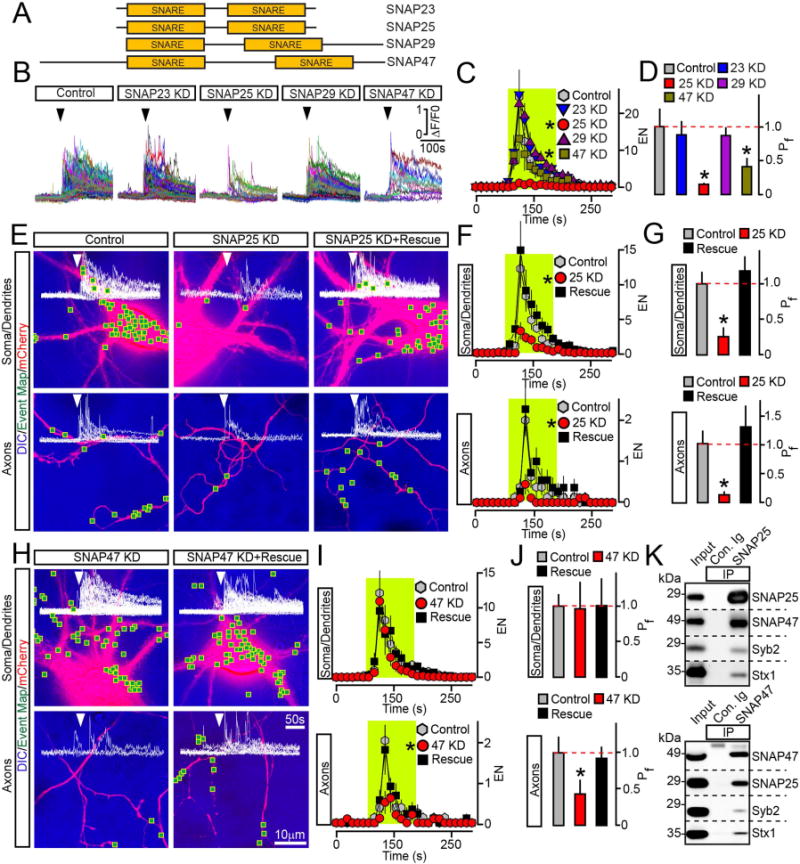
(A) Domain structures of vertebrate SNAREs of the SNAP family.
(B to D) Outcomes of global ablation of SNAPs on BDNF release. Cortical cultures were co-infected at DIV5 with viruses expressing BDNF-pHluorin and two shRNAs that target non-overlapping sequences of specific SNAP isoforms (KD). Exocytosis was monitored at DIV15 by TIRF imaging. (B) Sample traces reflecting all fusion evens in control and SNAP-deficient neurons. Arrows mark times of depolarization with KCl. (C) Averaged EN, plotted as a function of time. (D) Mean Pf across neuronal somas and processes. Control, n = 10; SNAP23 KD, n = 8; SNAP25 KD, n = 8; SNAP29 KD, n = 7; SNAP47 KD, n = 8. * p < 0.01 and < 0.05 for SNAP25 and SNAP47, respectively (ANOVA).
(E to J) Cell-autonomous effects of SNAP ablation on BDNF release. Exocytosis was monitored by TIRF microscopy in isolated neurons in DIV15 cultures that were sparsely co-infected with viruses encoding BDNF-pHluorin, mCherry and shRNAs against indicated SNAPs without or with corresponding rescue cDNAs. Panels show aligned DIC and mCherry images with mapped sites of depolarization-induced fusion, actual events of exocytosis in each site, EN plotted as a function of time, and Pf. n = 9–11/group. (E to G) Examples of exocytosis, averaged EN, and Pf in different subcellular compartments of control and SNAP25-deficient neurons. * p < 0.01 (ANOVA). (H to J) Examples of exocytosis, mean EN, and Pf in SNAP47-deficient neurons. * p < 0.05 (ANOVA). Control groups apply to both SNAP isoforms. Quantifications are represented as Mean ± S.E.M. See also Supplemental Figures 5 and 6.
(K) Solubilized proteins were co-immunoprecipitated (IP) from mouse forebrain extracts with control antibody (Ig) or antibodies against SNAP25 (top) or SNAP47 (bottom). Attached complexes were examined by immunoblotting with antibodies to SNAP25, SNAP47, Syb2 and Syntaxin1. Equal amounts of total protein were applied to each lane.
Previous in vitro studies have shown that SNAP47 is unable to fully substitute for SNAP25 in fusion of liposomes driven by SNARE complexes containing Syb2 and Syntaxin1 (Holt et al., 2006). SNAP47 has also been implicated in hippocampal LTP (Jurado et al., 2013), yet its role in secretion has remained unclear. The involvement of SNAP47 in exocytosis of BDNF vesicles is intriguing, since induction of Hebbian plasticity critically depends on BDNF signaling (Kang and Schuman, 1995; Park and Poo, 2013; Patterson et al., 1996). To gain more insight into functions of two SNAP isoforms in BDNF trafficking, we examined the cell-autonomous outcomes of their KDs on exocytosis of BDNF-pHluorin in single sparsely infected neurons. KD of SNAP25 uniformly abolished the depolarization-induced release in all subcellular domains. On the contrary, SNAP47-deficient cells only exhibited a prominent loss of exocytosis in axons, albeit we also detected a shift in kinetics of vesicle fusion across their entire surface (Figure 4E–J and Supplemental Figure 6). Consistent with these notions, both SNAP isoforms were distributed throughout neuronal processes (Supplemental Figure 5F–H). The phenotypes associated with each KD could be reversed by corresponding shRNA-insensitive cDNAs (Figure 4E–J, Supplemental Figures 5A–E and 6). Neither KD altered the distribution of internal BDNF vesicles making it unlikely that secretion was disrupted due to abnormal intracellular trafficking (Supplemental Figure 3C). To test whether SNAP47 and SNAP25 interact with each other, we immunoprecipitated these SNAREs with isoform-specific antibodies from mouse brain extracts. Immunoblotting showed that SNAP47 and SNAP25 are abundant in complexes that contain Syb2 and syntaxin1 (Figure 4K).
To mimic activity-dependent secretion using a more physiologically-relevant paradigm, we also induced exocytosis with bursts of action potentials. Cortical cultures were sparsely infected with BDNF-pHluorin and mCherry, and axonal secretion was monitored before, during and after 30 second stimulus trains that were triggered at various frequencies by brief injection of current through an extracellular electrode. Fusion events were detectable, yet, relatively rare at 10Hz, whereas 100Hz trains elicited robust responses. Nevertheless, TeNT or KD of SNAP47 potently suppressed EN and Pf at both frequencies (Figure 5A–D).
Figure 5. SNAP47 promotes the exocytosis of BDNF but not neurotransmitter vesicles.
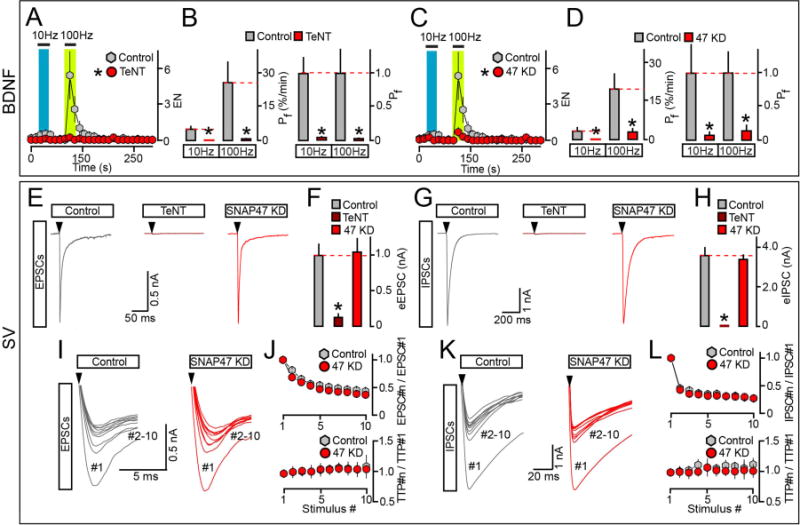
(A to D) Frequency-dependence of BDNF release in isolated wildtype and SNARE-deficient neurons. Cultures were sparsely infected with BDNF-pHluorin and mCherry lentiviruses alone or together with TeNT or shRNA against SNAP47 (KD). TIRF imaging was performed at DIV15. Exocytosis was elicited by electrical stimulation at 10 and 100 Hz (1 ms/10V/pulse) and monitored in mCherry-tagged axons. Each stimulus train was applied for 30 seconds. (A and B) Averaged EN and Pf. Colored boxes mark the times of action potential trains. Pf values are shown without normalization or as a fraction of control. Control, n = 14; TeNT, n = 10. * p < 0.001 (t-test). (C and D) EN and Pf were measured in control and SNAP47-deficient neurons and represented as described above. EN: Control, n = 14, this group is the same as for TeNT experiments; SNAP47 KD, n = 16. Pf: Control, n = 16; SNAP47 KD, n = 19. * p < 0.001 (ANOVA).
(E to L) Exocytosis of SVs in neurons carrying TeNT or shRNA against SNAP47. Cortical cultures were uniformly infected with corresponding lentiviruses. Evoked excitatory and inhibitory postsynaptic currents (eEPSCs and eIPSCs) were sampled at DIV15 in whole-cell voltage clamp mode. eEPSCs and eIPSCs were triggered by single action potentials or 10 Hz stimulus trains, and were pharmacologically isolated by addition of GABA or AMPA/NMDA receptor blockers. (E and F) Samples of individual eEPSCs (E, scale bars apply to all traces, arrows mark times of action potentials) and averaged eEPSC amplitudes (F). Control, n = 13; TeNT, n = 11; SNAP47 KD, n = 13. * p < 0.001 (ANOVA). (G and H) Samples of single eIPSCs (G) and averaged eIPSC amplitudes (H). Control, n = 19; TeNT, n = 3; SNAP47 KD, n = 14. * p < 0.001 (ANOVA). (I to L) Short-term plasticity of excitatory and inhibitory synapses in wildtype and SNAP47-deficient neurons. (I and K) On-sets of aligned EPSCs (I) and IPSCs (K) that were elicited by 10 Hz trains of 10 action potentials (arrows). (J and L) Top: averaged profiles of synchronous EPSC (J) and IPSC (L) depression during repetitive stimulation are presented as ratios of PSC amplitudes. Bottom: averaged profiles of EPSCs (J) and IPSC (L) de-synchronization are plotted as ratios to times to peak (TTP). EPSCs: Control, n = 12; SNAP47 KD, n = 8. IPSCs: Control, n = 16; SNAP47 KD, n = 12.
Data are plotted as Mean ± S.E.M. See also Supplemental Figure 7 and Table S2.
SNAP47-deficient neurons have normal exocytosis and recycling of SVs
Our experiments reveal largely promiscuous roles of Syb2 and SNAP25 in membrane fusion and identify SNAP47 as a critical regulator of vesicular release. To test if SNAP47 is necessary for exocytosis of SVs, we examined synapses of mature neurons in culture by confocal microscopy and electrophysiology. Uniform expression of TeNT or KD of Syb2, SNAP25 or SNAP47 did not disrupt the formation of glutamatergic and GABAergic terminals, consistent with previous notions that vesicular exocytosis is not necessary for gross synaptic differentiation in vitro (Harms and Craig, 2005; Schoch et al., 2001) (Supplemental Figure 7 and data not shown). We then performed whole-cell recordings to evaluate the properties of different synapse types. As expected, TeNT and hairpins against Syb2 and SNAP25 diminished the amplitudes of evoked excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs) and the rates of spontaneous SV fusion alike. However, synaptic strength was unaltered in neurons lacking SNAP47, despite their defects in exocytosis of BDNF (Figure 5E–H, Table S2, and data not shown). In addition, wildtype and SNAP47-deficient neurons had indistinguishable profiles of PSC depression and de-synchronization during action potential trains as well as kinetics of quantal AMPA- and GABA-type currents (Figure 5I–L). Hence, unlike Syb2 and SNAP25, SNAP47 appears to be unnecessary for SV exocytosis and recycling.
SNAP47 is essential for axon differentiation in vitro and in vivo
Targeted disruption of vesicular release in animal brains is becoming increasingly popular for studies of the role of neuronal activity in regulation of connectivity. For instance, several laboratories have shown that inducible expression of genetically encoded TeNT interferes with wiring of olfactory sensory neurons, photoreceptors, pyramidal neurons, and inhibitory GABAergic interneurons in the cerebral cortex and hippocampus (Kerschensteiner et al., 2009; Pieraut et al., 2014; Wang et al., 2007; Yu et al., 2004). While these phenomena are widely believed to reflect the instructive role of excitatory synaptic outputs, the involvement of BDNF or other secreted peptides cannot be ruled out. Indeed, BDNF is known to be necessary for axon growth and branching (Cao et al., 2007; Cheng et al., 2011; Marshak et al., 2007; Park and Poo, 2013) and we found that, by cleaving Syb2, TeNT directly blocks the release of fast neurotransmitters and BDNF. Notably, Syb2, SNAP25 and SNAP47 are expressed in the developing brain prior to the onset of synaptogenesis with SNAP47 turning on as early as embryonic day 10 (Holt et al., 2006). We therefore monitored secretion of BDNF in axons of differentiating SNAP47-deficient cortical neurons, and examined axon morphologies in culture and in the mouse brain in vivo. As demonstrated above, this strategy enabled us to interfere with axonal exocytosis of BDNF without affecting SVs. We initially imaged BDNF-pHluorin in isolated mCherry-tagged neurons in dissociated cultures. These cells were sparsely infected with viruses immediately after plating and analyzed six days later, when a marker of presynaptic boutons, synapsin1, remained diffusely distributed in axons. Immunostaining confirmed that endogenous SNAP47 and other SNAREs were present in axons at this stage (Supplemental Figure 8A–B). Differentiating neurons displayed robust exocytosis of BDNF-pHluorin during membrane depolarization (Figure 6A–C and Movie SM4). Secretion was blocked by TeNT or KD of SNAP47, but was restored in the presence of rescue SNAP47 cDNA (Figure 6D–H).
Figure 6. Analysis of BDNF release in differentiating axons.
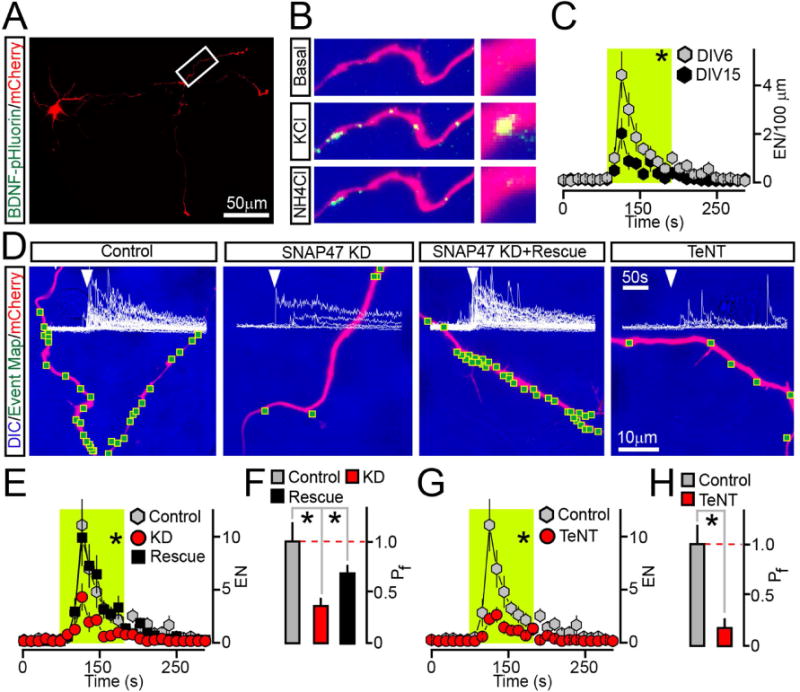
Cultured cortical neurons were sparsely co-infected at DIV0 with BDNF-pHluorin and mCherry lentiviruses alone (Control) or together with viruses encoding TeNT or shRNA against SNAP47 (without or with rescue cDNA). Axonal exocytosis was monitored by TIFR microscopy at DIV6.
(A) Typical low-magnification image shows axons of a neuron tagged with mCherry.
(B) Examples of BDNF-pHluorin fluorescence in a distal fragment of an axon under the normal conditions (Basal) during depolarization with KCl, and after vesicle de-acidification with NH4Cl.
(C) Averaged EN in axons of mature (DIV15) and differentiating (DIV6) wildtype neurons were normalized to axon length and plotted as a function of time. DIV6, n = 10; DIV15, n = 7.
(D) DIC and mCherry images with mapped sites of depolarization-induced fusion and actual BDNF-pHluorin responses detected in each site of control, SNAP47-deficient and TeNT-expressing neurons. (E–H) Averaged EN and Pf. * p < 0.02 (ANOVA). Note that in this case rescue is significant but incomplete, perhaps due to short time-course of cDNA overexpression.
See also Supplemental Figure 8A–B.
To determine how axonal exocytosis of BDNF impacts axon differentiation, a mix of two plasmids encoding a monomeric form of Venus and shRNA directed against SNAP47 was unilaterally introduced into the somatosensory cortex of E15.5 mouse embryos via in utero electroporation (Courchet et al., 2013). This method allows for selective targeting of callosally-projecting pyramidal neurons that populate the superficial layers 2/3. As the first step, the outcomes of SNAP47 KD in layer 2/3 neurons on their axon morphologies were assessed in cultures prepared from the cortices of electroporated embryos. SNAP47-deficient neurons (~1% of the entire pool, as determined by mVenus imaging, data not shown) had a slightly increased total axon length but exhibited a significant reduction in the number of terminal branches. The branching defect could be reversed with shRNA-resistant SNAP47 cDNA or by continuous incubation with recombinant BDNF added in the culture medium (Figure 7A–B).
Figure 7. SNAP47 is essential for BDNF-dependent axon branching in vitro and in vivo.
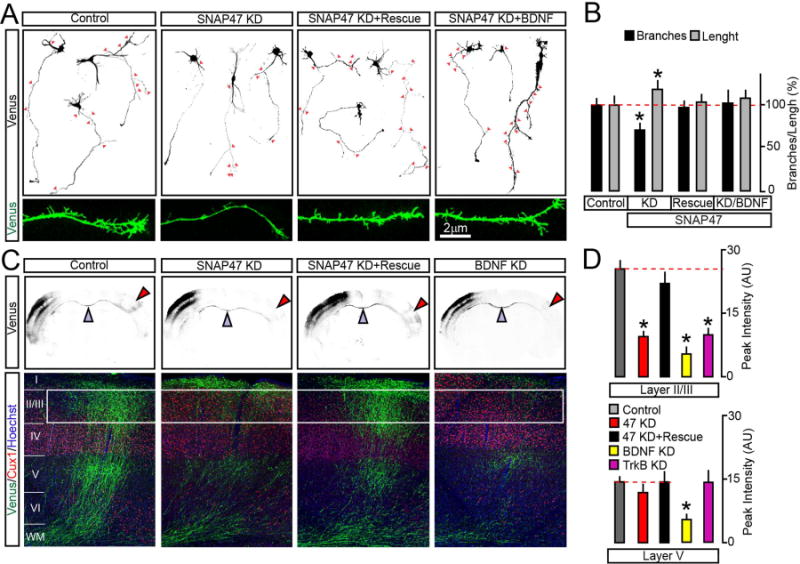
(A and B) Cortices of E15.5 mouse embryos were electroporated ex vivo with the pSCV2-mVenus plasmid together with control shuttle vector or vectors driving the expression of SNAP47 shRNA alone or with rescue cDNA. Morphologies of differentiating mVenus-positive axons were then examined in primary cultures prepared from these mice. Axon identities were confirmed by immunostaining for SMI312 and MAP2 (not displayed). (A) Typical images of DIV5 neurons carrying indicated constructs. Cultures were maintained in the normal medium or with addition of recombinant BDNF (50 ng/ml, applied at DIV1). Arrows mark each axonal branch. Panels on the bottom show enlarged fragments of axons. (B) Quantifications of total axon length and branch numbers. Data from three independent cultures were normalized to control in each case. Control, n = 62 axons; SNAP47 KD, n = 74; SNAP47 KD + Rescue, n = 51; SNAP47 KD + BDNF, n = 14. * p < 0.05 (ANOVA).
(C and D) E15.5 embryos were in utero electroporated into one cortical hemisphere with mVenus tracer together with control shuttle vector or vectors encoding indicated shRNAs. Axon branching was examined by imaging of brain slices at postnatal day 21. (C) Top: low-magnification images of coronal brain sections. mVenus signal is shown in black. Electroporated (ipsilateral) sides are on the left. Blue arrows mark corpus callosum. Red arrows point contralateral branching. Bottom: confocal images of contralateral sides of cortical sections stained for mVenus, a layer-specific marker, Cux1 and a nuclear marker, Hoechst. (D) Average surface areas of mVenus fluorescence in layers 2/3 and 5 of contralateral cortex. Control, n = 11 mice; SNAP47 KD, n = 8 mice; SNAP47 KD + Rescue, n = 5 mice. BDNF KD, n = 3 mice; TrkB KD, n = 8 mice. * p < 0.001 (ANOVA).
Data are plotted as Mean ± S.E.M. See also Supplemental Figure 8.
We then investigated the SNAP47 function in vivo, by imaging coronal cortical sections from mice that were electroporated at E15.5 and sacrificed at postnatal day 21 (P21). At P21, axons establish adult-like patterns of innervation in layers 2/3 and 5, both ipsi- and contralaterally (Courchet et al., 2013). On the contralateral side, the genetic manipulation was axon-specific since neurons projected into the normal environment. Immunolabeling of sections isolated from wildtype animals showed that native SNAP47 was abundant in callosal axons (Supplemental Figure 8C). Axons of mVenus-positive SNAP47-deficient neurons formed, grew, and successfully crossed the midline but had a drastically reduced terminal arborization in the contralateral layer 2/3. Again, the defect of terminal branching was alleviated by co-expression of SNAP47 shRNA with shRNA-insensitive cDNA (Figure 7C–D). Nonetheless, KD of SNAP47 had no detectable effect on density of dendritic spines (Supplemental Figure 8D–E), indicating that disruption of connectivity was likely restricted to axons. To ensure that these changes in callosal axon structure are attributed to abnormal BDNF signaling, we performed in utero electroporation with shRNAs against BDNF, or its receptor, TrkB. The morphological defects of contralateral axons of BDNF- and TrkB-deficient layer 2/3 neurons were similar to SNAP47-deficient neurons, albeit KD of BDNF produced a more robust effect with significant attenuation of branching in layer 5 (Figure 7C–D and Supplemental Figure 8F–G).
In summary, these studies elucidate the roles of SNAREs in vesicular exocytosis, BDNF secretion and neural circuit development. A large body of work shows that Syb2 and SNAP25 are essential for calcium-induced release of neurotransmitters and monoamines in central neurons and chromaffin cells, respectively (Sorensen et al., 2003b; Sudhof, 2013; Sudhof and Rothman, 2009). These SNAREs are thought to operate by priming vesicles and catalyzing fusion at specific sites, such as the presynaptic active zones. Remarkably, Syb2 and SNAP25 also trigger the release of BDNF from organelles that traffic and fuse with the plasma membrane across axons and dendrites, and do not form discrete pools that are primed for rapid exocytosis. By contrast, SNAP47 plays a more restricted role, perhaps as a part of a unique SNARE complex. SNAP47 interacts with SNAP25, promotes the exocytosis of BDNF in a cell-autonomous manner, but appears to be dispensable for synaptic release from SVs. Thus, two distinct types of neuronal vesicles share Syb2 and SNAP25, but their exocytosis is differentially controlled through combinatorial use of SNAPs.
Experiments with mutant animals carrying TeNT have led to a hypothesis that experience-driven refinement of connectivity is guided by neurotransmission between two relaying neurons in a given circuit (Okawa et al., 2014). Our findings challenge the generality of this model and underscore an alternative pathway that depends upon autocrine BDNF/TrkB signaling. First, we show unambiguously that, by cleaving Syb2, TeNT abolishes the release of neurotransmitters and BDNF. We further demonstrate that cell-autonomous ablation of SNAP47, which evidently does not affect SVs, leads to defects in layer-specific branching of callosal axons that are similar to those caused by KD of BDNF or its receptor, TrkB, and those previously detected upon expression of TeNT in pyramidal neurons in the same cortical area (Wang et al., 2007). Neurotransmitters and peptides may impact axon and synapse development by binding to their receptors in spatially-segregated micro-domains. Our results favor the scenario that axonally released BDNF promotes cortical axon branching via TrkB receptors localized on the same axon (Cao et al., 2007; Cheng et al., 2011).
Peptidergic vesicles contain diffusible cues other than BDNF (Lessmann et al., 2003), though their cargo may vary in different neuron classes and developmental stages. Identification of SNAREs essential for BDNF exocytosis will facilitate the further understanding of mechanisms of peptide secretion and will enable the design of new genetic tools to investigate the outcomes of secretion on neural circuit structure and information processing. In this regard, SNAP47 is a particularly attractive molecule due to its selectivity and unusually large size with putative sequences that may mediate protein-protein interactions (Holt et al., 2006). It would be of great interest to explore the interplay between this SNARE and other known and yet unidentified elements of membrane-trafficking machinery, including calcium sensors (Sadakata et al., 2012; Sudhof, 2013) and scaffolds that may recruit organelles to release sites.
EXPERIMENTAL PROCEDURES
Neuronal cultures
Cortices of P1 pups were dissociated by trypsin digestion; cells were plated on circle glass coverslips (Harvard Apparatus) coated with poly-D-Lysine (Sigma). Cultures were maintained for 4 days in MEM (Invitrogen) supplemented with FBS, B-27 (Invitrogen), glucose, transferrin and Ara-C (Sigma) followed by incubation in the serum-free medium. Neurons were seeded at 25,000–50,000 cells/cm2 for imaging and electrophysiological studies, and 150,000–175,000 cells/cm2 for immunoblotting. In both preparations, the densities of astrocytes did not exceed 2–3%, as determined by immunostaining for GFAP.
Virus infection
Recombinant lentiviruses were produced by co-transfection of human embryonic kidney 293T cells with corresponding shuttle vectors and pVSVg and pCMVΔ8.9 plasmids that encode the elementsessential for packaging viral particles. Transfections were performed using the FuGENE reagent (Promega). Secreted viruses were harvested 48 hours later and cleared by brief centrifugation. For uniform expression of fluorescent reporters and shRNA/rescue constructs, cultures were infected at DIV5 with 100 μl of viral supernatants per 1 ml of medium. This protocol was optimized to achieve >95% infection efficiency. For sparse infection, dissociated neurons were seeded onto Matrigel (BD Bioscience) coated 12-well plates and treated with viruses immediately after plating. 24 hours following infections, cells were washed three times with PBS, dissociated with trypsin, and mixed with non-infected neurons at a 1:20 ratio.
Immunocytochemistry
Neurons attached to the glass coverslips were rinsed once in PBS, fixed on ice in 4% paraformaldehyde (PFA), 4% sucrose in PBS and permeabilized for 5 minutes at room temperature in 0.2% Triton X-100 (Roche). Permeabilized neurons were incubated for 30 minutes in blocking solution containing 5% BSA (Sigma, fraction V) and 5% Normal Donkey Serum, followed by an overnight incubation with primary and corresponding fluorescently labeled secondary antibodies diluted in the same solution. The coverslips were mounted on glass slides with Aqua-Poly/Mount medium (Polysciences, Inc.). Images were collected under a Nikon C2 confocal microscope using 10× (N.A. 0.45), 20× (N.A. 0.75), and ×60 (N.A. 1.40) PlanApo objectives. Digital images were analyzed with the Nikon Elements software package. Digital manipulations were equally applied to all pixels. Analyses of organelle co-localization were performed with NIH ImageJ software.
Immunoprecipitations
Cortices of wildtype mice were homogenized in 20 mM HEPES-NaOH, pH7.4, 320 mM Sucrose, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml pepstatin. Membrane fractions were isolated by two sequential steps of centrifugation at 3,000 and 100,000g and solubilized for 1 hour in 20 mM HEPES-NaOH, pH7.4, 150 mM NaCl and 1% Triton X-100. Protein extracts were cleared by centrifugation at 100,000g and incubated overnight with sepharose beads (Sigma) coated with anti-SNAP25, anti-SNAP47 or control antibodies. Attached complexes were then washed 5 times with extraction buffer, eluted from the beads with an SDS gel loading buffer and used for immunoblotting.
Electrophysiology
Evoked synaptic release was triggered by 1 ms current injections with a local extracellular stimulating electrode (FHC, Inc.). Postsynaptic currents were monitored in whole-cell mode from randomly selected nearby neurons using a Multiclamp 700B amplifier (Axon Instruments, Inc.). The frequency, duration, and magnitude of extracellular stimuli were controlled with Model 2100 Isolated Pulse Stimulator (A-M Systems, Inc.). The whole-cell pipette solution contained 135 mM CsCl2, 10 mM HEPES-NaOH pH 7.4, 1 mM EGTA, 1 mM Na-ATP, 0.4 mM Na-GTP, and 1 mM QX-314. The resistance of filled pipettes varied between 3–5 mOhm. The bath solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 0.8 mM MgCl2, 10 mM HEPES-NaOH pH7.4, and 10 mM glucose. EPSCs and IPSCs were separated pharmacologically by addition of Picrotoxin (100 μM) or APV (50 μM) and CNQX (10 μM), respectively, to the bath solution. Spontaneous EPSCs and IPSCs were monitored under the same conditions but in the presence of Tetrodotoxin (1 μM). The currents were sampled at 10 kHz and analyzed offline using pClamp10 (Axon Instruments, Inc.) and Origin8 (Origin Lab) software.
Live imaging
Conventional fluorescence and TIRF imaging was carried out under the Nikon A1R inverted microscope controlled by the Nikon NIS-Elements software. Images were acquired at 1 Hz with a 12-bit cooled EMCCD camera (Andor, iXon) using ×10 PlanApo (N.A. 0.45), ×20 PlanApo (N.A. 0.75), ×60 PlanApo (N.A. 1.40) and ×100 PlanApo TIRF (N.A. 1.49) objectives. Neurons were bathed in the custom-build chamber perfused at 3–4 ml/min with the NRS solution containing 10 mM HEPES-NaOH, pH7.4, 140 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 2 mM CaCl2 and 10 mM Glucose. The temperature was maintained at 37°C with the lens and stage heaters. Direct depolarization was induced by application of NRS buffer in which 45 mM NaCl was substituted with KCl. Action potentials were elicited with a pair of parallel platinum electrodes connected to Model 2100 Isolated Pulse Stimulator (A-M Systems, Inc.). Toxins and inhibitors were diluted in the NRS. The intracellular pool (IP) of BDNF-pHluorin-positive vesicles was measured after perfusion of de-acidification buffer composed of 10 mM HEPES-NaOH, pH7.4, 50 mM NH4Cl, 90 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 2 mM CaCl2 and 10 mM Glucose.
Time-lapse image data analysis
Image stacks were aligned with TurboReg and analyzed with NIH ImageJ/Fiji plugins. Sites of activity-dependent exocytosis (e.g. regions exhibiting a rapid increase in BDNF-pHluorin or SyP-pHluorin fluorescence followed by a characteristic decay) were manually mapped in fames of a fixed size by assigning 0.8μm × 0.8μm regions of interest (R.O.I). Probabilities of evoked fusion (Pf) were calculated as ratios of total numbers of new fusion events (EN) observed prior to or during excitation to densities of internal reporter-positive vesicles that were automatically counted with FindMaxima NIH ImageJ/Fiji plugin at the end of each trial (following de-acidification with NH4Cl). Quantifications of exocytosis in specific subcellular domains were performed in sparsely infected neurons co-expressing BDNF-pHluorin and mCherry. A detailed description of TIRF imaging is enclosed in Supplemental information.
Statistical analyses were performed with InStat, Prism (Graphpad Inc.) and Origin8 (Origin Lab) software. Two groups of data were subjected to Student’s t-test and Mann-Whitney test. Multiple groups were compared by oneway ANOVA followed by Dunnett’s post hoc test or Kruskal-Wallis test.
Supplementary Material
Highlights.
BDNF is a secreted protein that regulates neuronal development and plasticity Vesicular exocytosis of BDNF is driven by SNAREs, Syb2, SNAP25 and SNAP47 Unlike Syb2 and SNAP25, SNAP47 appears to be unnecessary for neurotransmission Loss of BDNF or SNAP47 in callosal neurons diminishes branching of their axons
Acknowledgments
We thank the TSRI/DNC faculty members for advice and stimulating discussions; Drs. Thomas Südhof and Martyn Goulding for providing mouse strains and antibodies; Dr. Michael Ehlers for sharing the TfR-pHluorin expression vector; Dr. Kathy Spencer for expert technical assistance; and Elisabeth Rebboah for editing the manuscript. This study was supported by the NIH grant MH085776 (A.M.), Novartis Advanced Discovery Institute (A.M.), The Baxter Foundation (A.M.), Japanese Society for Promotion of Science (M.S.), NIMH Pre-doctoral National Research Service Award (R.S.) and Helen Dorris Postdoctoral Fellowship (S.P.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
A.M., and M.S. conceived the experiments. M.S. performed all studies of vesicle trafficking, secretion and SNARE interaction. J.C. analyzed axon branching in vitro and in vivo. S.P. together with M.S. performed electrophysiological recordings. N. T-R. and R.S. assisted with generation of expression vectors and data analysis. F.P. supervised axon branching studies. A.M. wrote the manuscript.
References
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, Greenberg ME. The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature. 2013;503:121–125. doi: 10.1038/nature12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Dhilla A, Mukai J, Blazeski R, Lodovichi C, Mason CA, Gogos JA. Genetic modulation of BDNF signaling affects the outcome of axonal competition in vivo. Current biology: CB. 2007;17:911–921. doi: 10.1016/j.cub.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM. Self-amplifying autocrine actions of BDNF in axon development. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18430–18435. doi: 10.1073/pnas.1115907108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchet J, Lewis TL, Jr, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510–1525. doi: 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Shin OH, Machius M, Tomchick DR, Sudhof TC, Rizo J. Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nature structural & molecular biology. 2004;11:844–849. doi: 10.1038/nsmb817. [DOI] [PubMed] [Google Scholar]
- de Wit J, Toonen RF, Verhage M. Matrix-dependent local retention of secretory vesicle cargo in cortical neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:23–37. doi: 10.1523/JNEUROSCI.3931-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nature neuroscience. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Liu H, Staudt T, Stahlberg MA, Vingill S, Buckers J, Kamin D, Engelhardt J, Jackson MB, Hell SW, et al. Distinct subsets of Syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:5398–5413. doi: 10.1523/JNEUROSCI.4515-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. The Journal of cell biology. 2012;196:775–788. doi: 10.1083/jcb.201201038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Giraudo CG, Garcia-Diaz A, Eng WS, Chen Y, Hendrickson WA, Melia TJ, Rothman JE. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science. 2009;323:512–516. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harata NC, Choi S, Pyle JL, Aravanis AM, Tsien RW. Frequency-dependent kinetics and prevalence of kiss-and-run and reuse at hippocampal synapses studied with novel quenching methods. Neuron. 2006;49:243–256. doi: 10.1016/j.neuron.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Harms KJ, Craig AM. Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. The Journal of comparative neurology. 2005;490:72–84. doi: 10.1002/cne.20635. [DOI] [PubMed] [Google Scholar]
- Holt M, Varoqueaux F, Wiederhold K, Takamori S, Urlaub H, Fasshauer D, Jahn R. Identification of SNAP-47, a novel Qbc-SNARE with ubiquitous expression. The Journal of biological chemistry. 2006;281:17076–17083. doi: 10.1074/jbc.M513838200. [DOI] [PubMed] [Google Scholar]
- Johansson JU, Ericsson J, Janson J, Beraki S, Stanic D, Mandic SA, Wikstrom MA, Hokfelt T, Ogren SO, Rozell B, et al. An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS genetics. 2008;4:e1000278. doi: 10.1371/journal.pgen.1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Sudhof TC, Malenka RC. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron. 2013;77:542–558. doi: 10.1016/j.neuron.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner D, Morgan JL, Parker ED, Lewis RM, Wong RO. Neurotransmission selectively regulates synapse formation in parallel circuits in vivo. Nature. 2009;460:1016–1020. doi: 10.1038/nature08236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Progress in neurobiology. 2003;69:341–374. doi: 10.1016/s0301-0082(03)00019-4. [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nature reviews Neuroscience. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- Marshak S, Nikolakopoulou AM, Dirks R, Martens GJ, Cohen-Cory S. Cell-autonomous TrkB signaling in presynaptic retinal ganglion cells mediates axon arbor growth and synapse maturation during the establishment of retinotectal synaptic connectivity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:2444–2456. doi: 10.1523/JNEUROSCI.4434-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Sudhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Sudhof TC. Cellubrevin is a ubiquitous tetanus-toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature. 1993;364:346–349. doi: 10.1038/364346a0. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Yamasaki M, Uchigashima M, Matsushima A, Watanabe M. Cellular expression and subcellular localization of secretogranin II in the mouse hippocampus and cerebellum. The European journal of neuroscience. 2011;33:82–94. doi: 10.1111/j.1460-9568.2010.07472.x. [DOI] [PubMed] [Google Scholar]
- Okawa H, Hoon M, Yoshimatsu T, Della Santina L, Wong RO. Illuminating the multifaceted roles of neurotransmission in shaping neuronal circuitry. Neuron. 2014;83:1303–1318. doi: 10.1016/j.neuron.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nature reviews Neuroscience. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Peng X, Parsons TD, Balice-Gordon RJ. Determinants of synaptic strength vary across an axon arbor. Journal of neurophysiology. 2012;107:2430–2441. doi: 10.1152/jn.00615.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieraut S, Gounko N, Sando R, 3rd, Dang W, Rebboah E, Panda S, Madisen L, Zeng H, Maximov A. Experience-dependent remodeling of basket cell networks in the dentate gyrus. Neuron. 2014;84:107–122. doi: 10.1016/j.neuron.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T, Kakegawa W, Shinoda Y, Hosono M, Katoh-Semba R, Sekine Y, Sato Y, Tanaka M, Iwasato T, Itohara S, et al. CAPS1 deficiency perturbs dense-core vesicle trafficking and Golgi structure and reduces presynaptic release probability in the mouse brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:17326–17334. doi: 10.1523/JNEUROSCI.2777-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T, Shinoda Y, Oka M, Sekine Y, Sato Y, Saruta C, Miwa H, Tanaka M, Itohara S, Furuichi T. Reduced axonal localization of a Caps2 splice variant impairs axonal release of BDNF and causes autistic-like behavior in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21104–21109. doi: 10.1073/pnas.1210055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Stein A, Jahn R, Neher E. Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science. 2005;309:491–494. doi: 10.1126/science.1112645. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Fernandez-Chacon R, Sudhof TC, Neher E. Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+ The Journal of general physiology. 2003a;122:265–276. doi: 10.1085/jgp.200308855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003b;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Su Q, Mochida S, Tian JH, Mehta R, Sheng ZH. SNAP-29: a general SNARE protein that inhibits SNARE disassembly and is implicated in synaptic transmission. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14038–14043. doi: 10.1073/pnas.251532398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Terashima A, Petralia RS, Wenthold RJ, Isaac JT, Roche KW, Roche PA. A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nature neuroscience. 2010;13:338–343. doi: 10.1038/nn.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CL, Zhang L, Zhou Y, Zhou J, Yang XJ, Duan SM, Xiong ZQ, Ding YQ. Activity-dependent development of callosal projections in the somatosensory cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:11334–11342. doi: 10.1523/JNEUROSCI.3380-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Brose N. Regulation of membrane fusion in synaptic excitation-secretion coupling: speed and accuracy matter. Neuron. 2007;55:11–24. doi: 10.1016/j.neuron.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Baumeister A, Binz T, Blasi J, Link E, Cornille F, Roques B, Fykse EM, Sudhof TC, Jahn R, et al. Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. The Journal of biological chemistry. 1994;269:12764–12772. [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O’Donnell S, Brown HE, Osborne J, Axel R, Gogos JA. Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron. 2004;42:553–566. doi: 10.1016/s0896-6273(04)00224-7. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, et al. V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron. 2008;60:84–96. doi: 10.1016/j.neuron.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.