

Abstract
Novel, single nucleotide mutations were identified in the mitochondrial methionyl amino-acyl-tRNA synthetase gene (MARS2) via whole exome sequencing in two affected siblings with developmental delay, poor growth, and sensorineural hearing loss. We show that compound heterozygous mutations c.550C>T:p.Gln184* and c.424C>T:p.Arg142Trp in MARS2 lead to decreased MARS2 protein levels in patient lymphoblasts. Analysis of respiratory complex (RC) enzyme activities in patient fibroblasts revealed decreased Complex I and IV activities. Immunoblotting of patient fibroblast and lymphoblast samples revealed reduced protein levels of NDUFB8 and COXII, representing Complex I and IV respectively. Additionally, overexpression of wild-type MARS2 in patient fibroblasts increased NDUFB8 and COXII protein levels. These findings suggest that recessive single nucleotide mutations in MARS2 are causative for a new mitochondrial translation deficiency disorder with a primary phenotype including developmental delay and hypotonia. Identification of additional patients with single nucleotide mutations in MARS2 is necessary to determine if pectus carinatum is also a consistent feature of this syndrome.
Keywords: MARS2, mitochondrial amino-acyl tRNA synthetase, mitochondrial translation
Mitochondrial amino-acyl-tRNA synthetases (mtARSs) are nuclear encoded proteins that are required for mitochondrial protein synthesis and thus the translation of specific oxidative phosphorylation (OXPHOS) system subunits. Each mtARS functions to catalyze a two-step reaction whereby an individual tRNA molecule is charged with its cognate amino acid. In recent years, multiple disease-causing mutations have been identified in several mtARSs and resulting clinical phenotypes are varied, although most of these conditions have included multi-system involvement (Bayat, et al., 2012; Belostotsky, et al., 2011; Edvardson, et al., 2007; Elo, et al., 2012; Gotz, et al., 2011; Pierce, et al., 2011; Pierce, et al., 2013; Riley, et al., 2010; Scheper, et al., 2007; Steenweg, et al., 2012). MARS2 (MIM# 609728), or methionyl tRNA synthetase 2, is a class Ia synthetase that covalently links methionine to mitochondrial tRNAmethionine (Spencer, et al., 2004). Complex copy number rearrangements in MARS2 have previously been noted to cause the neurodegenerative disease Autosomal Recessive Spastic Ataxia with Leukoencephalopathy (ARSAL or autosomal recessive spastic ataxia, type 3) (MIM# 611390) (Bayat, et al., 2012). All ARSAL patients display ataxia, spasticity, and hyperreflexia, while variable features include dysarthria, dysmetria, nystagmus, dystonia, scoliosis, urinary urgency, leukodystrophy, and cerebellar atrophy (Bayat, et al., 2012). In this study, we report two siblings presenting for genetics evaluation with history of developmental delay, growth failure, sensorineural hearing loss, and pectus carinatum who were found to have disease-causing compound heterozygous, single nucleotide mutations in MARS2. Our report is the first notation of single nucleotide changes in MARS2 resulting in human disease. In addition, the clinical features of these patients expand the phenotypic spectrum of disease associated with MARS2 mutations.
The proband (II.1) is a male born to healthy, non-consanguineous parents (Figure 1A). He was delivered at 32 weeks gestation via Cesarean section due to preeclampsia. Birth weight was 3 pounds (3–10th%) and birth length was 17 inches (50th%). Pectus carinatum was noted at birth. He was hospitalized for 2 months due to respiratory issues and poor weight gain. He passed his newborn hearing screen. Head ultrasound was notable for possible intraventricular hemorrhage. Early medical history was significant for poor weight gain and slow growth. A G-tube was placed at 4 years of age to maximize caloric intake and Nissen fundoplication was subsequently completed at 5 years of age due to multiple episodes of aspiration pneumonia. History is notable for chronic constipation and growth hormone deficiency with a peak value of 2.1 ng/mL of human growth hormone on Arginine/Glucagon stimulation test. He has slowly responded to growth hormone therapy. He also has bilateral moderate sensorineural hearing loss with a conductive component for which he wears hearing aids. There was a concern for seizures in the newborn period, but no specific information on type of seizures is available. At 5 years of age he has an abnormal EEG with mild background slowing and he is on oxcarbazepine. The proband has global developmental delay. He is currently 5 years and 6 months, can walk with a walker, is able to cruise independently, and has approximately 5 words used appropriately.
Figure 1.
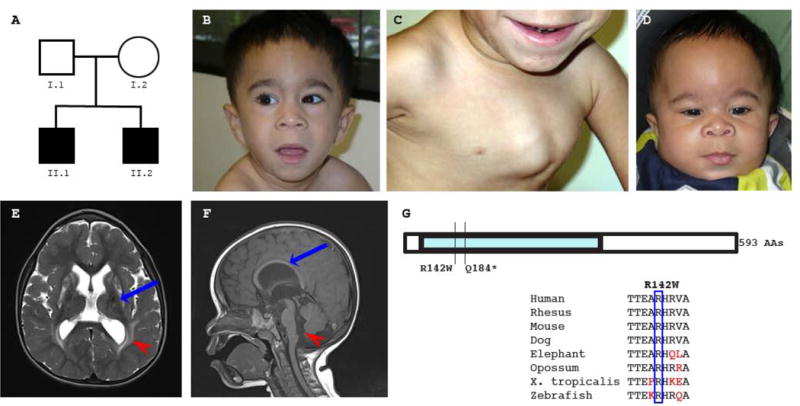
A. Family Pedigree. Squares denote males, circles denote females, and shaded symbols denote affected individuals.
B. Face photograph of affected sibling II.1. Dysmorphic features include a prominent forehead midline ridge, low-set ears, a short nose with a flat nasal bridge and broad nasal tip, and a long, smooth philtrum.
C. Chest photograph of affected sibling II.1 showing significant pectus carinatum.
D. Face photograph of affected sibling II.2. Dysmorphic features include a prominent forehead midline ridge, low-set ears, mild hypertelorism, a short nose with a broad and flat nasal bridge and anteverted nares, thick ala nasi, and a long philtrum.
E. Head MR imaging of affected sib II.1 (T2 TSE axial image). The red arrowhead indicates increased T2 signal within the periventricular white matter, which was far more prominent on other sections. Lamellae of the sagittal strata traverse the high signal zone. There is reduced size and abnormal, patchy increased T2 signal in the caudate and putamen bilaterally (blue arrow).
F. Head MR imaging of affected sib II.2 (T1 SE mid-sagittal image). The corpus callosum shows a highly-arched contour (blue arrow) and posterior thinning. The red arrowhead indicates the wide foramen of Magendie and vallecula.
G. MARS2 gene structure. MARS2 is a single exon gene that encodes a protein consisting of 593 amino acids. MARS2 amino acid R142 is conserved in a wide variety of species.
Prior imaging studies have included a normal echocardiogram and an unremarkable skeletal survey except for pectus carinatum. Recent head MRI was notable for prominent quadriventricular dilatation, but small triangular temporal horns. There was cerebral atrophy with marked bilateral symmetrical homogenous T2 signal increase throughout the cerebral white matter, completely sparing the subcortical “U” fibers, corpus callosum, and internal capsules. The corpus callosum was high-arched and thin posteriorly. The striatum showed symmetrical atrophy with high T2 signal in the caudate and putamen (Figure 1E, Supp. Table S1). Synechiae cross both lateral ventricles. The pons was slightly hypoplastic. The cerebellar vermis had a normal complement of vermian lobules, but did not approximate the posterior surface of the medulla, leaving a wide foramen of Magendie and vallecula. There was no evidence of calcification or current evidence of hemorrhage. Prior genetic and metabolic testing has included unremarkable chromosome analysis and SNP chromosomal microarray. Serum lactate levels have been occasionally mildly elevated (to 2.6 mmol/L). CPK level has been normal. Qualitative urine organic acid analysis revealed mild elevation in 3-methylglutaconic acid.
On recent exam at 5 ½ years of age, head circumference was 49 cm (10th%), weight was 15.85 kg (<1st%, 50th% for a 3.5 year old), and length was 86.5 cm (<1st%, 50th% for a 22 month old). Body mass index was 62nd%. Head appears large for body size. Forehead is notable for a prominent midline ridge. Eyes are without significant hypotelorism. Ears were low-set. The nose was short with a flat nasal bridge, a broad nasal tip, and thickened ala nasi. The philtrum was long and smooth (Figure 1B). Chest examination was notable for significant pectus carinatum (Figure 1C). Foot examination was notable for bilateral 2,3 syndactyly. He also had thin arms and legs and moderate generalized hypotonia.
The proband’s younger brother (II.2) presented with similar clinical features. He was born at 33 weeks gestation by Cesarean section due to preeclampsia. Birth weight was 3.5 pounds (25th%) and length was 15 inches (<3rd%). He was hospitalized for 6 weeks after delivery. He did not need significant ventilator support and no neurological issues were raised. A head MRI found larger than expected connatal cysts, but no other significant abnormalities. He had silent aspiration pneumonia and received Nissen fundoplication and G-tube placement prior to leaving the hospital. He failed his newborn hearing screen.
Currently at 2 years and 5 months of age, all feeds are via G-tube due to continuing aspiration. He has been diagnosed with growth hormone deficiency and is receiving growth hormone therapy. He has developed bilateral moderate sensorineural hearing loss with a conductive component and has hearing aids bilaterally. He has also developed progressive pectus carinatum. He has no history of seizures, but development is globally delayed. Recent head MRI has revealed quadriventricular dilatation, small triangular temporal horns, and symmetrical white matter changes highly similar to those of the older brother. Cerebral atrophy and a high-arched, posteriorly thin corpus callosum were noted (Figure 1F, Supp. Table S1). The basal ganglia and thalamus appeared normal. Mild pontine hypoplasia and changes of the cerebellar vermis were very similar to those described in the older sibling II.1 (Figure 1F).
On recent physical exam, head circumference is 47.5 cm (14th%), weight is 10.11 kg (<1st%, 50th% for 11 month old), and length is 74 cm (<1st%, 50th% for 11 month old). Dysmorphic features included low-set ears, mild hypertelorism, a short nose with a broad and flat nasal bridge and anteverted nares, thickened ala nasi, and a long philtrum (Figure 1D). There is marked pectus carinatum and mild to moderate generalized hypotonia.
In an effort to identify a possible causative genetic etiology for the clinical features of the affected siblings in this family, the family was enrolled in our Mount Sinai IRB-approved genetic research study. Written informed consent was obtained and all investigations were conducted in accordance with the principles of the Declaration of Helsinki.
DNA from persons II.1 and II.2 were processed for whole exome sequencing (WES) in the Mount Sinai Genomics Core Facility. Individual WES samples were barcoded and pooled with three other samples prior to enrichment with the Nimblegen SeqCap EZ Human Exome Library v3.0. Sequencing was performed on a HiSeq 2500 instrument (Illumina, San Diego, CA) with a 100 base-pair (bp) paired-end protocol. Alignment and variant calling was completed using an in-house GATK-based pipeline (Linderman, et al., 2014). 64,840,670 reads were generated for WES sample II.1 and 61,715,214 reads were generated for sample II.2. The targeted mean coverage for sample II.1 was 45.348 and 93.719% of the target had ≥10× coverage. The targeted mean coverage for sample II.2 was 43.365 and 93.643% of the target had ≥10× coverage.
Called variants were filtered with Ingenuity Variant Analysis (Qiagen Redwood City, http://www.ingenuity.com). 171,797 total variants in 17,696 genes were identified in samples II.1 and II.2. Variants were filtered based on confidence (call quality of ≥20, passed upstream pipeline filtering, and outside top 3% most exonically variable 100 base windows and/or 3% most exonically variable genes in healthy public genomes included); frequency (variants excluded if frequency was at least 0.5% in the 1000 Genomes Project or NHLBI ESP exomes); predicted deleteriousness (missense and not predicted tolerated by SIFT or PolyPhen-2, frame-shift, in-frame indel, or start/stop codon change, and likely splice site loss up to 2 bases into intron included); and genetic analysis (compound heterozygous, homozygous, or hemizygous variants in 2 of 2 case samples included). This filtering strategy resulted in the identification of 49 variants in 30 genes (Supp. Table S2). All of these variants were considered and knowledge of gene function was reviewed. An additional biological context filter was then also used; variants in genes known or predicted to affect genes implicated in the phenotype “neurological abnormality” were retained. This filtering strategy resulted in 14 variants in 9 genes requiring further investigation (Supp. Table S3).
Upon review of our list of variants and candidate genes, we noted two variants of interest in MARS2 (MIM# 609728), or methionyl t-RNA synthetase 2, a nuclear-encoded mitochondrial gene. The proband and his affected sibling have clinical features consistent with a mitochondrial disorder including neuromuscular abnormalities, multi-system involvement, failure to thrive, and involvement of hearing, endocrine, and gastrointestinal systems. In fact, although the family of the proband has declined muscle biopsy, he meets Mitochondrial Diagnostic Criteria (MDC) for a probable respiratory chain disorder based on clinical and neuroimaging information alone (Wolf and Smeitink, 2002). Both affected siblings II.1 and II.2 were compound heterozygous for MARS2 (RefSeq NM_138395.3) variants c.424C>T;p.Arg142Trp and c.550C>T:p.Gln184*. These variants have been submitted to the Leiden Open Variation Database (http://www.lovd.nl/MARS2). The MARS2 c.424C>T:p.Arg142Trp change has not been reported in dbSNP, 1000 Genomes Project, or NHLBI Exome Variant Server, and this missense change is predicted damaging by SIFT v1.03 (Sorting Intolerant From Tolerant, http://sift.jcvi.org/) with a score of 0.00 (SIFT scores ≤0.05 are predicted damaging). This variant is also predicted probably damaging by PolyPhen-2 v2.2.2.2r398 (http://genetics.bwh.harvard.edu/pph2/) with a HumVar score of 0.990. (Polyphen-2 HumVar scores range from 0 to 1 and a score is predicted probably damaging if it is ≥0.909, possibly damaging if it is ≤0.908 and ≥0.447, and benign if it is ≤ 0.446.) Additionally, this variant is noted to have a high functional impact score of 3.87 with MutationAssessor, Release 2 (http://www.MutationAssessor.org) and the corresponding scaled CADD score is 18.82 (Kircher, et al., 2014). The MARS2 c.550C>T variant results in a premature stop codon at amino acid 184 and is also not a known SNP in dbSNP, 1000 Genomes Project, or NHLBI Exome Variant Server. Sanger sequencing confirmed the genotypes of these variants in individuals II.1 and II.2. Additionally, parental samples were genotyped by Sanger sequencing and the father (I.1) was noted to be heterozygous for only the MARS2 c.550C>T:p.Gln184* change and the mother (I.2) was noted to be heterozygous for only the MARS2 c.424C>T:p.Arg142Trp change, consistent with an autosomal recessive mode of inheritance.
To evaluate if the identified variants in MARS2 may have a functional impact and to determine if the clinical symptoms may be due to mitochondrial pathology, we assessed respiratory chain enzyme activities. A skin biopsy was obtained from affected sibling II.2 and a fibroblast culture was established according to standard practices. We measured the enzyme activity of complexes I, II, III, IV, and V and citrate synthase spectrophotometrically in a mitochondrial-enriched fraction from individual II.2 fibroblasts as previously described (Rodenburg, 2011). Complex I enzyme activity was 116 mU/U CS (control range 163–599 mU/UCS), Complex II enzyme activity was 403 mU/U CS (control range 335–999 mU/UCS), Complex III enzyme activity was 669 mU/U CS (control range was 570–1383 mU/UCS), Complex IV enzyme activity was 188 (control range 288–954 mU/U CS), Complex V enzyme activity was 503 (control range 193–819 mU/U CS), and CS activity was 385 (control range 151–449 mU/mg). The decreased activities of complexes I and IV are consistent with a mitochondrial translation defect (Konovalova and Tyynismaa, 2013). Complex I, or NADH:ubiquinone oxidoreductase, is composed of 44 polypeptide chains, of which, seven are encoded by the mitochondrial genome (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6). ND4L contains 10.20% methionine, while the other mitochondrial encoded polypeptides have a methionine content of 4.31 to 6.96% (Bayat, et al., 2012). Complex IV, or cytochrome c oxidase, is composed of 14 polypeptides, including 3 polypeptides (Cox1, Cox2, and Cox3) that are synthesized in the mitochondria. These 3 polypeptides have methionine contents ranging from 4.21 to 6.24% (Bayat, et al., 2012). Additionally, to exclude variants in mitochondrial DNA as a cause of the mitochondrial pathology, commercial clinical mitochondrial genome sequencing and deletion analysis was completed, and no disease-causing mutation was identified.
To assess MARS2 and protein expression, we next performed immunoblot analysis. Fibroblast samples from individual II.2 and two control cell lines and lymphoblast samples from individuals I.1, I.2, II.1, and II.2 were utilized and processed as described (Houten, et al., 2014). The membrane was probed with mouse anti-OXPHOS cocktail at 1:1000 (MitoSciences), rabbit anti-MARS2 at 1:1000 (Sigma-Aldrich, HPA035589), and with rabbit anti-GAPDH at 1:2500 (Abnova) used as a loading control. Immunoblot analysis revealed decreased protein levels of NDUFB8 and COXII, which are selected subunits of Complex I and IV respectively, in fibroblasts from affected individual II.2 compared to control samples (Figure 2A). Additionally, in lymphoblasts decreased protein levels of NDUFB8 and COXII were noted in affected patients II.1 and II.2 compared to parental samples I.1 and I.2 (Figure 2B). MARS2 protein expression was also decreased in lymphoblast samples from affected siblings compared to parental samples (Figure 2B). MARS2 signal was low in fibroblast samples and expression could not be accurately quantified. To assess if overexpression of wild-type MARS2 could rescue the decreased protein levels of NDUFB8 and COXII in patient fibroblasts, we transfected patient II.2 fibroblasts with human MARS2 cDNA clone (Origene, RC224966) using the Lonza Nucleofector Kit for human dermal fibroblasts. MARS2 overexpression increased the protein levels of NDUFB8 and COXII (Figure 2C) further demonstrating causality of the MARS2 variants.
Figure 2.
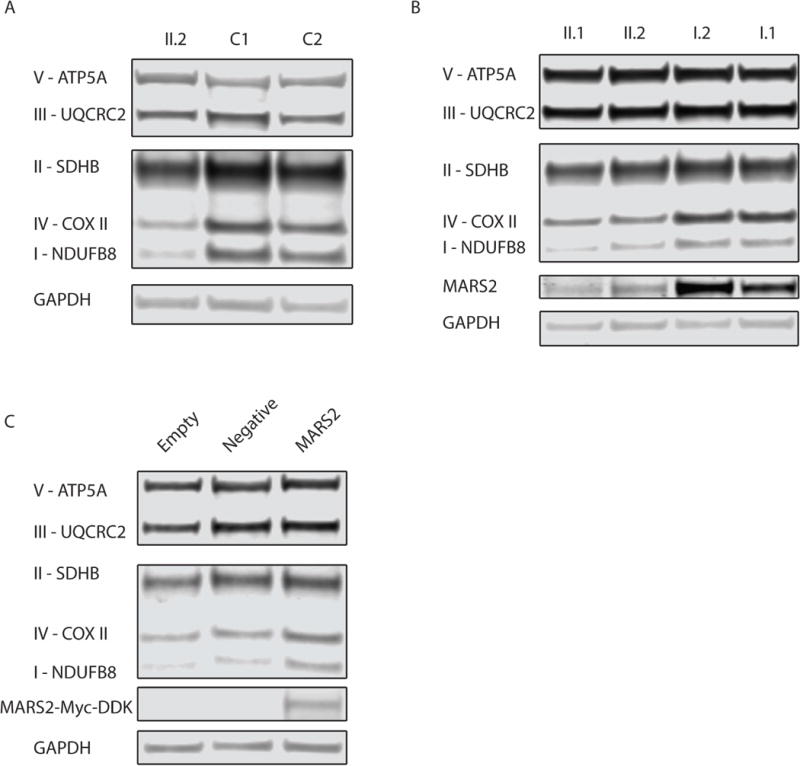
A. Western blot analysis to assess the levels of subunits of the 5 different oxidative phosphorylation complexes in fibroblast samples from affected sib II.2 compared to two controls. Protein content of NDUFB8 and COXII, which are subunits of Complex I and IV respectively, are decreased in II.2 compared to healthy, unrelated individuals. Samples were prepared in duplicate and the experiment was completed three times with consistent results. The signal intensity of NDUFB8 in patient fibroblasts compared to controls calculated after correcting for differences in protein loading by normalizing to GAPDH intensity was 14.1–16.0%. Similarly, the signal intensity for COXII in patient fibroblasts compared to controls was 18.8–19.3%.
B. Western blot analysis to assess the levels of subunits of the 5 different oxidative phosphorylation complexes and MARS2 expression in lymphoblast samples from affected siblings II.1 and II.2 and unaffected parents. Protein content of NDUFB8 and COXII, which are subunits of Complex I and IV respectively, are decreased in the affected siblings compared to parental samples. The signal intensity of NDUFB8 in patient II.1 lymphoblasts compared to parental samples computed after correcting for differences in protein loading by normalizing to GAPDH intensity was 42.1–55.6%. For patient II.2, the signal intensity of NDUFB8 in lymphoblasts compared to parental samples was 45.6–55.6%. Similarly, the signal intensity of COXII in patient II.1 lymphoblasts compared to parental samples was 41.7–43.5%. For patient II.2, the signal intensity of COXII in lymphoblasts compared to parental samples was 32.0–36.0%. Additionally, MARS2 expression is reduced in individuals II.1 and II.2. For patient II.1, the signal intensity of MARS2 in lymphoblasts after normalizing to GAPDH intensity ranges from 8.62–16.6% of parent I.2 and 17.6–30.1% of parent I.1. Similarly, for patient II.2, the signal intensity of MARS2 ranges from 16.5–20.3% of parent 1.2 and 33.7–36.7% of parent I.1. Samples were prepared in duplicate and the experiment was completed three times with consistent results.
C. Western blot analysis to assess the levels of subunits of the 5 different oxidative phosphorylation complexes in patient II.2 fibroblast samples after overexpression of MARS2 by transfection. 5 ug of human cDNA MARS2 ORF clone pCMV6-MARS2 or empty control vector pCDNA3 were transfected in patient II.2 fibroblasts and cells were harvested after 4 days. A negative control was also performed which included transfection without plasmid. A MARS2 antibody was used to visualize exogenous MARS2. A band was seen at ~64 kDa representing MARS2 with a C-terminal Myc-DDK tag in the MARS2 transfected samples. Four independent MARS2 transfections were completed in which 3 experiments produced an average 2.1 and 2.4-fold increase in NDUFB8 and COXII, respectively.
Here we report siblings with compound heterozygous single nucleotide variants in MARS2 presenting with a phenotype that is distinct from ARSAL and includes symptoms of global developmental delay, growth failure, hypotonia, sensorineural hearing loss, and pectus carinatum (Supp. Table S4). Additionally, symptoms in the presented siblings are apparent at birth and profound by late infancy. MARS2 mutations are expected to impact mitochondrial translation, which was confirmed by decreased activity of Complex I and IV in fibroblasts and decreased Complex I and IV protein in fibroblasts and lymphoblasts. These findings are consistent with the consequences of other gene defects impacting mitochondrial translation.
ARSAL is a recessive ataxia caused by recessive genomic rearrangements in MARS2. Age of diagnosis is variable and ranges from 2 to 59 years with an average of 15.0 years (Thiffault, et al., 2006). Patients with ARSAL are noted to have increased MARS2 mRNA levels, reduced MARS2 protein expression, and reduced Complex I activity in lymphoblasts. In contrast to the affected siblings presented here, Complex IV activity and Complex IV protein levels are normal in patients with ARSAL (Bayat, et al., 2012).
Similar to the published MR images of ARSAL patients, the affected siblings we present here are also noted to have bilateral, symmetrical increased T2 signal in the cerebral white matter (Thiffault, et al., 2006). However, the signal abnormalities are much more severe and confluent in the affected siblings we present here with recessive, single nucleotide mutations in MARS2. The striatal changes and ventricular synechiae seen in the older sibling (II.1) may be related to intraventricular hemorrhage as a neonate rather than intrinsic disease. The alternative possibility is that these changes may be age-related.
MRI findings commonly seen in mitochondrial respiratory chain disorders include abnormalities of the five brain areas: basal ganglia (hyperintensities on T2 imaging or calcifications), cerebellum (hyperintensities on T2 imaging or atrophy), brainstem (hyperintensities on T2 imaging or atrophy), white matter changes (leukoencephalopathy), and cortex (supratentorial atrophy). However, the diverse respiratory chain disorders display variable involvement of these areas. For example, Kearns-Sayre syndrome may have involvement of all five areas, while ubiquinone deficiency is associated with involvement of only 1 of the 5 areas (the cerebellum). Disorders caused by mutations in RARS2, FARS2, DARS2, or EARS2, may have involvement of between two and four areas (Bricout, et al., 2014). In the affected siblings with recessive single nucleotide variants in MARS2, we note similar involvement within three of these five brain areas as well as a differing form of abnormality in the cerebellum.
Aats-met is the homolog for MARS2 in Drosphila with 44% identity and 75% similarity. HV and FB Drosophila mutants identified via forward genetic screen for aberrant electroretinograms are partial loss of function mutants of Aats-met. The HV mutant is homozgyous for the Aats-met c.125T>A, p.V42D change and the FB mutant is homozygous for the c.671C>T, p.S224L change. Analysis of HV/FB flies by transmission electron microscopy revealed progressive loss of photoreceptors and degeneration of glia with lipid droplet accumulation. HV/FB and HV/HV flies appear morphologically normal, but have decreased life spans and inability to fly. Assessment of fly myofibrils by transmission electron microscopy revealed aberrant mitochondria and fragmented muscle. Enzyme activities of HV/Df and FB/Df flies revealed decreased Complex I activity. In contrast to the affected siblings presented here, the mutant flies were noted to have increased Citrate Synthase activity and normal Complex IV activity (Bayat, et al., 2012). The affected siblings presented here report no vision complaints, and therefore, electroretinogram has not been pursued. Interestingly, because aberrant Complex I activity may result in increased levels of reactive oxygen species (ROS), HV Drosophila mutants were supplemented with the antioxidant Vitamin E, which resulted in improvement in eye morphology and size (Bayat, et al., 2012). Antioxidant therapy may represent a potential treatment for these affected siblings, but further investigation is required.
Interestingly, both affected siblings in this family were noted to have profound pectus carinatum, a symptom that has not been associated with mitochondrial disease. Identification of additional persons affected with autosomal recessive MARS2 single nucleotide mutations is necessary to determine if pectus carinatum is part of this MARS2 clinical syndrome. An alternative explanation for the presence of pectus carinatum in these affected siblings is that a second genetic change may be causative of this symptom. Our whole exome sequencing results did not reveal good candidate genes for this scenario.
In conclusion, this is the first report of pathogenic single nucleotide mutations in MARS2. We have identified a new clinical disorder presenting with features of developmental delay, growth failure, and sensorineural hearing loss. Identification of additional affected individuals is necessary to determine if pectus carinatum is also a clinical feature of this condition.
Supplementary Material
Acknowledgments
We are grateful to the family for participation in this research study. We thank Carrie Crain, MS, CGC for her help with referral of this family to our study. Whole exome sequencing was completed at the Genomics Core Facility at the Icahn School of Medicine at Mount Sinai. Dr. Webb is supported by NIH T32 GM082773. Funding for this study was provided by the Icahn Institute for Genomics and Multi-Scale Biology. This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Contract Grant Sponsors: NIH (T32 GM082773)
Footnotes
Disclosure statement: The authors declare no conflict of interest.
References
- Bayat V, Thiffault I, Jaiswal M, Tetreault M, Donti T, Sasarman F, Bernard G, Demers-Lamarche J, Dicaire MJ, Mathieu J, Vanasse M, Bouchard JP, et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10(3):e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, Feinstein S, Zeligson S, Segel R, Elpeleg O, Nassar S, Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88(2):193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricout M, Grevent D, Lebre AS, Rio M, Desguerre I, DeLonlay P, Valayannopoulos V, Brunelle F, Rotig A, Munnich A, Boddaert N. Brain imaging in mitochondrial chain deficiency: combination of brain MRI features as a useful tool for genotype/phenotype correlations. J Med Genet. 2014;51(7):429–435. doi: 10.1136/jmedgenet-2013-102256. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, Saada A, Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81(4):857–62. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elo JM, Yadavalli SS, Euro L, Isohanni P, Gotz A, Carroll CJ, Valanne L, Alkuraya FS, Uusimaa J, Paetau A, Caruso EM, Pihko H, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet. 2012;21(20):4521–9. doi: 10.1093/hmg/dds294. [DOI] [PubMed] [Google Scholar]
- Gotz A, Tyynismaa H, Euro L, Ellonen P, Hyotylainen T, Ojala T, Hamalainen RH, Tommiska J, Raivio T, Oresic M, Karikoski R, Tammela O, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88(5):635–42. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Denis S, Te Brinke H, Jongejan A, van Kampen AH, Bradley EJ, Baas F, Hennekam RC, Millington DS, Young SP, Frazier DM, Gucsavas-Calikoglu M, et al. Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia. Hum Mol Genet. 2014;23(18):5009–16. doi: 10.1093/hmg/ddu218. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konovalova S, Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab. 2013;108(4):206–11. doi: 10.1016/j.ymgme.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Linderman MD, Brandt T, Edelmann L, Jabado O, Kasai Y, Kornreich R, Mahajan M, Shah H, Kasarskis A, Schadt EE. Analytical validation of whole exome and whole genome sequencing for clinical applications. BMC Med Genomics. 2014;7:20. doi: 10.1186/1755-8794-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, Opitz JM, Li W, Klevit RE, King MC. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A. 2011;108(16):6543–8. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, Malach D, Klevit RE, King MC, Levy-Lahad E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet. 2013;92(4):614–20. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley LG, Cooper S, Hickey P, Rudinger-Thirion J, McKenzie M, Compton A, Lim SC, Thorburn D, Ryan MT, Giege R, Bahlo M, Christodoulou J. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet. 2010;87(1):52–9. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenburg RJ. Biochemical diagnosis of mitochondrial disorders. J Inherit Metab Dis. 2011;34(2):283–92. doi: 10.1007/s10545-010-9081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, Muravina TI, Serkov SV, Uziel G, Bugiani M, Schiffmann R, Krageloh-Mann I, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39(4):534–9. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- Spencer AC, Heck A, Takeuchi N, Watanabe K, Spremulli LL. Characterization of the human mitochondrial methionyl-tRNA synthetase. Biochemistry. 2004;43(30):9743–54. doi: 10.1021/bi049639w. [DOI] [PubMed] [Google Scholar]
- Steenweg ME, Ghezzi D, Haack T, Abbink TE, Martinelli D, van Berkel CG, Bley A, Diogo L, Grillo E, Te Water Naude J, Strom TM, Bertini E, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135(Pt 5):1387–94. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- Thiffault I, Rioux MF, Tetreault M, Jarry J, Loiselle L, Poirier J, Gros-Louis F, Mathieu J, Vanasse M, Rouleau GA, Bouchard JP, Lesage J, et al. A new autosomal recessive spastic ataxia associated with frequent white matter changes maps to 2q33–34. Brain. 2006;129(Pt 9):2332–40. doi: 10.1093/brain/awl110. [DOI] [PubMed] [Google Scholar]
- Wolf NI, Smeitink JA. Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology. 2002;59(9):1402–5. doi: 10.1212/01.wnl.0000031795.91814.d8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.