

Abstract
Acthar gel is indicated for the treatment of acute exacerbations of multiple sclerosis (MS) in adults. Its effects on immune cells during a relapse are unknown. This study investigated the effects of Acthar in an animal model of relapsing-remitting MS, using SJL/J mice sensitized with myelin peptide. All animal studies were reviewed and approved by the University of Utah Institutional Animal Care and Use Committee and conducted in accordance with the guidelines prepared by the Committee on Care and Use of Laboratory Animals, Institute of Laboratory Animals Resources, National Research Council. Mice injected with Acthar to treat the second attack had a significantly lower mean clinical score during relapse and a significantly reduced cumulative disease burden compared to Placebo gel-treated mice. Furthermore, Acthar treatment ameliorated inflammation/demyelination in the spinal cord and markedly suppressed ex vivo myelin peptide-induced CD4+ T cell proliferation.
Keywords: ACTH, demyelination, experimental autoimmune encephalomyelitis, inflammation, T cell
Introduction
H.P. Acthar Gel® (Acthar), a highly purified repository gel preparation of porcine adrenocorticotropic hormone [ACTH(1-39)], is an FDA-approved therapeutic used to treat acute multiple sclerosis (MS) exacerbations in adults [1–3]. ACTH(1-39) is derived from the cleavage of pro-opiomelanocortin along with other melanocortin peptides, including α-melanocyte-stimulating hormone (α-MSH), β-MSH and γ-MSH [4]. ACTH(1-39) binds to all five melanocortin receptor (MCR) subtypes that are differentially expressed on multiple immune and non-immune cell populations [4]. Therefore, it has been proposed that Acthar inhibits immunologic diseases as a result of its principal active component, porcine ACTH(1-39), acting at MCRs expressed on leukocytes (reviewed in [5]). However, the effect of Acthar on immune cells and resident central nervous system (CNS) glial cells (microglia and astrocytes) during a clinical relapse in MS has not been elucidated.
Experimental autoimmune encephalomyelitis (EAE) is an animal model that is used to test the efficacy of new potential therapies for MS (reviewed in [6,7]). For example, glatiramer acetate (copaxone), mitoxantrone and natalizumab were discovered using EAE models (reviewed in [8,9]). A study using EAE in guinea pigs showed that a purified hormone preparation of ACTH inhibited EAE [10,11]. In addition, Acthar and peptides [α-MSH(1-13)] derived from ACTH were able to inhibit EAE in myelin oligodendrocyte glyco-protein (MOG)-induced EAE when administered orally starting prior to EAE induction [12,13]. However, these studies have not demonstrated the efficacy of ACTH when administered after the onset of EAE or determined the effect of ACTH on glial cells.
One of the murine EAE models used to test MS therapeutics is the SJL/J mouse sensitized with synthetic myelin proteolipid protein 139-151 peptide (PLP139-151) in adjuvant. These mice develop a relapsing-remitting EAE (RR-EAE), which is the most common disease course in patients with MS [14]. Furthermore, this EAE model is mediated by CD4+ T cells. These myelin-specific CD4+ T helper (TH) cells are activated by the major histocompatibility complex (MHC) class II (IAs) molecules presenting PLP139-151 peptide on the surface of antigen-presenting cells, such as monocytes and dendritic cells. Engagement of the T cell receptor and peptide-MHC complex leads to the activation of these myelin-specific CD4+ T cells, which then proliferate and secrete proinflammatory cytokines [15].
The present studies investigated the ability of Acthar to inhibit RR-EAE in a blinded manner using a Placebo gel control. More specifically, Acthar or Placebo gel were each injected subcutaneously at the onset of an EAE exacerbation (2nd attack, equivalent to the first exacerbation) or during an exacerbation (day 5 after onset of 2nd attack) in separate studies. Histological examination of spinal cords was performed to determine the extent of pathological changes that were inhibited in the CNS due to each treatment. Functional assays of T cell responses to myelin antigens ex vivo were performed to determine if Acthar was affecting myelin-specific CD4+ T cells leading to suppression of EAE. Lastly, immunophenotyping by flow cytometry was used to quantify the effect of Acthar treatment on immune cells implicated in EAE pathogenesis.
Methods
Experimental design
All members of the Fujinami lab were blinded for the entire duration of the experiments and analyses of data (flow cytometry data analysis and histology scoring) presented in this study. The program SigmaPlot (Systat Software, Inc., Chicago, IL) was used for all statistical analyses performed. The t test was performed for pairwise comparisons between each treatment group and the no treatment group. Analysis of variance (ANOVA), followed when necessary by the appropriate post hoc test, was performed to determine group differences.
Animal experiments
All animal studies were reviewed and approved by the University of Utah Institutional Animal Care and Use Committee and conducted in accordance with the guidelines prepared by the Committee on Care and Use of Laboratory Animals, Institute of Laboratory Animals Resources, National Research Council. SJL/J female mice (Jackson Laboratory, Bar Harbor, ME) were sensitized at 4–6 weeks of age [16]. Briefly, mice were injected subcutaneously in the flanks with 200 μL of 1 mM PLP139–151 peptide (DNA/Peptide Synthesis Core Facility, University of Utah) emulsified with reconstituted complete Freund’s adjuvant (CFA), composed of Freund’s incomplete adjuvant (Pierce Biotechnology, Rockford, IL) containing Mycobacterium tuberculosis H37 Ra (2 mg/mL) (Difco Laboratories, Detroit, MI). Mice were intravenously (i.v.) injected with 100 μL Bordetella pertussis (BP) toxin (List Biological Laboratories, Inc., Campbell, CA) at 0.2 μg per mouse on days 0 and 2 following sensitization. Mice developed a relapsing-remitting clinical course (RR-EAE). Mice were weighed and scored daily for clinical signs. Clinical scoring was as follows: 0, no clinical disease; 1, loss of tail tonicity; 2, presents with mild hind leg paralysis with no obvious gait disturbance; 3, mild leg paralysis with gait disturbance and paralysis; 4, hind limbs are paralyzed; and 5, moribund or dead.
Treatments
Acthar gel and Placebo gel formulations were provided by Mallinckrodt Pharmaceuticals (formerly Questcor Pharmaceuticals), Hayward, CA. Acthar is a highly purified preparation of adrenocorticotropic hormone [ACTH(1-39)] derived from porcine pituitary and formulated into a repository gel for prolonged release. Acthar was supplied at 80 U/ mL, and based on its use in another inflammatory animal model [17], each animal was injected subcutaneously with 240 U/kg per mouse every other day at the indicated time during two separate EAE studies. Treatment in the first study was initiated immediately upon onset of relapse, and treatment in the second study was initiated 5 days after onset of relapse (mice had a clinical score of ≥1 for 2 consecutive days).
Histology
Mice were euthanized and perfused with phosphate-buffered saline (PBS), followed by 4% paraformaldehyde phosphate-buffered solution. Spinal cords were harvested, divided into 12 transverse portions, embedded in paraffin and cut into 4 μm thick tissue sections. To visualize myelin, sections were stained with Luxol fast blue. For scoring of inflammation (perivascular cuffing) and demyelination in spinal cord sections, each spinal cord segment was divided into four quadrants: the anterior and posterior funiculi, and each lateral funiculus. Any quadrant containing perivascular cuffing or demyelination was given a score of 1 in that pathologic class. The total number of positive quadrants for each pathologic class was determined, then divided by the total number of quadrants present on the slide and multiplied by 100 to give the percent involvement for each pathologic score.
Astrocytes were detected on paraffin sections using glial fibrillary acidic protein (GFAP) antibody (DAKO Corp., Carpinteria, CA) [18]. The slides were labeled using the avidin-biotin peroxidase complex technique with 3,3′-diami-nobenzidine tetrahydrochloride (Sigma, St. Louis, MO) in 0.01% hydrogen peroxide (Sigma) in PBS. Gliosis was semi-quantitatively assessed by scoring GFAP+-activated astrocytes in the spinal cords of EAE mice. The spinal cords were divided into four segments (dorsal, lateral funiculi and ventral). Both the gray and white matters were scored. Gliosis was given a graded score as follows: score 0, no damage (<50 activated astrocytes present); score 1, mild (50–350 activated astrocytes present); score 2, moderate (351–700 astrocytes present); and score 3, severe (>700 activated astrocytes present) [18]. A score was given for each of the four areas of the spinal cord and then summed for each tissue section. For each mouse, we scored segments from each area of the spinal cord, approximately six tissue sections per mouse, and the scores were averaged.
Ricinus communis agglutinin (RCA)-I lectin histochemistry was used to detect activated microglia and macrophages [18]. Tissue sections were stained with biotinylated RCA-I. The slides were then labeled using the avidin-biotin peroxidase complex technique with 3,3′-diaminobenzidine tetra-hydrochloride in 0.01% hydrogen peroxide in PBS. RCA-I+ cells were enumerated over the entire spinal cord in more than six spinal cord tissue sections per mouse and averaged.
T cell proliferation assays
Spleens were collected from mice prior to fixation. The spleens were mechanically disrupted, cells were isolated over a Percoll gradient, and proliferation assays (3H-thymidine uptake assays) were performed using the isolated leukocytes at the indicated times post-sensitization [16,19]. Briefly, isolated spleen cells were suspended at 2 × 107 cells/mL in complete media [RPMI-1640 media (Mediatech, Manassas, VA) supplemented with 1% L-glutamine (Mediatech), 1% antibiotics (Mediatech), 50 μM 2-mercaptoethanol (Sigma) and 10% Cosmic calf serum (Hyclone, Logan, UT)]. PLP139-151 in 100 μL of complete media was added into culture in a dose-dependent manner. Cells were incubated at 37 °C, 5% CO2 for 72 h in the presence of the indicated peptide doses. 16–18 h prior to harvesting cultures, the cells were pulsed with 1 μCi/well of tritiated thymidine (3H-TdR) (PerkinElmer, Boston, MA). The cells were harvested onto glass fiber filters (PerkinElmer) for measurement of radiolabel incorporation using a liquid scintillation counter (PerkinElmer).
Spinal cord leukocyte phenotyping
Mouse spinal cords were obtained at the peak of the 2nd attack, 5 days after onset of treatment. Spinal cords were mechanically disrupted in Hibernate A (Life Technologies, Grand Island, NY) using a stainless steel sieve, which was followed by enzymatic dissociation in 1 mL of Trypsin LE (Life Technologies) for 30 min at 37 °C. Cells were washed and resuspended in 10 mL of Percoll of ρ = 1.03 which was underlain in Percoll of ρ = 1.095 and centrifuged for 30 min at 500μg and 4 °C with slow acceleration and no brake. The top layer containing myelin was discarded, and leukocytes were collected from the interface between ρ = 1.03 and ρ = 1.095. After washing, cells were treated with Fc block (BD Bioscience, San Jose, CA), stained with the indicated anti-mouse antibodies [anti-CD45-v500 and 7-AAD (obtained from BD Bioscience), anti-CD3e-FITC, anti-CD4-PE, anti-CD19-efluor450 and anti-CD11b-APC (all obtained from eBioscience, San Diego, CA)] for 30 min at 4 °C, and analyzed on a BD FACS Canto II flow cytometer. Spinal cord-derived cells were stained and analyzed by pooling two to three mouse spinal cord cells per tube. Gating was determined by fluorescence-minus-one (FMO) with isotype-matched immunoglobulin control. Flow cytometry data analysis was performed using FlowJo software (Tree Star, Inc., Ashland, OR).
Results
Acthar significantly dampens RR-EAE
Since the majority, if not all, of the individuals who seek treatment for MS have already experienced clinical symptoms of the disease, we first tested if Acthar was able to ameliorate PLP139-151-induced EAE after the first and immediately upon onset of the 2nd EAE attack (Figure 1A, day 25). At the start of EAE-relapse, mice were treated with either Acthar or Placebo gel and compared to no treatment (control). We monitored clinical score and weight of sensitized mice as biological markers related to the general health of the mice. The mice treated with Acthar had a significantly lower mean clinical score on days 35 through 46 (Figure 1A) and a significantly lower peak clinical score over the course of the relapse (Table 1), when compared to both Placebo gel treatment and no treatment groups. The cumulative disease index was used to determine the overall effect of Acthar during the entire 2nd attack. The cumulative disease index for the Acthar-treated mice (9.69 ± 6.05) was significantly lower in comparison to Placebo-treated mice (23.59 ± 5.8) and untreated mice (30.39 ± 5.81) (Table 1). Furthermore, Acthar was able to improve the general health of the sensitized mice that were undergoing EAE-relapse, as indicated by little or no weight loss in comparison to the control mice (data not shown).
Figure 1.

Acthar ameliorates EAE. SJL/J female mice were immunized with PLP139-151 peptide in CFA. (A) Upon relapse, at day 24 (start of 2nd attack, break in line), the mice were grouped by clinical score and treated with Acthar or Placebo gel (starting on day 25) every other day or untreated between days 25 and 46. (B) Upon relapse (start of 2nd attack on day 26), with a clinical score of ≥1 for 2 consecutive days, day 30 (break in line), the mice were treated with Acthar or Placebo gel (starting on day 31) every other day or untreated between days 31 and 43. Data represent mean clinical score ± standard error of the mean (SEM). Analysis of clinical scores can be found in Tables 1 and 2. *p<0.05, t test.
Table 1.
Effect of Acthar treatment, initiated upon onset of relapse, on EAE clinical disease in comparison to Placebo and no treatment in SJL/J female mice.
| Treatment | n | Mean day of onset | Peak clinical score on relapsea | Cumulative disease indexb |
|---|---|---|---|---|
| No treatment | 14 | 10.5 ± 0.17 | 3.25 ± 0.30 | 30.39 ± 5.81 |
| Acthar | 13 | 11.15 ± 0.15 | *1.85 ± 0.37 | *9.69 ± 6.05 |
| Placebo gel | 11 | 10.27 ± 0.27 | 3.27 ± 0.30 | 23.59 ± 5.80 |
Mice were immunized with PLP139-151 and treated every other day.
Peak clinical score = the peak score for each mouse for all mice within a group over the entire 2nd attack were averaged.
Cumulative disease index = all of the scores for all of the mice within a group over the entire 2nd attack were averaged. Data represent the mean ± standard error of the mean (SEM).
p<0.05, t test.
To further test Acthar’s in vivo effect on RR-EAE, in separate groups of animals, we started treatment of mice after the clinical EAE-relapse was already in progress (mice had a clinical score of μ1 for 2 consecutive days, which occurred on day 5 after onset of the 2nd attack) (Figure 1B, day 31). We again monitored clinical score and weight change. All groups of mice showed little weight change over the course of the relapse (data not shown). Comparison of the Acthar-treated mice versus the no treatment mice demonstrated that EAE mice treated with Acthar had a significantly lower mean clinical score for days 33 through 40 (Figure 1B) and a significantly lower peak clinical score over the course of the relapse (Table 2). The cumulative disease index for the Acthar-treated mice (7.78 ± 2.1) was significantly lower in comparison to Placebo-treated mice (16.72 ± 2.79) and untreated mice (16.65 ± 3.33) (Table 2). Therefore, Acthar treatment led to a significant reduction in clinical score regardless of whether Acthar was administered starting at the onset of relapse or while the relapse was in progress (clinical score ≥1).
Table 2.
Effect of Acthar treatment, initiated following onset of relapse, on EAE clinical disease in comparison to Placebo and no treatment in SJL/J female mice.
| Treatment | n | Clinical score day before treatment (day 30) | Clinical score on treatment day 1 (day 31) | Peak clinical score on relapsea | Cumulative disease indexb |
|---|---|---|---|---|---|
| No treatment | 17 | 0.76 ± 0.18 | 1.24 ± 0.18 | 2.29 ± 0.27 | 16.65 ± 3.33 |
| Acthar | 16 | 1.09 ± 0.06 | 1.06 ± 0.06 | *1.44 ± 0.22 | *7.78 ± 2.10 |
| Placebo gel | 16 | 1.53 ± 0.22 | 1.69 ± 0.22 | 2.41 ± 0.30 | 16.72 ± 2.79 |
Mice were immunized with PLP139-151 and treated every other day.
Peak clinical score = the peak score for each mouse for all mice within a group over the entire 2nd attack were averaged.
Cumulative disease index = all of the scores for all of the mice within a group over the entire 2nd attack were averaged. Data represent the mean ± SEM.
p<0.05, t test.
Acthar ameliorates inflammation and demyelination
Histological examination of the spinal cords from these mice was performed to determine the amounts of both inflammation (perivascular cuffing) and demyelination, compared to the untreated mice. Pathology was scored in the spinal cord tissue sections from groups of mice in which treatment was initiated at two different time points, i.e. at or after the onset of clinical relapse (as shown in Figure 1). Untreated RR-EAE mice and Placebo gel-treated RR-EAE mice showed extensive infiltration of inflammatory cells (perivascular cuffing as indicated by arrows) and demyelination (arrowhead) in comparison to Acthar-treated RR-EAE and naïve mice (Figure 2A). The Acthar-treated mice had statistically significantly lower pathology scores (p<0.05, t test) for both perivascular cuffing and demyelination, compared to Placebo gel-treated mice and untreated RR-EAE mice, when Acthar was administered at the onset of relapse (Figure 2B) and while the relapse was in progress (Figure 2C). Taken together, these results demonstrate that Acthar is able to limit inflammation and demyelination during clinical relapse in this experimental model.
Figure 2.
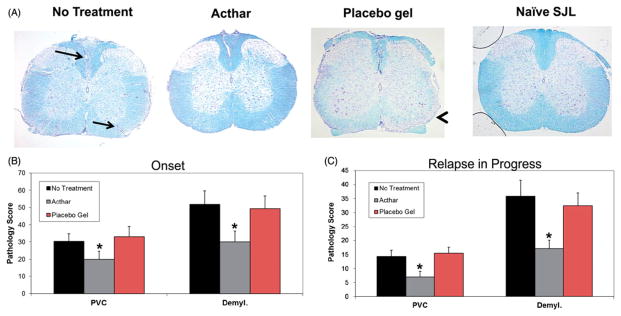
Acthar treatment protects EAE mice from inflammation and demyelination. (A) Representative Luxol fast blue-stained sections of mouse spinal cords. Arrows perivascular cuffs; Arrowhead: demyelination. Images shown at 4× magnification. (B) Pathology scoring of spinal cord tissue sections obtained from mice in which treatment was initiated at the onset of relapse (n = 15 for no treatment, 12 for Acthar treatment, 10 for Placebo gel treatment). (C) Pathology scoring of spinal cord tissue sections obtained from mice in which treatment was initiated after the onset of relapse (n = 16 for all groups of mice). Pathology scoring was performed as described in the “Methods” section. Values represent the mean ± SEM (at least six spinal cord tissue sections were scored for each mouse). PVC perivascular cuffing; Demyl. demyelination. *p<0.05, t test.
Acthar suppresses ex vivo PLP139-151-specific T cell responses
We sought to determine if Acthar had suppressive effects on PLP139-151-specific CD4+ T cells obtained directly from treated RR-EAE animals. Spleens were collected from mice in which Acthar treatment was initiated at the onset of relapse and after the onset of relapse. Mononuclear cells were isolated from the spleens of these mice at the indicated days and stimulated ex vivo with PLP139-151 in a dose-dependent manner (Figure 3). T cell proliferative assays showed that lymphocytes from the Acthar-treated mice had a markedly lower PLP139-151 recall response in comparison to T cells from control mice regardless of whether Acthar was administered at the onset of relapse (Figure 3A) or while the relapse was in progress (Figure 3B). Mouse spleen cells were pooled for proliferation assays, therefore statistics were not performed.
Figure 3.
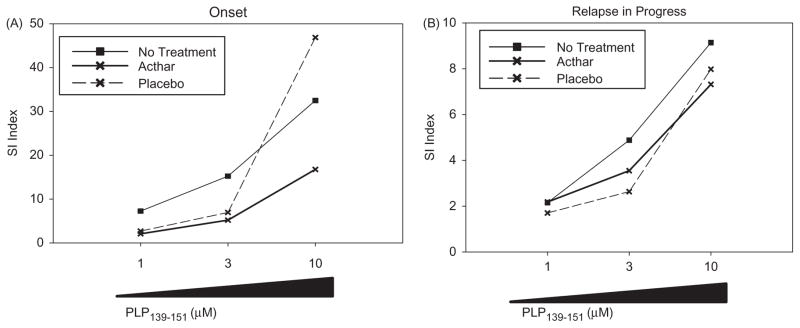
Acthar suppresses recall responses to PLP139-151 peptide ex vivo. SJL/J mice were immunized with PLP139-151 for the induction of EAE, as described in the “Methods” section. (A) Spleen cells were obtained on day 49 from mice in which treatment was initiated at the onset of relapse. (B). Spleens were obtained on day 43 from mice in which treatment was initiated after the onset of relapse. Cells were cultured in vitro with increasing concentrations of PLP139-151 (1, 3 and 10 μM) or medium alone. The proliferative response was measured by 3H-thymidine incorporation for the last 16–18 h of a 72-h assay. The stimulation index (SI) was used for proliferation analysis, in which the mean of the experimental data was divided by the mean of the background for each culture condition. Mouse spleen cells were pooled for proliferation assays, therefore statistics were not performed.
Acthar reduces gliosis
Glia cells (astrocytes and microglia) have been implicated in the initiation of EAE and MS. More specifically, evidence suggests that astrocytes play a role in MS pathogenesis (reviewed in [20]). Astrocytes are identified based on their morphology and expression of GFAP [21]. Therefore, to determine if Acthar was able to inhibit astrocyte activation in EAE mice, spinal cord tissue sections from mice receiving Acthar or Placebo gel treatment, either at the onset of relapse or after the onset of relapse, or untreated RR-EAE mice were stained and scored for GFAP+ cells (Figure 4A). The pathology scoring from mice in which treatment was initiated at the onset of relapse (Figure 4B) or after the onset of relapse (Figure 4C) demonstrated that although the Placebo gel-treated mice had a significantly lower GFAP score in the spinal cords in comparison to untreated mice, the Acthar-treated mice had a significantly lower GFAP score, compared to both Placebo gel-treated and untreated mice (Figure 4B and C), suggesting that Acthar treatment dampens astrocyte activation in vivo.
Figure 4.

Acthar dampens astrogliosis in the spinal cords of EAE mice. (A) Representative spinal cord tissue sections stained for GFAP, as described in the “Methods” section. Images shown at 20× magnification and insets at 10× magnification. (B) Quantification of GFAP scoring (described in the “Methods” section) of spinal cord tissue sections obtained from mice in which treatment was initiated at the onset of relapse (n = 15 for no treatment, 12 for Acthar treatment and 10 for Placebo gel treatment). *p<0.05, one-way ANOVA followed by Dunn’s test. (C) Quantification of GFAP scoring of spinal cord sections obtained from mice in which treatment was initiated after the onset of relapse (n = 16 for all groups of mice). **p<0.01, ANOVA followed by Holm–Sidak test. Values represent the mean ± SEM (at least six spinal cord tissue sections were scored for each mouse). NT no treatment.
The ability of Acthar to inhibit microglial activation and infiltration of activated macrophages in EAE mice was assayed using RCA-1 staining of spinal cord tissue sections from mice receiving Acthar or Placebo gel treatment, either at the onset of relapse or after the onset of relapse, or untreated mice (Figure 5A). Acthar-treated mice had significantly fewer numbers of RCA-1+ cells in the spinal cords in comparison to untreated and Placebo gel-treated mice whether the treatment was initiated at the onset of relapse (Figure 5B) or after the onset of relapse (Figure 5C). In addition, the Placebo gel-treated mice had significantly fewer RCA-1+ cells in the spinal cords in comparison to untreated RR-EAE mice when the treatment was initiated after the onset of relapse (Figure 5C). Therefore, Acthar treatment reduced the numbers of microglia and macrophages in the spinal cords of EAE mice as determined by RCA-1 staining and quantification. Taken together, these results demonstrate that Acthar treatment dampens glial cell activation in the spinal cords of EAE mice.
Figure 5.

Acthar reduces the numbers of activated microglia/macrophages in the spinal cords of EAE mice. (A) Representative spinal cord tissue sections stained for RCA-1, as described in the “Methods” section. Images shown at 20× magnification and insets at 10× magnification. (B) Enumeration of RCA-1+ cells from spinal cord tissue sections obtained from mice in which treatment was initiated at the onset of relapse (n = 15 for no treatment, 12 for Acthar treatment and 8 for Placebo gel treatment). **p<0.01, ANOVA followed by Dunn’s test. (C) Enumeration of RCA-1+ cells from spinal cord tissue sections obtained from mice in which treatment was initiated after the onset of relapse (n = 16 for all groups of mice). **p<0.01, ANOVA followed by Tukey’s test. Values represent the mean ± SEM (at least six spinal cord tissue sections were scored for each mouse). NT no treatment.
Phenotypic analysis of immune cells infiltrating the spinal cords of EAE mice
To determine if Acthar treatment altered the distribution of mononuclear or resident microglial cells in the spinal cords of EAE mice, cells were isolated from mouse spinal cords, stained with antibodies specific for T and B cells, and microglia and macrophages and flow cytometric analyses were performed. For this study, treatment was initiated after the onset of relapse (mice had a clinical score of ≥1 for 2 consecutive days) and animals were sacrificed at the peak of the 2nd attack (5 days after onset of treatment). Microglia (resident CNS cells) and peripheral macrophages were differentiated by CD45 expression, i.e. microglia were phenotypically characterized as CD45int/lo CD11b+ (R1) and macrophages as CD45hi CD11b+ (R2) (Figure 6A) [22]. Leukocytes (CD45+ total) were markedly reduced in the spinal cords of Acthar-treated mice (Table 3). In addition, Acthar-treated RR-EAE mice had markedly fewer R2 cells migrating to the spinal cord at the peak of the 2nd attack (Figure 6A; Table 3). There was no difference in the frequency of total T cells (CD45+ CD3+), suggesting that T cells are still able to migrate from the periphery into the spinal cords of Acthar-treated mice. However, the frequency of CD45+ CD3+ CD4+ T cells in the spinal cords of RR-EAE mice was markedly lower in Acthar-treated mice in comparison to no treatment or Placebo gel-treated mice (Figure 6B; Table 3). Taken together, these results suggest that Acthar treatment is able to significantly reduce EAE exacerbation (2nd attack), even when administered after the onset of relapse, by dampening CD4+ T cells and macrophages.
Figure 6.

Representative flow cytometric plots of cells isolated from the spinal cords of EAE mice euthanized 5 days after onset of treatment. (A) Macrophages (R2) are markedly lower in Acthar-treated mice in comparison to no treatment and Placebo gel-treated mice. R region. (B) Frequency of T cells (CD45+ CD3+ CD4+) is markedly lower in Acthar-treated mice. Quantification of flow cytometric data is shown in Table 3.
Table 3.
Percentage of cells in the spinal cords of SJL/J female mice at the peak of the 2nd EAE attack.
| Cell populationa | NT (%) | Acthar (%) | Placebo gel (%) |
|---|---|---|---|
| CD45+ total | 57.5 ± 3.9 | 39.4 ± 15.4 | 54.7 ± 11.9 |
| CD45+ CD3+ | 78.6 ± 4.3 | 86.9 ± 2.2 | 85.2 ± 2.6 |
| CD45+ CD3+ CD4+ | 55.6 ± 6.1 | 40.3 ± 6.8 | 59.4 ± 0.9 |
| CD45+ CD19+ | 0.146 ± 0.1 | 0.1 ± 0.1 | 0.3 ± 0.2 |
| R1 | 18.0 ± 2.9 | 50.6 ± 18.7 | 17.6 ± 1.7 |
| R2 | 32.5 ± 1.2 | 19.3 ± 9.6 | 22.4 ± 3.1 |
| R1/R2 | 0.55 | 2.62 | 0.79 |
Cell populations were gated on 7-AAD negative (viable) cells.
NT no treatment.
Spinal cord cells from to 2–3 mice/group were pooled for a total of three pooled groups for each treatment.
Data represent the mean ± SEM.
Discussion
Using a well-established murine model for MS, we showed that Acthar gel treatment significantly improved clinical outcomes in RR-EAE, which correlated with a marked reduction in inflammation and demyelination in the spinal cord when compared to untreated and Placebo gel controls. Importantly, to test Acthar’s efficacy, treatment was initiated after the onset of the 2nd EAE exacerbation, which mirrors what occurs in patients with MS. Taken together, the results of the clinical scoring, histological analysis of spinal cords and ex vivo assays demonstrate that Acthar gel dampened the myelin-specific immune response leading to improved clinical course in EAE mice.
Porcine ACTH(1-39) is the principal active component in Acthar, and synthetic analogs of ACTH(1-39) inhibit the pro-inflammatory response of both the innate and adaptive immune systems (reviewed in [5]). One accepted mechanism of action of ACTH(1-39) is to induce endogenous cortico-steroids, leading to dampening of inflammation during an MS exacerbation. Serum corticosteroid levels were not measured in the present study, as it was anticipated that random levels would vary significantly based on multiple factors besides Acthar administration, including animal handling and disease activity. Also, the assessment of corticosterone pharmaco-dynamics following Acthar administration was beyond the scope of the study. Although the relative contribution of steroid-dependent and -independent properties of Acthar on efficacy was not evaluated in this study, both published and preliminary data in lymphocytes and neural cell models have suggested that ACTH and/or other melanocortin peptides may modulate the immune response independent of cortisol production [23,24]. Published literature suggests that plasma corticosteroid levels were significantly lower in human subjects treated with a dose of intramuscular ACTH [25] used clinically to treat MS exacerbation as compared with a clinically efficacious dose of i.v. prednisolone [26].
The ability of ACTH(1-39) to activate all five MCR subtypes suggests this drug could also have direct effects on a wide variety of cells expressing these receptors, including T cells, monocytes, microglia and astrocytes. ACTH(1-39) shares the first 13 amino acids with α-MSH that can bind multiple melanocortin receptors. Taylor and Kitaichi [27] systemically injected α-MSH peptide into SJL mice that had PLP139-151-induced EAE. α-MSH treatment inhibited EAE and α-MSH-treated myelin-specific splenic T cells functioned as regulatory T cells. However, the authors did not assess the activation of myelin-specific T cells from the CNS or the effects of α-MSH on glial cells, such as microglia and astrocytes.
Myeloid progenitors give rise to both microglia and macrophages [28–30]. These cells have been implicated in EAE and MS. Resident CNS microglia expressing CD11b are activated early in EAE disease, suggesting that these cells play an active role in the pathogenesis of EAE and MS [31]. Acthar-treated RR-EAE mice had a significantly lower number of RCA-1+ cells in the spinal cords in comparison to Placebo gel-treated and untreated mice, suggesting that Acthar is able to suppress multiple myeloid-lineage cells (Figure 5). To further support the immunohistochemistry results, flow cytometric analysis of these cells was performed. Microglia and macrophages, derived from different myeloid progenitor cell lineages, can be identified by differential expression of CD45 and CD11b [22,32,33]. Although another group has found that a subset of activated microglial cells contributes (37%) to the total pool of CD45hi CD11b+ cells, this was determined using a passive, adoptive transfer, myelin basic protein-induced EAE model in B10.PL mice [31], which may differ greatly from our active PLP139-151-induced EAE model. In addition, the authors admitted that the level of CD45 expression on the activated microglia was lower than that observed on infiltrating macrophages [31], which could correspond to what we see as CD45int cells [part of our CD45int/lo CD11b+ (R1) cell population]. Our work using green fluorescence protein (GFP)-expressing chimeric mice to study an inflammatory disease of the CNS demonstrated that CD45hi CD11b+ GFP+ activated microglial cells contributed only approximately 8–9% to the total pool of CD45hi CD11b+ (R2) cells; the remainder were CD45hi CD11b+ GFP+ infiltrating macrophages [22]. Therefore, using this same gating scheme for the present work, we define activated microglia as having low-to-intermediate expression of CD45 and high-CD11b expression (Figure 6A, R1), and macrophages as expressing high levels of both CD45 and CD11b on the cell surface (Figure 6A, R2). Acthar treatment led to a significant reduction in macrophages (R2 cells) in the spinal cords of EAE mice in comparison to no treatment and Placebo gel treatment, demonstrating that Acthar reduces microglial and/or macrophage activation and supporting the immunohistochemistry results.
Our flow cytometric data suggested that Acthar treatment did markedly lower the total percent of CD45+ CD3+ CD4+ T cells found in the spinal cords, however, this was not statistically significant (Figure 6; Table 3). Furthermore, immunohistochemical staining of spinal cord tissue sections did not detect differences in CD3+ T cell frequency among Acthar, untreated and Placebo gel treatment groups from mice in which treatment was initiated after the onset of relapse (data not shown), suggesting that Acthar did not affect T cell migration from the periphery into the CNS. However, a unique aspect of the second study is that Acthar treatment was not started until the mice had a clinical score of 1 or greater during the 2nd attack, which could explain the apparent lack of effect of Acthar on T cell migration. Therefore, finding a marked difference in clinical score, inflammation and overall presence of CD45+ leukocytes demonstrates that Acthar is suppressing the myelin-specific inflammatory responses in the CNS. Further, inhibition of T cell proliferation to myelin peptide by Acthar indicates that a suppressive effect on T cells is induced. Further investigation is needed to determine if Acthar is shifting CD4+ T cell differentiation from a pro-inflammatory to a suppressive phenotype. Work by Brod and Hood [12] demonstrated that ACTH-fed mice had a two-fold increase in T regulator cells (Tregs) in the periphery of MOG-induced C57BL/6 EAE mice when compared to the control group, suggesting that ACTH is able to shift the Th1/Th17 CD4+ T cells to the Th2/Treg phenotype. However, Acthar’s mechanism(s) of action is unknown and future experiments are needed.
In conclusion, Acthar had a beneficial immunomodulatory effect and prevented demyelination in EAE mice, while also promoting significant reductions in activated astrocytes and microglia/monocytes in the CNS. Taken together, these results support Acthar’s continued use for the treatment of exacerbations in patients with MS.
Acknowledgments
We would like to thank Daniel J. Doty for technical assistance and Daniel J. Harper for the outstanding preparation of the manuscript.
This work was supported by Mallinckrodt Pharmaceuticals (formerly Questcor Pharmaceuticals). M.F.C. was supported by NIH grant T32AI055434. L.O. and S.J. are employed by Mallinckrodt Pharmaceuticals (formerly Questcor Pharmaceuticals).
Footnotes
Declaration of interest
L.O. and S.J. are recipients of stock options from Mallinckrodt Pharmaceuticals (formerly Questcor Pharmaceuticals).
References
- 1.Miller H, Newell DJ, Ridley A. Multiple sclerosis. Treatment of acute exacerbations with corticotrophin (A.C.T.H.) Lancet. 1961;2:1120–1122. doi: 10.1016/s0140-6736(61)91030-3. [DOI] [PubMed] [Google Scholar]
- 2.Rose AS, Kuzma JW, Kurtzke JF, et al. Cooperative study in the evaluation of therapy in multiple sclerosis. ACTH vs. placebo – final report. Neurology. 1970;20:1–59. doi: 10.1212/wnl.20.5_part_2.1. [DOI] [PubMed] [Google Scholar]
- 3.Thompson AJ, Kennard C, Swash M, Summers B, et al. Relative efficacy of intravenous methylprednisolone and ACTH in the treatment of acute relapse in MS. Neurology. 1989;39:969–971. doi: 10.1212/wnl.39.7.969. [DOI] [PubMed] [Google Scholar]
- 4.Catania A, Gatti S, Colombo G, Lipton JM. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol Rev. 2004;56:1–29. doi: 10.1124/pr.56.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Arnason BG, Berkovich R, Catania A, et al. Mechanisms of action of adrenocorticotropic hormone and other melanocortins relevant to the clinical management of patients with multiple sclerosis. Mult Scler. 2013;19:130–136. doi: 10.1177/1352458512458844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Libbey JE, Tsunoda I, Fujinami RS. Studies in the modulation of experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:168–175. doi: 10.1007/s11481-010-9215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsunoda I, Fujinami RS. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. J Neuropathol Exp Neurol. 1996;55:673–686. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Libbey JE, Fujinami RS. Experimental autoimmune encephalomyelitis as a testing paradigm for adjuvants and vaccines. Vaccine. 2011;29:3356–3362. doi: 10.1016/j.vaccine.2010.08.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- 10.Field EJ, Miller H. Studies in the inhibition of experimental allergic encephalomyelitis. Arch Int Pharmacodyn Ther. 1961;134:76–88. [PubMed] [Google Scholar]
- 11.Moyer AW, Jervis GA, Black J, et al. Action of adrenocorticotropic hormone (ACTH) in experimental allergic encephalomyelitis of the guinea pig. Proc Soc Exp Biol Med. 1950;75:387–390. doi: 10.3181/00379727-75-18207. [DOI] [PubMed] [Google Scholar]
- 12.Brod SA, Hood ZM. Ingested (oral) ACTH inhibits EAE. J Neuroimmunol. 2011;232:131–135. doi: 10.1016/j.jneuroim.2010.10.030. [DOI] [PubMed] [Google Scholar]
- 13.Brod SA, Hood ZM. Ingested (oral) alpha-MSH inhibits acute EAE. J Neuroimmunol. 2008;193:106–112. doi: 10.1016/j.jneuroim.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Tuohy VK, Lu Z, Sobel RA, et al. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–1527. [PubMed] [Google Scholar]
- 15.Ahmed R. Immunological memory against viruses. Semin Immunol. 1992;4:105–109. [PubMed] [Google Scholar]
- 16.Cusick MF, Libbey JE, Trede NS, et al. Human T cell expansion and experimental autoimmune encephalomyelitis inhibited by Lenaldekar, a small molecule discovered in a zebrafish screen. J Neuroimmunol. 2012;244:35–44. doi: 10.1016/j.jneuroim.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Decker D, Grant C, Oh L, et al. Immunomodulatory effects of H.P. Acthar Gel on B cell development in the NZB/W F1 mouse model of systemic lupus erythematosus. Lupus. 2014;23:802–812. doi: 10.1177/0961203314531840. [DOI] [PubMed] [Google Scholar]
- 18.Kirkman NJ, Libbey JE, Wilcox KS, et al. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia. 2010;51:454–464. doi: 10.1111/j.1528-1167.2009.02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trede NS, Heaton W, Ridges S, et al. Discovery of biologically active oncologic and immunologic small molecule therapies using zebrafish: overview and example of modulation of T cell activation. Curr Protoc Pharmacol. 2013;Chapter 14(Unit14):24. doi: 10.1002/0471141755.ph1424s60. [DOI] [PubMed] [Google Scholar]
- 20.Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci. 2008;65:2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eng LF. Glial fibrillary acidic protein (GFAP): the major protein of glial intermediate filaments in differentiated astrocytes. J Neuroimmunol. 1985;8:203–214. doi: 10.1016/s0165-5728(85)80063-1. [DOI] [PubMed] [Google Scholar]
- 22.Cusick MF, Libbey JE, Patel DC, et al. Infiltrating macrophages are key to the development of seizures following virus infection. J Virol. 2013;87:1849–1860. doi: 10.1128/JVI.02747-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delgado R, Carlin A, Airaghi L, et al. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J Leukoc Biol. 1998;63:740–745. doi: 10.1002/jlb.63.6.740. [DOI] [PubMed] [Google Scholar]
- 24.Taylor AW, Namba K. In vitro induction of CD25+CD4+ regulatory T cells by the neuropeptide alpha-melanocyte stimulating hormone (alpha-MSH) Immunol Cell Biol. 2001;79:358–367. doi: 10.1046/j.1440-1711.2001.01022.x. [DOI] [PubMed] [Google Scholar]
- 25.Besser GM, Butler PW, Plumpton FS. Adrenocorticotrophic action of long – acting tretracosactrin compared with corticotrophingel. Br Med J. 1967;4:391–394. doi: 10.1136/bmj.4.5576.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coburg AJ, Gray SH, Katz FH, et al. Disappearing rates and immunosuppression of intermittent intravenously administered prednisolone in rabbits and human beings. Surg Gynecol Obstet. 1970;131:933–942. [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor AW, Kitaichi N. The diminishment of experimental autoimmune encephalomyelitis (EAE) by neuropeptide alpha-melanocyte stimulating hormone (alpha-MSH) therapy. Brain Behav Immun. 2008;22:639–646. doi: 10.1016/j.bbi.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beers DR, Henkel JS, Xiao Q, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKercher SR, Torbett BE, Anderson KL, et al. Targeted disruption of the PU.1 gene results in multiple hemato-poietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 31.Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- 32.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- 33.Sedgwick JD, Schwender S, Imrich H, et al. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]