

Summary
Cell cycle progression is regulated by the cyclin-dependent kinase (Cdk) family of protein kinases, so named because their activation depends on association with regulatory subunits known as cyclins [1]. Cyclin E normally accumulates at the G1/S boundary, where it promotes S phase entry and progression by activating Cdk2. In normal cells, cyclin E/Cdk2 activity is associated with DNA replication-related functions [2]. However, deregulation of cyclin E leads to inefficient assembly of pre-replication complexes [3], replication stress [4], and chromosome instability [5]. In malignant cells, cyclin E is frequently overexpressed, correlating with decreased survival in breast cancer patients [6, 7]. Transgenic mice deregulated for cyclin E in the mammary epithelia develop carcinoma [8], confirming that cyclin E is an oncoprotein. However, it remains unknown how cyclin E-mediated replication stress promotes genomic instability during carcinogenesis. Here we show that deregulation of cyclin E causes human mammary epithelial cells to enter into mitosis with short unreplicated genomic segments at a small number of specific loci, leading to anaphase anomalies and ultimately deletions. Incompletely replicated regions are preferentially located at late-replicating domains, fragile sites and breakpoints, including the mixed-lineage leukemia breakpoint cluster region (MLL BCR). Furthermore, these regions are characterized by a paucity of replication origins or unusual DNA structures. Analysis of a large set of breast tumors shows a significant correlation between cyclin E amplification and deletions at a number of the genomic loci identified in our study. Our results demonstrate how oncogene-induced replication stress contributes to genomic instability in human cancer.
Results
Ongoing DNA replication in mitotic cells
Cyclin E-mediated replication stress results in depressed origin firing [9], slowed fork progression [10], and aberrant fork architecture [11]. However, the molecular mechanisms that link replication stress to genomic instability remain poorly understood. We hypothesized that cyclin E deregulation expands the time interval required for DNA replication, causing cells to enter into mitosis with incompletely-replicated genomes. To test this idea, recombinant cyclin E-expressing adenoviruses were used to increase cyclin E levels in immortalized human mammary epithelial cells (HME1) (Figure 1A). MDA-MB-157 [12] and SUM149PT [13], breast cancer-derived cell lines that overexpress cyclin E, were used as controls. Transduction multiplicities that recapitulated cyclin E levels observed in the high cyclin E breast cancer cell lines (Figure 1A) were used in all subsequent experiments. To compare the rate of S phase progression in cells deregulated for cyclin E expression and controls, HME1 cells were transduced with cyclin E and control viruses and released from a double-thymidine block for 8 hours (Figure 1B). Flow cytometric analysis revealed that cyclin E deregulation reduced the rate of progression through S phase (control = 20% versus cyclin E = 62% remaining in S phase after 8 hours). Cells expressing deregulated cyclin E required ∼12-16 hours to complete S phase (Figure S1A). To determine whether cells could enter into mitosis with ongoing replication, strong phosphorylation of histone H3 on serine 10 was used as a marker for late G2/M phase, while ongoing replication was scored by incorporation of BrdU during a short pulse (Figure S1B and S1C). A significant fraction of cyclin E-deregulated cells that stained strongly positive for phospho-H3 also stained positive for BrdU incorporation (cyclin E = 16.4%, n = 286; Figures 1C and 1D). However, double-positive cells were completely absent in controls (n = 526; Figure 1D). Increased transduction multiplicities correlated with higher frequencies of double-positive cells, reaching almost 50% of the total (Figure 1E). These data indicate that a fraction of cells experiencing cyclin E deregulation are near or in mitosis while DNA replication is ongoing.
Figure 1. Ongoing DNA duplication in mitosis upon cyclin E deregulation.
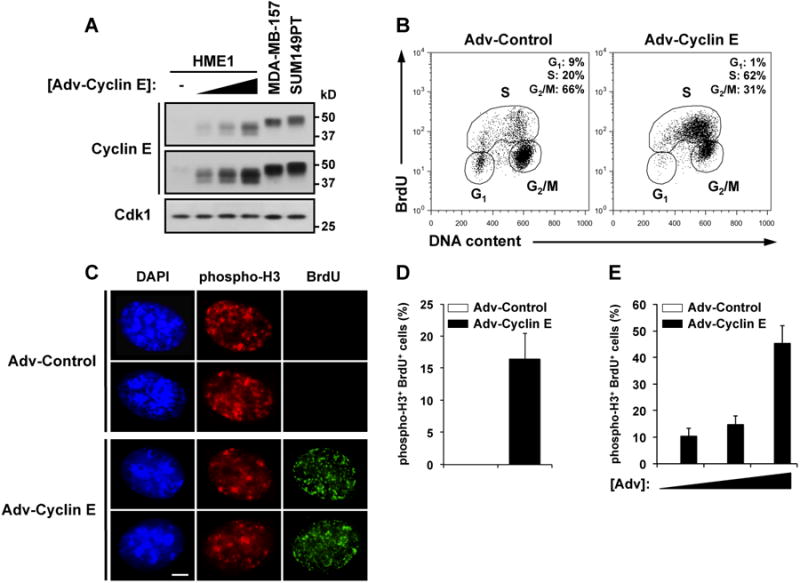
(A) Immunoblot analysis of HME1, MDA-MB-157, and SUM149PT cells. HME1 cells were transduced with control adenovirus (-) or increasing amounts of cyclin E adenovirus (1-10 × 103 particles/cell). Upper blot, long exposure; Lower blot, short exposure.
(B) Flow cytometry analysis of HME1 cells transduced as in (A), synchronized with a double-thymidine block protocol, and released for 8 hours.
(C) Immunofluorescence of HME1 cells treated as in (B), and pulsed with BrdU for 10 min 8 hr after release.
(D and E) Frequency of double-positive cells (phospho-H3+ and BrdU+) transduced and treated as in (B) (D) or transduced with increasing amounts of cyclin E (1-10 × 103 particles/cell) (E).
Results are representative of at least three independent experiments. Error bars represent one standard deviation. Scale bar, 5 μm.
See also Figure S1.
Cyclin E deregulation causes aberrant anaphases
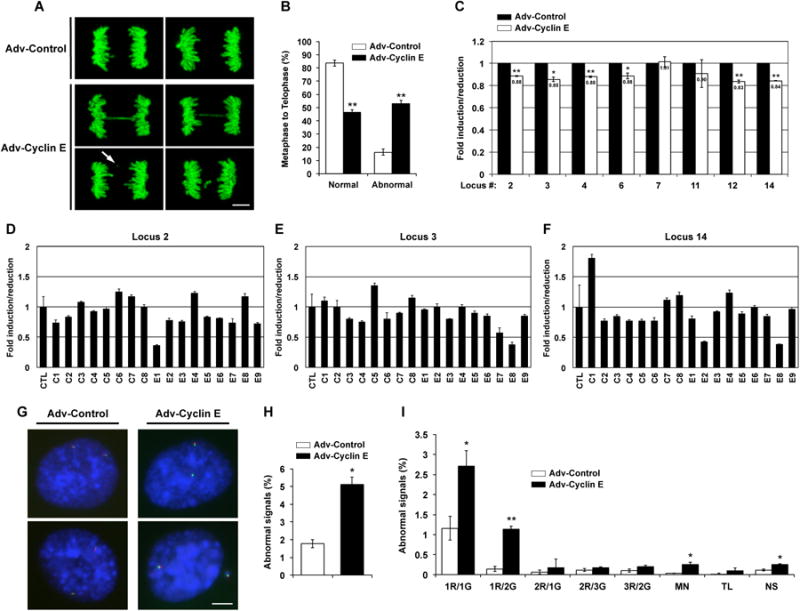
Persistence of unreplicated DNA into mitosis is expected to cause abnormalities during chromosome segregation. We therefore screened cyclin E-deregulated HME1 cells for aberrant mitotic chromosome dynamics by live cell microscopy (Figure 2A). Cyclin E deregulation caused a 3.2-fold increase in abnormal metaphase-to-telophase transitions (control = 16.3% versus cyclin E = 53.2%; n > 100, P = 2.9 × 10-5, unpaired t-test; Figure 2B), including the formation of anaphase bridges, lagging chromosomes, and micronuclei (AB, P = 0.0037; LC, P = 0.0009; MN, P = 0.0025, unpaired t-test; Figure S2A; Movies S1 and S2). These abnormal anaphases are consistent with attempted chromosome segregation in the presence of incompletely-replicated DNA [14-16].
Figure 2. Cyclin E deregulation causes loss of MLL BCR locus and chromosome aberrations in mitosis.

(A) Laser scanning confocal microscopy of live HME1 cells expressing GFP-H2B and transduced with control (Adv-Control) or cyclin E adenovirus (Adv-Cyclin E).
(B) Frequency of normal/abnormal metaphase-to-anaphase transitions in HME1 cells transduced with control (white bars) or cyclin E adenovirus (black bars).
(C) Real-time PCR analysis of eight genomic loci. HME1 cells were transduced with control (black bars) or cyclin E adenovirus (white bars) twice a week for three weeks.
(D-F) Real-time PCR analysis of three genomic loci in individual cells. HME1 cells were transduced twice with control (C) or cyclin E adenovirus (E), individually isolated using a FACS, processed for whole genome amplification and analyzed for loci 2 (D), 3 (E), and 14 (F). CTL indicates the mean of the 8 control clones. Error bars for individual genomes correspond to PCR quadruplicate reactions.
(G) FISH analysis of MLL BCR locus in HME1 cells transduced with control or cyclin E adenovirus. Probes are located to the N-terminus (green signal) or C-terminus (red signal) of the MLL BCR.
(H) Frequency of abnormal FISH signals in HME1 cells transduced with control (white bars) or cyclin E adenovirus (black bars).
(I) Frequency of abnormal FISH signals at the MLL BCR locus in HME1 cells transduced with control (white bars) or cyclin E adenovirus (black bars). R, red probe; G, green probe; MN, micronucleus; TL, translocation; NS, no signal. All results are representative of at least two independent experiments.
All error bars represent one standard deviation. Asterisks indicate significance levels compared to control samples (*, P < 0.05; **, P < 0.005). Scale bars, 5 μm.
See also Figure S2 and Movies S1 and S2.
Cyclin E causes replication failure at a limited number of specific genomic loci
To determine whether specific loci are incompletely replicated in mitotic cells after one cell cycle during which cyclin E was deregulated, we carried out comparative genome hybridization (CGH) array analysis on DNA from prometaphase cells that were first synchronized by a double-thymidine block, and released into nocodazole (see Supplemental Experimental Procedures). CGH arrays indicated ∼20-fold more underreplicated chromosomal segments than overreplicated segments upon cyclin E deregulation (8,334 versus 341 probes in at least one sample; Figure S3A and Table S1). We then focused on chromosomal segments that were scored as underreplicated in at least two of the three experiments, and were detected by at least 5 contiguous probes. Surprisingly, decreased signals, indicating underreplication, were concentrated at only 16 genomic loci distributed over many chromosomes, and varied in size from a few Kb to over 100 Kb (Table 1 and Figure S3B). These loci were frequently, but not always, located in late-replicating domains based on replication timing of IMR-90 human fibroblasts (Table 1) [17]. Furthermore, some loci were located in pericentromeric or subtelomeric regions, as well as fragile sites and breakpoint or recombination hotspots (Table 1) [18]. Of note, one of the sites encompasses the breakpoint cluster region of the mixed-lineage leukemia gene (MLL BCR; Tables 1 and S1), often rearranged in leukemias [19] but also deleted in breast cancer (see below).
Table 1. Analysis of DNA underreplication in mitotic cells with deregulated cyclin E.
| Locus | Chrom a | Region | Size (bp) | Inferred copy number | Replication timing | Location b | Fragile site | Breakpoint/Recombination | Gene |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 3p26.1 | 7,396 | -0.1029 | Late | Subtelomeric | |||
| 2 | 3 | 3q11.2 | 102,906 | -0.0956 | Late | Pericentromeric | EPHA6 | ||
| 3 | 3 | 3q26.1 | 14,758 | -0.0973 | Late | Large LRD (9 Mb) | |||
| 4 | 3 | 3q26.1 | 35,313 | -0.1146 | Late | Large LRD (9 Mb) | |||
| 5 | 3 | 3q29 | 2,636 | -0.1930 | Late | Subtelomeric | Breakpoint | ||
| 6 | 11 | 11q23.3 | 4,331 | -0.1617 | Early | ERD (1.5 Mb) | FRA11B/G | MLL | |
| 7 | 16 | 16q21 | 4,100 | -0.1067 | Late | Large LRD (7 Mb) | |||
| 8 | 17 | 17q21.2 | 228 | -0.3222 | Early | Large ERD (15 Mb) | Breakpoint | ||
| 9 | 18 | 18p11.21 | 48,333 | -0.0995 | Late | Pericentromeric | ANKRD30B | ||
| 10 | 18 | 18q21.2-q21.31 | 13,443 | -0.1449 | Early | ERD (1.5 Mb) | FRA18B | Recomb. hotspot | |
| 11 | 18 | 18q22.1 | 18,363 | -0.1136 | Late | LRD (3.5 Mb) | FRA18C | Breakpoint | |
| 12 | 21 | 21q21.1 | 11,690 | -0.1346 | Late | Large LRD (9.5 Mb) | Breakpoint | ||
| 13 | 21 | 21q21.1 | 8,809 | -0.1153 | Late | Large LRD (9.5 Mb) | NCAM2 | ||
| 14 | 21 | 21q21.1 | 56,443 | -0.1105 | Late | Large LRD (9.5 Mb) | Recomb. hotspot | MAPK6PS2 | |
| 15 | 21 | 21q21.2 | 72,026 | -0.0951 | Late | Large LRD (9.5 Mb) | Recomb. hotspot | ||
| 16 | 22 | 22q12.3 | 56,452 | -0.0772 | Late | Late-replicating island |
Sources: UCSC Genome Browser, Assembly NCBI Build 36.1/hg18; Database of Genomic Variants, The Center for Applied Genomics, Build 36; Replication Domain Database, The Florida State University.
Chromosome;
LRD, Late-replicating domain; ERD, Early-replicating domain. See also Figure S3 and Table S1.
To determine whether underreplication after one pulse of cyclin E correlates with eventual genomic loss in proliferating populations, HME1 cells were transduced twice a week for three weeks with control or cyclin E adenovirus, after which average copy number at eight loci that exhibited underreplication in the population was interrogated by real-time PCR (Figure 2C). Significant genomic losses were detected for 6 of the 8 loci analyzed, with signal decreases varying from 12% to 17%, confirming a link between cyclin E-mediated underreplication and subsequent genomic loss. For a heterogeneous diploid cell population, a signal decrease of 15% indicates that 1 in every 3 cells has experienced a deletion at the interrogated locus.
To determine the frequency of cells experiencing genomic losses at specific loci, single cells were analyzed for deletion at three loci by real-time PCR. HME1 cells were transduced twice with control or cyclin E adenovirus, and individual cells were isolated by FACS. Genomes from eight control and nine cyclin E cells were amplified and analyzed for deletions at loci 2, 3, and 14 (Figures 2D-2F). Deletions were detected in 1 of the 9 cyclin E cells at locus 2 (cell E1, Figure 2D) and 2 of the 9 cyclin E cells at loci 3 and 14, respectively (cells E7 and E8, Figure 2E; cells E2 and E8, Figure 2F). The occurance of 5 deletions at 3 loci for 9 cyclin E cells compared to none for 8 control cells was significant (P = 0.032, Fisher's exact test).
Cyclin E deregulation causes loss of the MLL BCR locus
We then specifically addressed deletion at the MLL BCR locus by fluorescence in situ hybridization (FISH) (Figure 2G). Cyclin E deregulation caused an almost 3-fold increase in aberrant FISH signals at this locus (control = 1.77% versus cyclin E = 5.11%; n > 5,000 cells, P = 0.0104, unpaired t-test; Figure 2H). The primary signal differences observed were loss of the distal region (1R/2G = loss of one red signal; control = 0.153% versus cyclin E = 1.147%, P = 0.0040, unpaired t-test; Figure 2I) and the loss of one entire copy of the locus through micronucleus formation (MN, control = 0.034% versus cyclin E = 0.269%, P = 0.0231, unpaired t-test; Figure 2I). These data are consistent with persistence of unreplicated DNA during mitosis, leading to chromosome breaks and subsequent loss of genetic material [14-16]. Consistent with this, a recent study showed that cyclin E deregulation frequently causes deletion of most or all of the right arm of chromosome 3 [20], which contains several of the loci identified in this study.
Origin paucity correlates with genomic loss
One feature of genomic regions that are sensitive to replication stress is paucity of replication origins, which forces DNA replication forks to travel long distances, increasing the probability of fork collapse [21], and which also reduces the likelihood of rescue by adjacent forks [22]. We therefore addressed DNA replication origin (ORI) density at the 16 genomic loci found underreplicated in CGH experiments using two databases of human genome-wide origin distribution [23, 24] (Figures 3A, and S2B). It is noteworthy that ORI mapping data in mammalian cells have been characterized by low reproducibility. However, analysis of a 1 Mb region surrounding each locus showed that many of the loci are located in origin-poor regions (Locus # 2, 3, 4, 7, 9-15; Figure 3A), and the total number of ORIs in the 1 Mb neighborhoods surrounding the 16 genomic loci is significantly lower than the total number of ORIs in the 1 Mb neighborhoods of 16-randomly selected genomic locations (94 versus 161 ORIs [23]; P < 0.025, Permutation-based enrichment test; Table S2). Remarkably, among the 16 genomic loci, seven are located > 400 Kb away from the closest ORI, including four loci that are more than 600 Kb from the closest ORI (Locus # 3, 11, 12 and 14; Table S2). In comparison, only three out of 16-randomly selected genomic loci are located > 400 Kb away from the closest ORI [23]. Although these data are only correlative and not derived from HME1 cells, they suggest that paucity of replication origins may contribute to the replication impairment and eventual loss of specific regions in response to cyclin E deregulation.
Figure 3. Paucity of replication origins and unusual DNA structures underlies cyclin E-mediated genomic loss.
(A) Schematic representation of the 16 CNV loci identified in this study along with the ORIs previously identified on human chromosomes [23]. The black ticks mark the locations of the ORIs, the blue triangles indicate the locations of the 16 loci, and the red lines plot the estimated ORI probability density (see Supplemental Experimental Procedures).
(B) Schematic representation of the MLL BCR N terminus containing two putative stemloop-forming sequences (MLL BCR) or the same DNA sequence lacking the two stemloops (Control) cloned into the pCEP4 plasmid. Diagrams are not drawn to scale.
(C) Cell survival analysis in U2OS cells transfected with pCEP4 plasmid encoding a fragment of the MLL BCR N terminus (MLL BCR, black squares) or a control fragment lacking the stemloop structures (Control, open circles). Cells were transduced with control adenovirus (-) or increasing amounts of cyclin E adenovirus (see Figure S2 for non-normalized data and also Supplemental Experimental Procedures).
(D) Real-time PCR analysis of DpnI-treated pCEP4 plasmids on day 0 (white bars) and day 10 (black bars) containing part of the MLL BCR sequence (MLL BCR) or a control sequence lacking the two stemloop-forming regions (Control). Data represent relative plasmid levels (Control or MLL BCR) extracted from cyclin E-deregulated cells normalized to plasmid levels extracted from control cells (see Figure S2 for non-normalized data and also Supplemental Experimental Procedures).
Results are representative of at least two independent experiments. Error bars represent one standard deviation. Asterisks indicate significance of difference from control samples (*, P = 0.012; **, P = 0.0019; ***, P = 9.7 × 10-5).
See also Figure S2 and Table S2.
Unusual DNA structures can contribute to cyclin E-mediated genomic loss
Another feature of regions that are sensitive to replication stress is DNA sequences that are predicted to form stemloops [25]. The MLL BCR, identified in our study, is not in an origin-poor or late-replicating region. However, analysis of DNA folding of the MLL BCR N terminus, underreplicated in our CGH experiments, showed two putative stemloop structures located in introns 7 and 8; ∆G = - 7.15 kcal and - 9.69 kcal, respectively (Figures S2C and S2D). The stemloop structures consist of 94 bp and 67 bp, respectively, and are separated by approximately 500 bp. To test the idea that cyclin E deregulation promotes deletions by impairing replication of such structures, we cloned part of the MLL BCR N terminus containing the two stemloop structures into the episomal plasmid pCEP4 (Figure 3B and see Supplemental Experimental Procedures). As a control, the same sequence missing the stemloops was inserted into the vector. Cyclin E deregulation had no effect on the control plasmid but conferred a dose-dependent impairment of maintenance and transmission of the plasmid containing the two stemloop-forming structures, as measured by relative number of hygromycin-resistant cells (Low cyclin E, P = 0.012; High cyclin E, P = 9.7 × 10-5, unpaired t-test; Figures 3C, S2E, and S2F) or of relative replicated plasmid levels, as determined by real-time PCR (P = 0.0019, unpaired t-test; Figures 3D, S2G, and S2H).
Cyclin E amplification correlates with loss of specific genomic regions in breast cancer
We next evaluated the relevance of our in vitro findings to breast cancer. CGH arrays from 1,962 primary fresh-frozen breast cancer specimens [18] were interrogated for association between cyclin E1 amplification and copy number losses at the genomic regions established in vitro by CGH (see Supplemental Experimental Procedures). We observed a significant correlation between cyclin E1 gain and genomic region loss in 6/16 analyzed loci (Locus # 2, 3, 4, 11, 12, 14; P < 0.05, Fisher exact test; Figure S3C). Furthermore, three additional loci (Locus # 6, 7, 16; Figure S3C), including the N terminus of the MLL BCR locus, were lost in more than 10% of breast cancer patients (Locus # 6 = 16.6%, n = 326/1,962 [CNV- patients/all patients]), even though these losses did not correlate with cyclin E1 amplification (Figure S3C). It is therefore likely that other mechanisms contribute to copy number losses at these genomic loci, masking the effects of cyclin E1 overexpression. Taken together, our data show that cyclin E deregulation causes cells to enter into mitosis with incompletely-replicated genomes, leading to chromosome segregation anomalies and copy number losses originating at specific unreplicated loci, and thus contributing to the genomic instability observed in breast cancer.
Discussion
Oncogene expression has been linked to replication stress, leading to stalled replication forks, DNA damage and, ultimately, impaired progression through S phase [4, 26-30]. It has been assumed that a cell cycle checkpoint prevents cells with expanded replication times from entering mitosis prior to completion of DNA replication. However, in the case of cyclin E deregulation, cells are not prevented from entering mitosis with a small number of short unreplicated segments. These data suggest that a minimal amount of unreplicated DNA is required to trigger this checkpoint and that levels below a threshold can evade surveillance [14, 31].
It is not clear why a small select group of loci are particularly sensitive to cyclin E-mediated replication failure. Nevertheless, these loci appear to subdivide into two classes, suggesting that at least two different mechanisms may be involved. The first class, which includes most of the loci identified, is characterized by location within late-replicating domains with a paucity of replication origins. This suggests a mechanism whereby fork collapse caused by cyclin E-mediated replication stress cannot be repaired prior to mitotic entry because of late timing of origin initiation and large inter-origin distances prohibiting rescue by adjacent forks [32, 33]. However, not all origin-poor late replicating regions were identified in our study. It is therefore likely that other properties of these regions contribute to their sensitivity to cyclin E deregulation. One possible compounding factor may be related to the observation that cyclin E overexpression interferes with pre-replication complex assembly [3], exacerbating replication stress by preventing the formation of dormant origins required to rescue collapsed forks [22]. Thus, it may be that the cyclin E sensitive loci identified in this study are particularly impaired in formation of dormant origins when cyclin E is overexpressed.
The second class of loci, typified by the MLL BCR, might be characterized by structured DNA that is likely to impose a barrier to replication fork progression. The likely mechanism of replication failure is that cyclin E-mediated replication stress sensitizes forks so that they tend to stall or collapse when they encounter such structures. However, many such structures have been identified in the genome [32, 34]. Yet, most of these were not identified as cyclin E sensitive in the current study. Again, other factors must contribute to sensitization by cyclin E overexpression. One possible explanation is that the MLL BCR contains two predicted stemloops within a short distance (500 bp).
Interestingly, one well-defined MLL breakpoint in intron 8 localizes at the base of the putative stemloop structure identified here, which also contains a binding site for topoisomerase II and recombinase [35]. On the other hand, deletion of the MLL BCR in breast cancer, which is quite frequent, did not correlate with cyclin E amplification, suggesting that stemloop structures are sensitive to multiple forms of replication stress. Nevertheless, a study of MLL rearragements in leukemia patients identified elevated cyclin E levels as a major driver [36], indicating that in some cancers, cyclin E-mediated replication stress is critical for damage at this locus.
Our findings suggest a mechanism for how replication stress contributes to genomic instability in cancer. In particular, we demonstrate a likely mechanism to explain specific genetic losses that occur during mammary tumorigenesis mediated by cyclin E deregulation. It will be interesting to determine whether the same mechanisms apply in other cell types and cancers driven by cyclin E, as well as other oncoproteins.
Supplementary Material
Highlights.
Cyclin E deregulation allows cells to enter into mitosis with ongoing DNA duplication
Cyclin E causes replication failure at specific genomic loci (MLL BCR)
Origin paucity and unusual DNA structures contribute to genomic loss
Cyclin E1 amplification correlates with specific genomic losses in breast cancer
Acknowledgments
We thank M. Henze, TSRI flow cytometry (particularly Matthew Haynes for help with single cell isolation) and microscope core facilities for assistance, J. Cogswell, S. Neill, and J. Albrecht for reagents, E. Denchi and J. Erwin for help with FISH, and V. Liberal for comments. L.K.T. acknowledges support from the Pew Latin American Fellows Program in the Biomedical Sciences. This work was supported by NIH grants CA078343 to S.I.R., CA138215, GM082802, CA160034, CA138293 to P.W., and CA140972, CA102361 to X.W.3
Footnotes
Supplemental Information: Supplemental Information includes four figures, two movies, two tables, and Supplemental Experimental Procedures.
Author Contributions: L.K.T. and S.I.R. conceived the study and designed the experiments. L.K.T performed and analyzed most cellular and molecular experiments, X.W.2 and P.W. analyzed CGH arrays and ChIP-seq data, Y.L., and X.W.3 performed and analyzed plasmid retention experiments, S.E.-R. and S.I.R. performed and analyzed single cell genome amplification experiments, and S.I.R. supervised all aspects of the project. L.K.T. and S.I.R. wrote the manuscript with contributions from all co-authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 2.Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- 3.Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol. 2004;165:789–800. doi: 10.1083/jcb.200404092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LF, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 5.Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 6.Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, Roberts JM. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med. 1997;3:222–225. doi: 10.1038/nm0297-222. [DOI] [PubMed] [Google Scholar]
- 7.Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347:1566–1575. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 8.Bortner DM, Rosenberg MP. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol. 1997;17:453–459. doi: 10.1128/mcb.17.1.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liberal V, Martinsson-Ahlzen HS, Liberal J, Spruck CH, Widschwendter M, McGowan CH, Reed SI. Cyclin-dependent kinase subunit (Cks) 1 or Cks2 overexpression overrides the DNA damage response barrier triggered by activated oncoproteins. Proc Natl Acad Sci USA. 2012;109:2754–2759. doi: 10.1073/pnas.1102434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neelsen KJ, Zanini IM, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci USA. 1993;90:1112–1116. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strohmaier H, Spruck CH, Kaiser P, Won K, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 14.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 15.Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- 16.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell. 2011;41:543–553. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weddington N, Stuy A, Hiratani I, Ryba T, Yokochi T, Gilbert DM. ReplicationDomain: a visualization tool and comparative database for genome-wide replication timing data. BMC Bioinformatics. 2008;9:530. doi: 10.1186/1471-2105-9-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curtis C, Shah SP, Chin S, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol Mech Dis. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343:88–91. doi: 10.1126/science.1243211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Letessier A, Millot GA, Koundrioukoff S, Lachages A, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 22.Blow JJ, Gillespie PJ. Replication licensing and cancer - a fatal entanglement? Nat Rev Cancer. 2008;8:799–806. doi: 10.1038/nrc2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin MM, Ryan M, Kim R, Zakas AL, Fu H, Lin CM, Reinhold WC, Davis SR, Bilke S, Liu H, et al. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011;21:1822–1832. doi: 10.1101/gr.124644.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dellino GI, Cittaro D, Piccioni R, Luzi L, Banfi S, Segalla S, Cesaroni M, Mendoza-Maldonado R, Giacca M, Pelicci PG. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013;23:1–11. doi: 10.1101/gr.142331.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell. 2011;43:122–131. doi: 10.1016/j.molcel.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 26.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 27.Gorgoulis VG, Vassiliou LF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, DiTullio RA, Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 28.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 29.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hills SA, Diffley JFX. DNA replication and oncogene-induced replicative stress. Curr Biol. 2014;24:R435–R444. doi: 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 32.Durkin SG, Glover TW. Chromosome Fragile Sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 33.Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. Common fragile sites: mechanisms of instability revisited. Trends Genet. 2012;28:22–32. doi: 10.1016/j.tig.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Ozeri-Galai E, Bester AC, Kerem B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012;28:295–302. doi: 10.1016/j.tig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Negrini M, Felix CA, Martin C, Lange BJ, Nakamura T, Canaani E, Croce CM. Potential topoisomerase II DNA-binding sites at the breakpoints of a t(9;11) chromosome translocation in acute myeloid leukemia. Cancer Res. 1993;53:4489–4492. [PubMed] [Google Scholar]
- 36.Accordi B, Espina V, Giordan M, VanMeter A, Milani G, Galla L, Ruzzene M, Sciro M, Trentin L, De Maria R, et al. Functional protein network activation mapping reveals new potential molecular drug targets for poor prognosis pediatric BCP-ALL. PLoS One. 2010;5:e13552. doi: 10.1371/journal.pone.0013552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.