

Abstract
Medulloblastoma (MB) and ependymoma (EP) are the most common pediatric brain tumors, afflicting 3000 children annually. Radiotherapy (RT) is an integral component in the treatment of these tumors; however, the improvement in survival is often accompanied by radiation‐induced adverse developmental and psychosocial sequelae. Therefore, there is an urgent need to develop strategies that can increase the sensitivity of brain tumors cells to RT while sparing adjacent healthy brain tissue. Apurinic endonuclease 1 (Ape1), an enzyme in the base excision repair pathway, has been implicated in radiation resistance in cancer. Pharmacological and specificity limitations inherent to small molecule inhibitors of Ape1 have hindered their clinical development. Here we report on a nanoparticle (NP) based siRNA delivery vehicle for knocking down Ape1 expression and sensitizing pediatric brain tumor cells to RT. The NP comprises a superparamagnetic iron oxide core coated with a biocompatible, biodegradable coating of chitosan, polyethylene glycol (PEG), and polyethyleneimine (PEI) that is able to bind and protect siRNA from degradation and to deliver siRNA to the perinuclear region of target cells. NPs loaded with siRNA against Ape1 (NP:siApe1) knocked down Ape1 expression over 75% in MB and EP cells, and reduced Ape1 activity by 80%. This reduction in Ape1 activity correlated with increased DNA damage post‐irradiation, which resulted in decreased cell survival in clonogenic assays. The sensitization was specific to therapies generating abasic lesions as evidenced by NP:siRNA not increasing sensitivity to paclitaxel, a microtubule disrupting agent. Our results indicate NP‐mediated delivery of siApe1 is a promising strategy for circumventing pediatric brain tumor resistance to RT.
Keywords: siRNA, Ape1, Medulloblastoma, Ependymoma, Nanomedicine, Base excision repair
Abbreviations
- Ape1
apurinic endonuclease 1
- EP
ependymoma
- MB
medulloblastoma
- NP
nanoparticle
- PEI
polyethyleneimine
- PEG
polyethylene glycol
- RT
radiotherapy
- siApe1
siRNA targeting Ape1
- siGFP
siRNA targeting GFP
1. Introduction
Pediatric primary brain tumors afflict approximately 3000 children annually and are the leading cause of cancer death in children (Dolecek et al., 2012). Effective treatments for medulloblastoma (MB) and ependymoma (EP), which comprise about 25% of pediatric brain tumors, remain elusive, and as a consequence these diagnoses contribute disproportionably to mortality. Due to the rarity of these childhood cancers, they are relatively understudied and there is a lack of novel treatment development. Radiotherapy (RT) is an integral component of the treatment for MB and the only effective adjuvant therapy for EP (Kilday et al., 2009; Mueller and Chang, 2009; Tamburrini et al., 2009; Witt et al., 2012). Despite advances in RT technique the 5‐year survival, especially for younger children, remains low (Dolecek et al., 2012), and effective therapies for recurrent disease have yet to be developed. Moreover, survival is frequently accompanied by one or more radiation‐induced adverse developmental and psychosocial sequelae as MB and EP most frequently occur in children less than 10 years old (Northcott et al., 2012). These considerations emphasize the need to develop new strategies to enhance the tumoricidal action of RT while sparing adjacent normal tissue.
The cytotoxic action of RT is primarily caused by the formation of double‐strand DNA breaks that arise as a consequence of oxidative free radical‐induced DNA lesions that impede DNA replication fork progression. Single‐strand breaks containing fragmented deoxyribose and abasic sites are the most common replication blocking lesions produced by radiation (Abbotts and Madhusudan, 2010; Demple and Harrison, 1994). The multifunctional DNA repair protein Ape1 initiates the excision of fragmented deoxyribose and intact abasic sites (hereafter referred to collectively as abasic lesions) and is essential for radiation resistance in human cells (Robertson et al., 2009). Suppression of Ape1 expression in MB and EP cell lines is accompanied by significantly elevated sensitivity to radiation as well as to abasic site inducing alkylating agents (Bobola et al., 2005, 2011). More importantly, overall and progression free survival following RT is inversely associated with the abasic lesion endonuclease activity of Ape1 in MB and EP tissue (Bobola et al., 2005, 2011). These findings strongly suggest that Ape1 promotes treatment resistance in MB and EP and that Ape1 is a target for anti‐resistance therapies.
There have been a number of chemical Ape1 inhibitors developed (Abbotts et al., 2014; Al‐Safi et al., 2012; Dorjsuren et al., 2012; Liu and Gerson, 2004; Srinivasan et al., 2012; Sultana et al., 2012), although only one, methoxyamine, an indirect inhibitor of Ape1 has progressed to Phase I and II clinical trials (Wilson and Simeonov, 2010). As opposed to chemical Ape1 inhibitors, delivery of short interfering RNAs (siRNAs) is advantageous owing to the potency and specificity of the RNA interference (RNAi) pathway that minimizes the off‐target effects inherent with small molecule drugs (Whitehead et al., 2009). However, clinical utilization of RNAi has been impeded by the lack of optimal siRNA delivery vehicles (Whitehead et al., 2009). Significant effort has been placed on the development of nanoparticle (NP) carriers of siRNA (Davis et al., 2010; Pecot et al., 2011; Whitehead et al., 2014). NPs for siRNA delivery must be able to bypass numerous physiological and cellular barriers to deliver siRNA to the intracellular site of action (Kievit and Zhang, 2011, 2011). First, NPs must efficiently condense and protect siRNA from degradation upon injection into the blood, while maintaining a size of between 10 and 100 nm to avoid clearance through the kidneys and reticuloendothelial system. Second, NPs must be able to extravasate from the blood across the blood–brain barrier, into the tumor. Third, the NPs must escape lysosomal degradation during cellular uptake by endocytosis, and traffic to the perinuclear region for siRNA to act with the RNA‐induced silencing complex to mediate gene knockdown.
Here we report on a NP delivery vehicle that effectively protects and transports siRNA to pediatric MB and EP cells. The NPs comprise an iron oxide (Fe3O4) core coated with a polymeric shell consisting of three materials: chitosan, low molecular‐weight polyethyleneimine (PEI), and polyethylene glycol (PEG). Chitosan is a biocompatible, biodegradable natural polymer that provides a stable coating on the NP and bears active sites for covalent binding of PEI and PEG. Polycationic PEI avidly binds nucleic acids electrostatically, enabling loading of large quantities of siRNA, and provides protection against degradation by serum nucleases. Although the use of PEI has been limited due to its cytotoxicity, our studies have shown that NPs coated with low molecular‐weight PEI (1200 Da) grafted to PEG‐chitosan demonstrate high gene delivery in brain cancer cells with no detectable cytotoxicity both in vitro and in vivo (Kievit et al., 2009, 2010, 2010). We show that siApe1 suppresses Ape1 expression and abasic lesion endonuclease activity in MB and EP cells. We also show that siApe1‐mediated suppression increases abasic lesion and double‐strand break abundance and reduces tumor cellular resistance to 137Cs‐γ‐rays. Our results suggest a new strategy to circumvent RT resistance in order to improve clinical outcome for MB and EP.
2. Materials and methods
2.1. Materials
All cell culture reagents were purchased from Invitrogen unless otherwise noted. All chemicals were purchased from Sigma unless otherwise noted. The MB‐derived UW228‐1 (Bobola et al., 2005) and EP‐derived Res196 (Bobola et al., 2011) cell lines were maintained in DMEM supplemented with 10% FBS and 1% antibiotic–antimycotic in a 37 °C humidified 95/5% air/CO2 incubator. siApe1 was purchased as a SMARTpool consisting of four validated siRNA sequences against APEX1 (Thermo Scientific). The anti‐green fluorescent protein siRNA (siGFP) was used as a non‐specific sequence control as previously described (Mok et al., 2010; Veiseh et al., 2010). Fluorophore labeled siRNA (siRNA‐Dy677) was purchased from Dharmacon.
2.2. Nanoparticle synthesis
Nanoparticles were synthesized with slight modification to previous methods (Kievit et al., 2009, 2010, 2010). Iron oxide nanoparticles (NP) with a core size of 4–6 nm coated with chitosan‐g‐PEG copolymer (CP) were prepared via a co‐precipitation method in the presence of copolymer as described (Veiseh et al., 2009) to generate NP–CP. NP–CP were then thiolated using Traut's reagent (Molecular Biosciences, Boulder, CO) at 5 mg Traut's reagent to 1 mg NPs (Fe equivalent) for 1.5 h in thiolation buffer (100 mM sodium bicarbonate, pH 8.0, 5 mM EDTA). Unreacted Traut's reagent was then removed by size exclusion chromatography using S‐200 Sephacryl (GE Healthcare). PEI (1.2 kDa) was activated with succinimidyl iodoacetate (SIA) at a 1:1 M ratio of PEI:SIA. PEI was diluted in thiolation buffer to 150 mg/mL and SIA dissolved in dimethylformamide (DMF) at 150 mg/mL was added and reacted for 30 min. Activated PEI was conjugated to thiolated NP–CPs at 62.5 mg PEI per 1 mg of NPs (Fe equivalent) for 2 h at room temperature and then at 4 °C overnight. Unreacted PEI was removed by S‐200 Sephacryl chromatography to generate NP–CP–PEI, called NP hereafter. NPs were stored at 4 °C in 10 mM HEPES, pH 7.4 and used within one week of synthesis.
2.3. Nanoparticle characterization
The sizes and zeta potentials of the NPs were determined using dynamic light scattering on a Zetasizer Nano (Malvern). Protection of siRNA was determined using a protection and release assay where NP‐bound or free siRNA was incubated with FBS for 30 min prior to releasing siRNA from the NP using heparin. Degradation was then assessed by polyacrylamide gel electrophoresis using unincubated NP‐bound or free siRNA as standards. Endosomal escape was evaluated using a calcein assay (Veiseh et al., 2010) in which cells were co‐incubated with NPs at 10 μg/mL and calcein at 0.25 mM for 2 h prior to imaging by fluorescence microscopy.
2.4. NP:siRNA treatment
NPs were loaded with siRNA (siApe1, siGFP, or siRNA‐Dy677) at a 10:1 NP:siRNA weight ratio and a nanoparticle concentration 200 μg Fe/mL Fe. NP:siRNA complexes were allowed to form for 30 min before treating cells. For siRNA‐Dy677 treatments, 100,000 cells were plated on 22 × 22 mm cover slips in 2 mL fully supplemented culture medium in 6‐well plates prior to 4 h treatment with 2 μg NP‐bound siRNA‐Dy677. After 24 h, cells were washed, fixed with 4% formaldehyde, counterstained with DAPI, and imaged by fluorescence microscopy. For siApe1 and siGFP treatments, cells were plated in 12‐well plates at 100,000 cells per well in 1 mL fully supplemented culture medium and treated with 1 μg NP‐bound siRNA (∼75 nM siRNA) prior to attachment. Three days after treatment, cells were harvested by trypsinization and washed pellets were stored at −80 °C prior to subsequent analyses.
2.5. Ape1 expression
2.5.1. mRNA
RNA was extracted from cells using the RNeasy mini kit (Qiagen) following the manufacturer's protocol. cDNA was prepared using the iScript cDNA synthesis kit (Bio‐Rad), Ape1 mRNA levels were determined by qRT‐PCR using CYBR green master mix (Bio‐Rad) and normalized to β‐actin mRNA content. Primers used for Ape1 were forward: CAACACACCCTATGCCTACA, reverse: GTAACAGAGAGTGGGACAA, and for β‐actin were forward: AGCGAGCATCCCCCAAAGTT, reverse: GGGCACGAAGGCTCATCATT.
2.5.2. Protein
Cell pellets were solubilized by incubation for 15 min on ice in 0.1% Triton X‐100 in PBS. Extracts were diluted 1:1 with Laemmli sample loading buffer containing 2% β‐mercaptoethanol. After heating at 100 °C for 5 min, 10 μg of extract protein was resolved by SDS‐PAGE and transferred onto nitrocellulose membranes. Membranes washed three times with TBS were incubated with 3% QuickBlocker (Chemicon) in TBS for 1 h at room temperature and then incubated overnight at 4 °C with 1 μg/mL antibody against Ape1 (rabbit polyclonal; Abcam, ab105081) or β‐actin (rabbit polyclonal; Abcam, ab75186) in TTBS containing 3% QuickBlocker. Membranes were washed with TTBS before being incubated for 1 h at room temperature with alkaline phosphatase‐conjugated goat anti‐rabbit secondary antibody (Bio‐Rad) diluted 1:3000 in 3% QuickBlocker. Membranes were then washed thrice with TTBS and antibody binding visualized by chemiluminescence (Immun‐Star detection kit; Bio‐Rad) and quantified using the ChemiDoc system running the Quantity One software package (Bio‐Rad).
2.6. Abasic endonuclease activity and abasic lesion quantitation
Abasic site endonuclease activity was measured in cleared supernatants of whole cell extracts using a sensitive assay that measures the conversion of acid‐treated, super‐coiled plasmid DNA to relaxed form caused by incision at abasic sites as detailed elsewhere (Bobola et al., 2005). This highly sensitive assay measures the abasic lesion endonuclease activity of cell extracts using supercoiled plasmid DNA substrate containing 1.5 abasic sites per plasmid (Bobola et al., 2001, 2005, 2011, 2002). In the presence of Ape1 activity, the cleaved abasic site relaxes the supercoiled plasmid to its open circular form (Figure 3a and b). Relaxed plasmid is quantitated by comparison with known amounts of linearized substrate allowing estimation of activity expressed as fmoles lesions incised per minute per cell (fmol/min/cell). Abasic lesion content in genomic DNA of radiation‐treated cells was measured by conjugation with an aldehyde reactive probe (Dojindo) that reacts with intact abasic sites (Kubo et al., 1992). Probe‐labeled DNA was stored at 4 °C overnight before quantification of abasic sites per the manufacturer's protocol.
Figure 3.
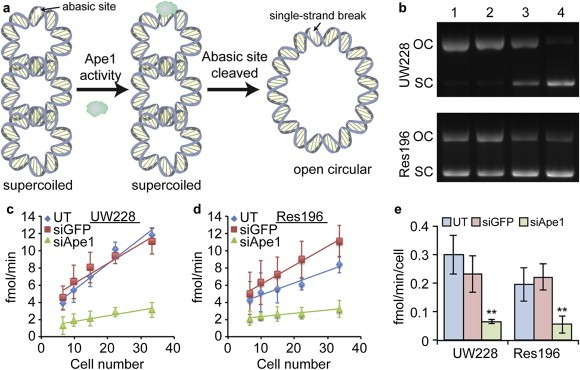
Knockdown of Ape1 activity. Ape1 activity assay showing reduced abasic site endonuclease activity 72 h after treatment with siApe1 loaded NPs. a) Principle of the Ape1 activity assay. Supercoiled (SC) plasmid DNA substrate containing a single abasic site relaxes to open circular (OC) form in the presence of active Ape1 enzyme. b) Agarose gel electrophoresis used to resolve the shift in abasic site containing SC plasmid substrate to OC form after treatment with UW228 (top) and Res196 (bottom) cell extracts. Lanes: 1. Untreated (UT) cells; 2. NP:siGFP treated cells; 3. NP:siApe1 treated cells; 4. Substrate only. c–d) Plot of numbers of abasic sites removed at various extract dilutions from c) UW228 and d) Res196 cells. Data is from three independent experiments. e) Quantification of Ape1 activity as determined from the slopes in (c–d). ** indicates a statistical difference from UT cells p < 0.001.
2.7. Radiation and drug sensitivity
The clonogenic assay was used for assessing radiation and drug sensitivity as it is the gold‐standard assay for assessing replicative cell death as it provides the best indication of long‐term cell death, as apposed to measuring changes in cell density or metabolism (e.g., XTT, Alamar blue, CellTiter Glo). NP‐treated cells were harvested by trypsinization, resuspended in medium and were immediately irradiated at 4 Gy/min with 137Cs‐γ‐rays. Six well trays were then inoculated with 250, 500 or 1000 irradiated cells in 2 mL supplemented medium. For drug exposure, cells were plated as above 66 h after initiating NP treatment and incubated for an additional 6 h to allow attachment and resumption of proliferation before treating with bleomycin or paclitaxel for 0.5 h and 4 h, respectively. After treatment, cells were changed to fresh medium, and incubation continued until colonies of ≥50 cells were formed after 10–14 days. Colonies were stained with 0.5% methylene blue in methanol/water (1:1 v/v) and counted using a dissecting microscope. Survival is the fraction of colonies formed by treated cells compared to untreated controls.
2.8. γH2AX immunostaining
After NP:siRNA and radiation treatment as described above, cells were plated on cover slips in 6‐well plates and allowed to attach for 24 h. After fixation with 4% formaldehyde and permeabilization with 0.1% Triton X‐100 in PBS, cells were incubated in PBS containing 10% FBS and 1% sodium azide (PSA) for 30 min. Cells were then incubated overnight at 4 °C with rabbit γH2AX monoclonal antibody (Thermo Scientific, 1:400 dilution) in PSA. After 3 washes with PSA, incubation was resumed with PSA containing FITC‐conjugated goat anti‐rabbit secondary antibody (Abcam, 1:1000 dilution). Washed cells were counterstained with DAPI and mounted onto slides using ProLong Gold antifade reagent (Invitrogen). Cells were visualized at 600× by fluorescence microscopy using a Nikon Ri1 Color Cooled Camera System (Nikon Instruments, Melville, NY). γH2AX foci were manually counted from at least 15 cells in each of five fields of view.
2.9. Statistical analyses
Data shown are mean ± standard deviation. Statistical significance was determined using Student's t‐test where p‐values of less than 0.05 were considered significant. Survival parameters were estimated from kill curves (log survival versus dose) by linear regression.
3. Results
3.1. NP bound siRNA is protected against degradation
NP physicochemical properties are important to ensure proper trafficking within the body and cell. Our NP comprises an iron oxide core coated with a biocompatible cationic copolymer of chitosan, PEG, and PEI (Figure 1a). The NP was approximately 40 nm as determined by dynamic light scattering (Figure 1b), and had a positive zeta potential, a measure of NP surface charge, of around 15 mV (Figure 1c). Binding and protection of siRNA against GFP was tested using a gel retardation, protection, and release assay (Figure 1d). Complete binding of siRNA was observed at a NP:siRNA weight ratio of 10:1 as evidenced by the lack of detectable free siRNA by gel electrophoresis (lane 4). Full‐length siRNA was released from the NP by incubation with heparin, an anionic molecule that competes for binding sites on the NP (lane 5). Importantly, the degradation of unbound siRNA by serum nucleases (lane 3) was not detectible for NP‐bound siRNA (lane 6), indicating that siRNA remains bound to the NP in serum and binding affords protection against serum nucleases. Earlier work from our laboratory revealed that NPs enter the cell via endocytosis (Fang et al., 2012; Veiseh et al., 2010). Uptake by and release from endosomes of NP:siRNA was evaluated by treatment of MB cells in the presence and absence of NP:siRNA with calcein, a fluorescent dye sequestered in intact endosomes (Figure 1e, left). In contrast, calcein fluorescence (green) was detected throughout cells treated concurrently with NP:siRNA, indicating endosomal release (Figure 1e, right). These results provide evidence of NP:siRNA uptake by endocytosis and subsequent release into the cell.
Figure 1.
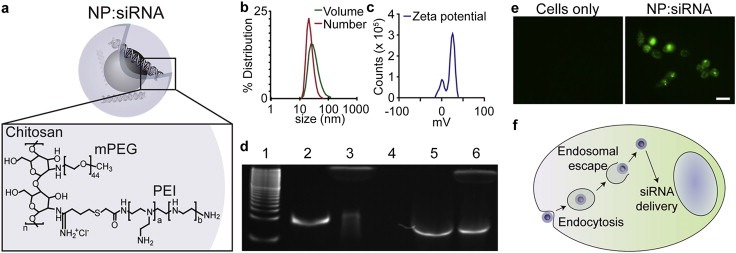
NP characteristics. a) Schematic illustration of NP with encapsulated siRNA. b) Hydrodynamic size of the NP as determined by DLS by volume and number average. c) Zeta potential of the NP. d) siRNA protection and release assay. Lanes of the polyacrylamide gel correspond to 1. 10 bp ladder, 2. siRNA, 3. siRNA + FBS, 4. NP:siRNA, 5. NP:siRNA + heparin, 6. NP:siRNA + FBS + heparin. e) fluorescence images of cells treated with calcein (left) and cells treated with NP:siRNA and calcein (right). Scale bar corresponds to 25 μm. f) Model of NP trafficking within the cell. NPs are taken up through endocytosis and escape the endosome to deliver siRNA to the perinuclear region.
3.2. NP:siRNA suppresses Ape1 expression and activity
To evaluate the efficacy of our NP as an siRNA delivery vehicle, we assayed suppression of Ape1 expression in UW228‐1 and Res196 incubated with NP:siRNA for 72 h. As illustrated in Figure 2, treatment reduced Ape1 mRNA abundance to 25 ± 6% (p < 0.001) and 15 ± 2% (p < 0.001) of that of untreated UW228‐1 and Res196 cells, respectively (Figure 2b). Treating cells with NP‐bound siRNA targeting green fluorescent protein (NP:siGFP, as control) had no significant effect on Ape1 mRNA levels in UW228‐1 (87 ± 11%) and in Res196 (99 ± 11%) cells. As shown in Figure 2c–d, Ape1 protein expression was also reduced relative to untreated controls in AP:siApe1‐treated UW228‐1 (11 ± 7.1%, p < 0.001) and Res196 (7.2 ± 1.4%, p < 0.001) cells while NP:siGFP had little effect on Ape1 protein content in UW228‐1 (107 ± 21%) and Res196 cells (82 ± 12%). These data provide strong evidence that NP:siApe1 protects siApe1 against lysosomal degradation after endocytosis and facilitates release to the intracellular site of action for RNAi (Figure 1f).
Figure 2.
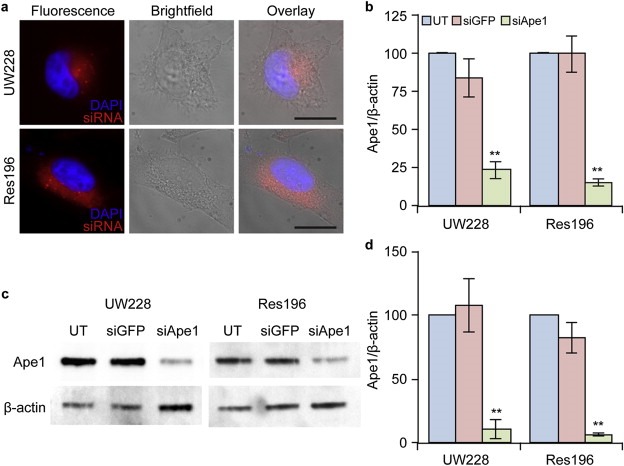
NP‐mediated knockdown of Ape1 expression in UW228 (MB) and Res196 (EP) cells. a) Fluorescence images of cells treated with NPs and NP‐mediated intracellular delivery of fluorophore labeled siRNA (red) to the perinuclear region of cells. The scale bars correspond to 10 μm. b) qRT‐PCR analysis showing knockdown of Ape1 mRNA 72 h after treatment with NPs where UT: untreated cells; siGFP: NP:siGFP treated cells; siApe1: NP:siApe1 treated cells. c) Western blot showing knockdown of Ape1 protein 72 h after treatment. d) Quantification of Western blot band density from three independent experiments. Data shown is normalized to UT. ** indicates a statistical difference from untreated (p < 0.001).
Suppression of Ape1 mRNA and protein content was accompanied by significant reduction in abasic endonuclease activity determined by a biochemical assay (Figure 3a). Both UW228‐1 and Res196 treated with NP:siApe1 displayed reduced incision at substrate abasic lesions compared to cells treated with NP:siGFP (Figure 3b) and decreased rate of abasic lesions cleaved/min with increasing cell number (Figure 3c–d). As a result, activity was approximately 4‐fold lower in both UW228‐1 (0.07 ± 0.007 vs 0.30 ± 0.007 fmol sites/cell/min; p ≤ 0.001) and Res196 (0.055 ± 0.03 vs 0.19 ± 0.05 fmol sites/cell/min; p ≤ 0.001). In contrast, treatment with NP:siGFP had little effect on activity in UW228‐1 (0.23 ± 0.07 vs 0.30 ± 0.007 fmol sites/cell/min) and Res196 (0.22 ± 0.05 vs 0.19 ± 0.05 fmol sites/cell/min). These results show that our NP can deliver a biologically active siRNA that suppresses both Ape1 expression and Ape1‐mediated cleavage at abasic lesions.
3.3. Nanoparticle‐mediated knockdown of Ape1 enhances the DNA damaging effects of radiation
UW228‐1 and Res196 cells treated with siRNA loaded NPs were subjected to 2 Gy γ‐irradiation, and abasic sites were quantified before, 30 min after, and 4 h after irradiation (Figure 4). Prior to irradiation, untreated, NP:siGFP treated, and NP:siApe1 treated cells all had similar numbers of abasic sites in their genomic DNA. There were 32 ± 17, 34 ± 8, and 44 ± 11 abasic sites per 105 bp for untreated, NP:siGFP treated, and NP:siApe1 treated UW228‐1 cells, respectively. There were 31 ± 20, 25 ± 15, and 32 ± 21 abasic sites per 105 bp for untreated, NP:siGFP treated, and NP:siApe1 treated Res196 cells, respectively. Although there was a slight increase in the numbers of abasic sites in cells treated with NP:siApe1, which would be expected for cells with a lowered ability to repair these sites, this increase was not statistically significant. Shortly after irradiation, an increase in the numbers of abasic sites in the DNA was observed as expected. There were 41 ± 29, 45 ± 9, and 65 ± 51 abasic sites per 105 bp for untreated, NP:siGFP treated, and NP:siApe1 treated UW228‐1 cells, respectively. There were 23 ± 16, 31 ± 19, and 54 ± 19 abasic sites per 105 bp for untreated, NP:siGFP treated, and NP:siApe1 treated Res196 cells, respectively. No significant increase was observed for UW228‐1 cells treated with NP:siApe1 as compared to untreated (p ≥ 0.27) and NP:siGFP (p ≥ 0.25) at this time point. Res196 cells treated with NP:siApe1 showed a significant increase in the number of abasic sites as compared to untreated (p < 0.05) and NP:siGFP (p < 0.01) at this time point. After four hours, abasic sites in untreated and NP:siGFP treated cells reached their baseline levels at 35 ± 27 and 29 ± 10 abasic sites per 105 bp, respectively for UW228‐1 and at 21 ± 14 and 21 ± 9 abasic sites per 105 bp, respectively for Res196. NP:siApe1 treated cells maintained a significantly higher number of abasic sites after four hours at 85 ± 45 (p < 0.05 vs untreated; p < 0.01 vs NP:siGFP) abasic sites per 105 bp for UW228‐1 and at 49 ± 22 (p < 0.05 vs untreated; p < 0.01 vs NP:siGFP) abasic sites per 105 bp for Res196.
Figure 4.
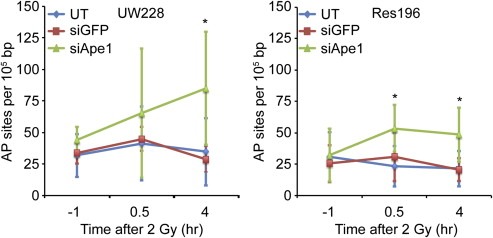
Abasic site quantification after UW228 (left) and Res196 (right) cells were exposed to 2 Gy 137Cs‐γ‐rays. Abasic sites were repaired in untreated and NP:siGFP treated cells with high Ape1 activity within 4 h, but abasic sites persisted for at least 4 h in NP:siApe1 treated cells with low Ape1 activity. Data is from three independent experiments. * indicates a statistical difference from untreated (UT) and NP:siGFP treated cells (p < 0.05).
The evolution of abasic sites into double‐strand breaks was probed through γH2AX immunostaining and foci counting (Figure 5). Untreated and NP:siGFP treated UW228‐1 cells had 1.7 ± 1.1 and 1.5 ± 0.8 foci per cell, respectively, 24 h after 2 Gy irradiation whereas NP:siApe1 treated UW228 cells had 6.5 ± 3.6 foci per cell. In Res196 cells, untreated and NP:siGFP treated cells had 2.5 ± 1.0 and 5.8 ± 2.5 foci per cell, respectively, 24 h after 2 Gy irradiation whereas NP:siApe1 treated cells had 9.3 ± 2.3 foci per cell.
Figure 5.
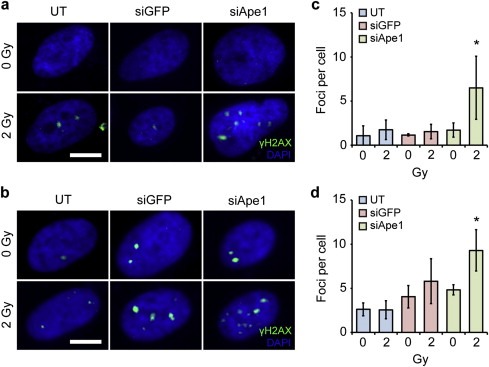
Double‐strand break quantification as determined by γH2AX foci counting. a–b) Fluorescence images of γH2AX immunostaining (green) with DAPI counterstaining (blue) in (a) UW228 and (b) Res196 cells. Scale bars correspond to 5 μm. c–d) Quantification of foci per cell through manual counting in (c) UW228 and (d) Res196 cells. Data shown are mean ± SD of greater than 15 cells per condition in at least three independent experiments. * indicates a statistical difference between pre and post‐irradiation (p < 0.05).
3.4. Nanoparticle‐mediated knockdown of Ape1 enhances the sensitivity of cells to low doses of radiation
The effect of NP:siApe1 on the radiosensitivity of UW228‐1 and Res196 was evaluated by clonogenic survival assays. As shown in Figure 6, both lines treated with NP:siApe1 displayed greater sensitivity to 137Cs‐γ‐rays compared to untreated and NP:siGFP‐treated cells. The effect of NP:siApe1 was most notable at doses less than 2 Gy, the standard fractionated dose in most RT treatment regimens (Lannering et al., 2012; Stuben et al., 1997). The detectible shoulder of resistance displayed by untreated and NP:siGFP‐treated cells was eliminated with NP:siApe1 treatment, indicating that repair of radiation damage by Ape1 was the predominant determinate of the insensitivity of UW228‐1 and Res196 at low doses. The effect of NP:siApe1‐mediated reduction of the shoulder of resistance is also reflected in the greater than 3‐fold reduction of LD50 (Table 1) compared to untreated (1.3 ± 0.5 vs 4.5 ± 0.9 Gy; p ≤ 0.01) and NP:siGFP‐treated (1.3 ± 0.5 vs 4.3 ± 0.3 Gy; p ≤ 0.001) in UW228‐1, and the approximately 3‐fold reduction of LD50 compared to untreated (0.9 ± 0.4 vs 2.7 ± 0.1 Gy; p ≤ 0.01) and NP:siGFP‐treated (0.9 ± 0.4 vs 3.0 ± 0.2 Gy; p ≤ 0.01) in Res196. We note that an appreciable fraction of NP:siApe1‐treated UW228‐1 and Res196 displace similar radiation sensitivity at doses greater than 2 Gy as untreated and NP:siGFP‐treated cells (Table 1). In light of the near complete reduction of Ape1 expression (2, 3), it is unlikely that this reflects failure of NP:siApe1 to suppress Ape1‐mediated repair, but rather reflects the dominant action of other repair activities to foster recovery (Lord and Ashworth, 2012).
Figure 6.
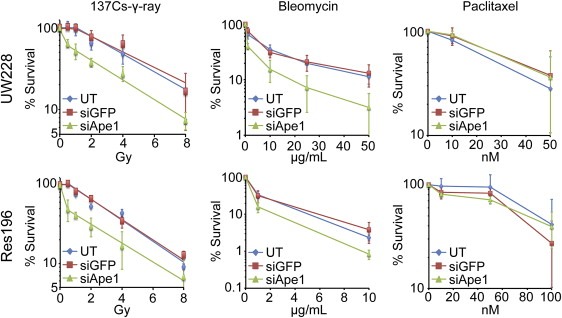
Clonogenic survival after exposure to 137Cs‐γ‐rays, bleomycin, and paclitaxel. UW228 and Res196 cells were left untreated or treated with siGFP or siApe1 loaded NPs for three days prior to γ‐ray, bleomycin, or paclitaxel exposure. NP‐mediated knockdown of Ape1 sensitized cells specifically to therapies that generate abasic sites in genomic DNA. Data is from three independent experiments.
Table 1.
NP‐mediated suppression of Ape1 activity in pediatric brain cancer cells increases radiosensitivity. Data is from three independent experiments.
| D10a | D37a | LD50a | ||
|---|---|---|---|---|
| UW228 | UT | 12 ± 2.7 | 5.9 ± 1.1 | 4.5 ± 0.9 |
| siGFP | 12 ± 2.5 | 5.7 ± 0.7 | 4.3 ± 0.3 | |
| siApe1 | 7.0 ± 0.6 | 2.4 ± 0.3 | 1.3 ± 0.5 | |
| Res196 | UT | 8.0 ± 0.7 | 3.7 ± 0.2 | 2.7 ± 0.1 |
| siGFP | 8.8 ± 0.6 | 4.0 ± 0.3 | 3.0 ± 0.2 | |
| siApe1 | 5.2 ± 1.0 | 1.7 ± 0.5 | 0.9 ± 0.4 | |
Gy.
Importantly, NP:siApe1 also increased the sensitivity of both lines to bleomycin, an oxidizing agent that also produces abasic sites and strand breaks containing fragmented deoxyribose moieties (Figure 6). In contrast, NP:siApe1 had no effect on sensitivity to paclitaxel, a microtubule disrupting agent (Figure 6). These results indicate the radiosensitization afforded by NP:siApe1 was specific to the suppression of Ape1‐mediated repair and not the consequence of a non‐specific sensitization to cytotoxic agents.
4. Discussion
The promise of RNAi to effect gene‐specific therapies in cancer therapy has been stymied by the lack of a means to circumvent numerous anatomic, biochemical, and physiological barriers to the delivery of biologically active siRNA to tumors. Our prototype NP was designed to circumvent these limitations in order to transport siRNA to pediatric brain tumors. Inclusion of PEI to the biocompatible copolymer of chitosan and PEG bound to an iron oxide core together with the positive zeta potential of the resulting NP promoted binding and protection of siRNA while preserving the ability of the polymer to facilitate cellular uptake and perinuclear localization (Kievit et al., 2009). Importantly, our NP protects siRNA from degradation by serum nucleases, an essential defense for active siRNA to survive passage through the circulation. Also critical, the small size of our NP, approximately 40 nm, promotes internalization by target cells and minimizes clearance from circulation (Chithrani and Chan, 2007; Chithrani et al., 2006; Wang et al., 2013; Zhang et al., 2009). In toto, these properties suggest our NP can function in vivo as a systemic delivery vehicle.
To evaluate the potential clinical utility of our NP, we examined the ability of NP:siApe1 to reduce radioresistance in cell lines derived from pediatric MB and EP. RT is an essential part of the standard adjuvant care for both tumors (Kilday et al., 2009; Tamburrini et al., 2009; Witt et al., 2012), but efficacy is frequently limited by intrinsic resistance and the potential of adverse effects that accompany treatment in the pediatric population (Mueller and Chang, 2009). Ape1 catalyzes the vast majority (∼95%) of abasic site endonuclease activity in human tumors, which are estimated to harbor 105–106 molecules/cell (Bobola et al., 2005, 2011, 2012). Other considerations guiding this study were the essential role Ape1 plays in the repair of radiation‐induced precursors of lethal double‐strand breaks (Kelley et al., 2012), and the strong inverse association between tumor abasic endonuclease activity and progression‐free survival following RT for both MB (Bobola et al., 2005) and EP (Bobola et al., 2011). Our results document that NP:siApe1 produces near quantitative suppression of Ape1 expression and abasic endonuclease activity in cell lines with activities greater than the mean observed in MB and EP tissue (Bobola et al., 2005, 2011). Importantly, reduced expression produced near total ablation of resistance to radiation at clinically relevant doses, resulting in a 3‐fold decrease in LD50 for both UW228‐1 and Res196 (Figure 6). Our data also indicate that increased sensitivity was specific to a diminished ability to repair damage substrates of Ape1 rather than non‐specific effects of NP:siApe1 on survival. We cannot, however, rule out that suppression of other Ape1 activities such as the redox function of Ref‐1 (Kelley et al., 2012) contributed to circumventing radiation resistance. Overall, our findings provide strong evidence that NP:siApe1 is an effective vehicle for delivering biologically active siRNA to pediatric brain tumor cells.
The survival curves for NP:siApe1‐treated UW228‐1 and Res196 are biphasic, both harboring a population of cells that show the same sensitivity to γ‐rays as untreated and siGFP‐treated cells, as shown by the similar slopes of the curves for the three groups at higher doses (Figure 6). The magnitude of reduction of gene expression and abasic endonuclease activity by NP:siApe1 strongly suggests that a large majority of treated cells received active siRNA. We note the possibility that a single treatment with NP:siApe1 was insufficient to suppress repair long enough in some cells for abasic lesions to be converted to lethal double‐strand breaks by blocking DNA replication. Importantly, it is estimated that 50% of double‐strand breaks result from incision by Ape1 at closely opposed lesions on opposite DNA strands (Georgakilas et al., 2004; Sutherland et al., 2000; Yang et al., 2006). As closely opposed lesions are more likely to form at higher doses, it is possible that suppression of Ape1 may reduce the burden of double‐strand breaks (Fung and Demple, 2011), facilitating survival via double‐strand break repair mechanisms. Suppressing these activities along with Ape1 would be expected to further increase radiosensitivity.
The clinical potential of NP:siApe1 will next be evaluated in animal models of MB and EP (Huse and Holland, 2009). To be effective, the NP must protect siRNA integrity while circumventing a variety of anatomic and physiological barriers to reach tumors in the central nervous system. Our previous experience is that iron core NPs with similar size and polymer compositions are not rapidly cleared from circulation (Kievit and Zhang, 2011b), and can effectively deliver nucleic acids to animal models of brain tumors (Kievit et al., 2009). Notably, attaching the targeting agent chlorotoxin, a peptide derived from the venom of the giant Israeli scorpion that binds to the vast majority of brain tumors (Lyons et al., 2002; Soroceanu et al., 1998; Veiseh et al., 2007), enhanced NP uptake and distribution throughout the tumor (Kievit et al., 2010). Importantly, we previously observed that chlorotoxin‐targeted chitosan‐based NPs cross the blood–brain barrier in a transgenic mouse model of MB (Veiseh et al., 2009). These results inspire confidence that NP:siApe1 will be able to deliver an effective dose of siApe1 to model MB and EP tumors in vivo.
Acknowledgments
This work was supported in part by NIH grants R01CA161953, R01CA134213, and R01EB006043, and Seattle Children's Hospital. F.M.K., Z.R.S., and J.G.S. acknowledge support through an NCI training grant T32CA138312. F.M.K. also acknowledges support from the American Brain Tumor Association Basic Research Fellowship in Honor of Susan Kramer. K.W. acknowledges support from the University of Washington College of Engineering Dean's Fellowship. We thank Jeffery Schwartz for use of the gamma irradiator.
Kievit Forrest M., Stephen Zachary R., Wang Kui, Dayringer Christopher J., Sham Jonathan G., Ellenbogen Richard G., Silber John R., Zhang Miqin, (2015), Nanoparticle mediated silencing of DNA repair sensitizes pediatric brain tumor cells to γ-irradiation, Molecular Oncology, 9, doi: 10.1016/j.molonc.2015.01.006.
Contributor Information
Richard G. Ellenbogen, Email: rge@uw.edu
Miqin Zhang, Email: mzhang@uw.edu.
References
- Abbotts, R. , Jewell, R. , Nsengimana, J. , Maloney, D.J. , Simeonov, A. , Seedhouse, C. , Elliott, F. , Laye, J. , Walker, C. , Jadhav, A. , Grabowska, A. , Ball, G. , Patel, P.M. , Newton-Bishop, J. , Wilson, D.M. , Madhusudan, S. , 2014. Targeting human apurinic/apyrimidinic endonuclease 1 (APE1) in phosphatase and tensin homolog (PTEN) deficient melanoma cells for personalized therapy. Oncotarget 5, 3273–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbotts, R. , Madhusudan, S. , 2010. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat. Rev. 36, 425–435. [DOI] [PubMed] [Google Scholar]
- Al-Safi, R.I. , Odde, S. , Shabaik, Y. , Neamati, N. , 2012. Small-molecule inhibitors of APE1 DNA repair function: an overview. Curr. Mol. Pharmacol. 5, 14–35. [PubMed] [Google Scholar]
- Bobola, M.S. , Blank, A. , Berger, M.S. , Stevens, B.A. , Silber, J.R. , 2001. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin. Cancer Res. 7, 3510–3518. [PubMed] [Google Scholar]
- Bobola, M.S. , Finn, L.S. , Ellenbogen, R.G. , Geyer, J.R. , Berger, M.S. , Braga, J.M. , Meade, E.H. , Gross, M.E. , Silber, J.R. , 2005. Apurinic/apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin. Cancer Res. 11, 7405–7414. [DOI] [PubMed] [Google Scholar]
- Bobola, M.S. , Jankowski, P.P. , Gross, M.E. , Schwartz, J. , Finn, L.S. , Blank, A. , Ellenbogen, R.G. , Silber, J.R. , 2011. Apurinic/apyrimidinic endonuclease is inversely associated with response to radiotherapy in pediatric ependymoma. Int. J. Cancer 129, 2370–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chithrani, B.D. , Chan, W.C. , 2007. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 7, 1542–1550. [DOI] [PubMed] [Google Scholar]
- Chithrani, B.D. , Ghazani, A.A. , Chan, W.C. , 2006. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 6, 662–668. [DOI] [PubMed] [Google Scholar]
- Davis, M.E. , Zuckerman, J.E. , Choi, C.H. , Seligson, D. , Tolcher, A. , Alabi, C.A. , Yen, Y. , Heidel, J.D. , Ribas, A. , 2010. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464, 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demple, B. , Harrison, L. , 1994. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63, 915–948. [DOI] [PubMed] [Google Scholar]
- Dolecek, T.A. , Propp, J.M. , Stroup, N.E. , Kruchko, C. , 2012. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro-oncology 14, (Suppl. 5) v1–v49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorjsuren, D. , Kim, D. , Vyjayanti, V.N. , Maloney, D.J. , Jadhav, A. , Wilson, D.M. , Simeonov, A. , 2012. Diverse small molecule inhibitors of human apurinic/apyrimidinic endonuclease APE1 identified from a screen of a large public collection. PLoS One 7, e47974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, C. , Kievit, F.M. , Veiseh, O. , Stephen, Z.R. , Wang, T.Z. , Lee, D.H. , Ellenbogen, R.G. , Zhang, M.Q. , 2012. Fabrication of magnetic nanoparticles with controllable drug loading and release through a simple assembly approach. J. Control. Release 162, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung, H. , Demple, B. , 2011. Distinct roles of Ape1 protein in the repair of DNA damage induced by ionizing radiation or bleomycin. J. Biol. Chem. 286, 4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas, A.G. , Bennett, P.V. , Wilson, D.M. , Sutherland, B.M. , 2004. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res. 32, 5609–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse, J.T. , Holland, E.C. , 2009. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol. 19, 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, M.R. , Georgiadis, M.M. , Fishel, M.L. , 2012. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 5, 36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit, F.M. , Veiseh, O. , Bhattarai, N. , Fang, C. , Gunn, J.W. , Lee, D. , Ellenbogen, R.G. , Olson, J.M. , Zhang, M. , 2009. PEI-PEG-chitosan copolymer coated iron oxide nanoparticles for safe gene delivery: synthesis, complexation, and transfection. Adv. Funct. Mater. 19, 2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit, F.M. , Veiseh, O. , Fang, C. , Bhattarai, N. , Lee, D. , Ellenbogen, R.G. , Zhang, M. , 2010. Chlorotoxin labeled magnetic nanovectors for targeted gene delivery to glioma. ACS Nano 4, 4587–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit, F.M. , Zhang, M. , 2011. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv. Mater. 23, H217–H247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit, F.M. , Zhang, M. , 2011. Surface engineering of iron oxide nanoparticles for targeted cancer therapy. Acc. Chem. Res. 44, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilday, J.P. , Rahman, R. , Dyer, S. , Ridley, L. , Lowe, J. , Coyle, B. , Grundy, R. , 2009. Pediatric ependymoma: biological perspectives. Mol. Cancer Res. – MCR 7, 765–786. [DOI] [PubMed] [Google Scholar]
- Kubo, K. , Ide, H. , Wallace, S.S. , Kow, Y.W. , 1992. A novel, sensitive, and specific assay for abasic sites, the most commonly produced DNA lesion. Biochemistry 31, 3703–3708. [DOI] [PubMed] [Google Scholar]
- Lannering, B. , Rutkowski, S. , Doz, F. , Pizer, B. , Gustafsson, G. , Navajas, A. , Massimino, M. , Reddingius, R. , Benesch, M. , Carrie, C. , Taylor, R. , Gandola, L. , Bjork-Eriksson, T. , Giralt, J. , Oldenburger, F. , Pietsch, T. , Figarella-Branger, D. , Robson, K. , Forni, M. , Clifford, S.C. , Warmuth-Metz, M. , von Hoff, K. , Faldum, A. , Mosseri, V. , Kortmann, R. , 2012. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J. Clin. Oncol. – Off. J. Am. Soc. Clin. Oncol. 30, 3187–3193. [DOI] [PubMed] [Google Scholar]
- Liu, L. , Gerson, S.L. , 2004. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr. Opin. Investig. Drugs 5, 623–627. [PubMed] [Google Scholar]
- Lord, C.J. , Ashworth, A. , 2012. The DNA damage response and cancer therapy. Nature 481, 287–294. [DOI] [PubMed] [Google Scholar]
- Lyons, S.A. , O'Neal, J. , Sontheimer, H. , 2002. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 39, 162–173. [DOI] [PubMed] [Google Scholar]
- Mok, H. , Veiseh, O. , Fang, C. , Kievit, F.M. , Wang, F.Y. , Park, J.O. , Zhang, M. , 2010. pH-Sensitive siRNA nanovector for targeted gene silencing and cytotoxic effect in cancer cells. Mol. Pharm. 7, 1930–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, S. , Chang, S. , 2009. Pediatric brain tumors: current treatment strategies and future therapeutic approaches. Neurotherapeutics – J. Am. Soc. Exp. NeuroTherapeutics 6, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott, P.A. , Jones, D.T. , Kool, M. , Robinson, G.W. , Gilbertson, R.J. , Cho, Y.J. , Pomeroy, S.L. , Korshunov, A. , Lichter, P. , Taylor, M.D. , Pfister, S.M. , 2012. Medulloblastomics: the end of the beginning. Nat. Rev. Cancer 12, 818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot, C.V. , Calin, G.A. , Coleman, R.L. , Lopez-Berestein, G. , Sood, A.K. , 2011. RNA interference in the clinic: challenges and future directions. Nat. Rev. Cancer 11, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, A.B. , Klungland, A. , Rognes, T. , Leiros, I. , 2009. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol. Life Sci. – CMLS 66, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber, J.R. , Bobola, M.S. , Blank, A. , Schoeler, K.D. , Haroldson, P.D. , Huynh, M.B. , Kolstoe, D.D. , 2002. The apurinic/apyrimidinic endonuclease activity of Ape1/Ref-1 contributes to human glioma cell resistance to alkylating agents and is elevated by oxidative stress. Clin. Cancer Res. 8, 3008–3018. [PubMed] [Google Scholar]
- Soroceanu, L. , Gillespie, Y. , Khazaeli, M.B. , Sontheimer, H. , 1998. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 58, 4871–4879. [PubMed] [Google Scholar]
- Srinivasan, A. , Wang, L. , Cline, C.J. , Xie, Z. , Sobol, R.W. , Xie, X.Q. , Gold, B. , 2012. Identification and characterization of human apurinic/apyrimidinic endonuclease-1 inhibitors. Biochemistry 51, 6246–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuben, G. , Stuschke, M. , Kroll, M. , Havers, W. , Sack, H. , 1997. Postoperative radiotherapy of spinal and intracranial ependymomas: analysis of prognostic factors. Radiother. Oncol. – J. Eur. Soc. Ther. Radiol. Oncol. 45, 3–10. [DOI] [PubMed] [Google Scholar]
- Sultana, R. , McNeill, D.R. , Abbotts, R. , Mohammed, M.Z. , Zdzienicka, M.Z. , Qutob, H. , Seedhouse, C. , Laughton, C.A. , Fischer, P.M. , Patel, P.M. , Wilson, D.M. , Madhusudan, S. , 2012. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int. J. Cancer 131, 2433–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, B.M. , Bennett, P.V. , Sidorkina, O. , Laval, J. , 2000. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. U. S. A. 97, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburrini, G. , D'Ercole, M. , Pettorini, B.L. , Caldarelli, M. , Massimi, L. , Di Rocco, C. , 2009. Survival following treatment for intracranial ependymoma: a review. Childs Nerv. Syst. – ChNS – Off. J. Int. Soc. Pediatr. Neurosurg. 25, 1303–1312. [DOI] [PubMed] [Google Scholar]
- Veiseh, M. , Gabikian, P. , Bahrami, S.B. , Veiseh, O. , Zhang, M. , Hackman, R.C. , Ravanpay, A.C. , Stroud, M.R. , Kusuma, Y. , Hansen, S.J. , Kwok, D. , Munoz, N.M. , Sze, R.W. , Grady, W.M. , Greenberg, N.M. , Ellenbogen, R.G. , Olson, J.M. , 2007. Tumor paint: a chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 67, 6882–6888. [DOI] [PubMed] [Google Scholar]
- Veiseh, O. , Kievit, F.M. , Fang, C. , Mu, N. , Jana, S. , Leung, M.C. , Mok, H. , Ellenbogen, R.G. , Park, J.O. , Zhang, M. , 2010. Chlorotoxin bound magnetic nanovector tailored for cancer cell targeting, imaging, and siRNA delivery. Biomaterials 31, 8032–8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiseh, O. , Sun, C. , Fang, C. , Bhattarai, N. , Gunn, J. , Kievit, F. , Du, K. , Pullar, B. , Lee, D. , Ellenbogen, R.G. , Olson, J. , Zhang, M. , 2009. Specific targeting of brain tumors with an optical/magnetic resonance imaging nanoprobe across the blood-brain barrier. Cancer Res. 69, 6200–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Kievit, F.M. , Veiseh, O. , Arami, H. , Stephen, Z.R. , Fang, C. , Liu, Y. , Ellenbogen, R.G. , Zhang, M. , 2013. Targeted cell uptake of a noninternalizing antibody through conjugation to iron oxide nanoparticles in primary central nervous system lymphoma. World Neurosurg. 80, 134–141. [DOI] [PubMed] [Google Scholar]
- Whitehead, K.A. , Dorkin, J.R. , Vegas, A.J. , Chang, P.H. , Veiseh, O. , Matthews, J. , Fenton, O.S. , Zhang, Y. , Olejnik, K.T. , Yesilyurt, V. , Chen, D. , Barros, S. , Klebanov, B. , Novobrantseva, T. , Langer, R. , Anderson, D.G. , 2014. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun. 5, 4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead, K.A. , Langer, R. , Anderson, D.G. , 2009. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, D.M. , Simeonov, A. , 2010. Small molecule inhibitors of DNA repair nuclease activities of APE1. Cell Mol. Life Sci. CMLS 67, 3621–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt, H. , Korshunov, A. , Pfister, S.M. , Milde, T. , 2012. Molecular approaches to ependymoma: the next step(s). Curr. Opin. Neurol. 25, 745–750. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Chaudhry, M.A. , Wallace, S.S. , 2006. Base excision repair by hNTH1 and hOGG1: a two edged sword in the processing of DNA damage in gamma-irradiated human cells. DNA Repair 5, 43–51. [DOI] [PubMed] [Google Scholar]
- Zhang, S. , Li, J. , Lykotrafitis, G. , Bao, G. , Suresh, S. , 2009. Size-dependent endocytosis of nanoparticles. Adv. Mater. 21, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]