

Abstract
Neuronal ceroid lipofuscinosis (NCL), commonly referred to as Batten disease, is a group of autosomal recessive neurodegenerative diseases of childhood characterized by seizures, blindness, motor and cognitive decline and premature death. Currently, there are over 400 known mutations in 14 different genes, leading to five overlapping clinical variants of NCL. A large portion of these mutations lead to premature stop codons (PTCs) and are predicted to predispose mRNA transcripts to nonsense-mediated decay (NMD). Nonsense-mediated decay is associated with a number of other genetic diseases and is an important regulator of disease pathogenesis. We contend that NMD targets PTCs in NCL gene transcripts for degradation. A number of PTC mutations in CLN1, CLN2 and CLN3 lead to a significant decrease in mRNA transcripts and a corresponding decrease in protein levels and function in patient-derived lymphoblast cell lines. Inhibiting NMD leads to an increased transcript level, and where protein function is known, increased activity. Treatment with read-through drugs also leads to increased protein function. Thus, NMD provides a promising therapeutic target that would allow read-through of transcripts to enhance protein function and possibly ameliorate Batten disease pathogenesis.
INTRODUCTION
Neuronal ceroid lipofuscinosis (NCL or Batten disease) is composed of a group of autosomal recessive neurodegenerative diseases of childhood (1). These disorders are characterized pathologically by the accumulation of autofluorescent storage material within lysosomes, and are considered lysosomal storage disorders (LSDs), with an estimated world-wide incidence of ∼1 in 100 000 live births (2). NCL typically presents with visual deficits due to retinal degeneration that gradually progresses to complete blindness. In addition, patients can suffer from seizures, motor abnormalities, ataxia, mental retardation leading to dementia and eventually premature death (3). The neurological symptoms of NCL are due to an extensive loss of neurons in the retina and central nervous system, predominantly in the cerebral and cerebellar cortices. Cortical atrophy is also accompanied by subcortical neuron loss, especially in the thalamus. Currently, there are over 400 known genetic mutations in at least 14 genes that lead to NCL (www.ucl.ac.uk/ncl/mutation.shtml), many of which have not been fully characterized (3–7). Mutations in many of these genes lead to a number of overlapping clinical variants, which have variable ages of onset and severity. There is no cure for any of the NCL subtypes, only long-term palliative care for the progressive symptomology. A large portion of NCL gene mutations lead to a premature termination codon (PTC, also called nonsense codon or premature stop codon) within the mRNA transcript (6). These various mutations range from base substitutions that directly introduce a nonsense mutation, insertions and deletions that shift the reading frame resulting in a PTC, and mutations within introns that cause aberrant splicing and subsequent frameshifts leading to PTCs. Early stop codons predispose NCL mRNAs to premature degradation by the nonsense-mediated decay (NMD) pathway. In previous work we have shown that the 1.02 kb deletion in CLN3 leads to a significant decrease in mRNA transcript abundance, most likely due to NMD (8).
NMD is a translational-dependent mRNA quality control mechanism in eukaryotes that recognizes and eliminates aberrant transcripts, such as those containing PTCs, for the prevention of abnormal protein production (9–16). This pathway is crucial for the post-transcriptional control of human gene expression, and has become an important therapeutic target for genetic diseases, such as cystic fibrosis, Duchenne and Becker muscular dystrophy, Usher syndrome type 1C and various LSDs (17–22). Approximately 75% of human genes produce pre-mRNA that undergoes alternative splicing, and over one-third of alternatively spliced mature mRNAs are targeted for degradation by the NMD pathway (23). In addition, ∼30% of all human genetic disorders arise as a consequence of nonsense or frameshift mutations that introduce a PTC (24). NMD plays a critical role in disease pathogenesis through the prevention of the potential dominant-negative effect of non-functional protein within the cell, as well as the prevention of abnormal protein accumulation and subsequent initiation of the ER stress response. It is therefore crucial for proper cell function that mRNA transcripts harboring PTCs are recognized early by NMD and degraded. Although NMD has been shown to modulate disease phenotype through the removal of mutated gene transcripts in several genetic disorders, it is becoming increasingly apparent that NMD can worsen disease phenotypes by preventing the translation of truncated protein that may yet retain function (14, 25–27).
Mutations in CLN1, which encodes palmitoyl-protein thioesterase 1 (PPT1), are the basis for infantile neuronal ceroid lipofuscinosis (INCL or classic INCL). Alternatively referred to as CLN1 disease (28). Classic INCL is the most severe form of NCL, presenting in children at 1–2 years of age, and typically leads to death by ages 8–11 (1). In addition, mutations in CLN1 are associated with other clinical variants, such as late-infantile, juvenile and adult NCL. To date, there are 61 CLN1 mutations that have been identified, consisting of 27 missense, 11 nonsense, 10 splice-site affecting, 4 insertions, 6 deletions and various others (6). The three most common mutations in CLN1 are the missense mutations p.Arg122Trp and p.Thr75Pro and the nonsense mutation p.Arg151X, which is the most commonly occurring CLN1 mutation worldwide (6, 29–31). PPT1 is a lysosomal enzyme that removes palmitoyl groups from modified cysteine residues in proteins targeted for degradation. However, the specific targets of this protein remain unknown (32). Mutations in CLN1 play a critical role in NCL pathology through decreased enzyme activity (6, 32, 33).
Mutations in CLN2, which encodes tripeptidyl-peptidase 1 (TPP1), are the basis for late-infantile neuronal ceroid lipofuscinosis (LINCL) or classic LINCL. Alternatively referred to as CLN2 disease (28). Classic LINCL presents in children at 2–4 years of age, and typically leads to death by 6–15 years of age (2). Mutations in CLN2 are also associated with infantile and juvenile NCL. To date, there are 89 mutations, consisting of 42 missense, 14 nonsense, 17 splice-site affecting, 11 deletions, 4 insertions and 1 deletion-insertion mutations (6). The two most common mutations in CLN2 are c.509–1G>C and p.Arg208X, which comprise 57% of all identified mutant alleles, and are found in ∼89% of disease cases (6, 34). TPP1 is a lysosomal enzyme that cleaves tripeptides from proteins undergoing degradation in the lysosome (35). The majority of CLN2 mutations lead to decreased enzyme activity, but there are also mutations that cause localization defects and subsequent retention within the ER (36).
Mutations in CLN3, which encodes the CLN3 protein, are the basis for juvenile neuronal ceroid lipofuscinosis (JNCL or classic JNCL). Alternatively referred to as CLN3 disease (28). Classic JNCL presents in children at 5–10 years of age, and typically leads to death in the second to third decade of life (3). Mutations in CLN3 are also associated with protracted and infantile NCL. To date, there are 59 mutations, consisting of 16 splice-site affecting, 13 nonsense, 12 missense, 11 deletions, 5 insertions and one mutation that affects the first methionine (6). The most common mutation is c.461–280_677 + 382del966 (1.02 kb deletion), which causes a frameshift and nonsense mutation, resulting in a truncated and non-functional protein, accounting for over 85% of disease cases (3, 6, 8, 37–39). CLN3 is a 438-amino acid transmembrane protein of unknown function that is comprised of six transmembrane domains with both N- and C- termini facing the cytoplasm. Many functional roles have been proposed for CLN3, such as lysosomal acidification, lysosomal arginine transport, vesicular trafficking, membrane fusion, autophagy and apoptosis (40), but further investigation is needed to accurately define its role within the cell.
Very little is currently known about NCL mRNA regulation and the subsequent effects of genetic mutations on their degradation. Understanding the role of NMD in NCL is crucial for understanding the molecular pathology of NCL genetic variants. Most importantly, NMD has emerged as a pathological mechanism for many genetic diseases, and may serve as a druggable therapeutic target for NCL. The frequency of patients diagnosed with NCL that have stop codon mutations as one of the disease causing alleles is 52.3% in INCL, 33.8% in LINCL and 2.8% in JNCL (17). In this work, the expression of CLN1, CLN2 and CLN3 mRNA in INCL, LINCL and JNCL lymphoblast cells was examined using quantitative real-time PCR (qPCR) to show that transcripts with PTCs have decreased abundance. PPT1 and TPP1 enzyme activity was measured in INCL and LINCL lymphoblast cells using fluorogenic enzyme assays to assess the effect of various mutations on protein function. In addition, siRNA-mediated knockdown of the NMD components UPF1 and eIF4A3 was performed to show that transcripts with PTCs increase in abundance when NMD is inhibited. Knockdown of UPF1 and eIF4A3 in INCL and LINCL cell lines increases PPT1 and TPP1 enzyme activity, respectively, providing the basis for further investigation using read-through drugs that inhibit the NMD pathway and provide a therapeutic option for these diseases.
RESULTS
It is currently unknown whether premature stop codons in CLN1, CLN2 or CLN3 in INCL, LINCL and JNCL lymphoblast cells, respectively, will lead to decreased mRNA levels. We have speculated that nonsense mutations lead to decreased levels of mutant NCL proteins through the regulation of mRNA transcripts within the cell. The most likely mechanism causing decreased mRNA levels is NMD, which is the most common mechanism through which mutant mRNAs are degraded within the cell, and has been associated with a number of genetic disorders. With this in mind, the expression of CLN1, CLN2 and CLN3 mRNA transcripts containing various nonsense mutations was measured (Fig. 1).
Figure 1.

Schematic illustration of CLN1, CLN2 and CLN3 genes and their respective proteins showing the relative location of the mutations used in this study. Exons are represented as gray boxes. Untranslated exon regions and introns are represented by white boxes and lines, respectively. (A) Four INCL cell lines were used with four mutations (two nonsense and two missense) in CLN1. The structure of PPT1 is shown below CLN1. The residues that comprise the catalytic triad of PPT1 are represented by light-gray circles. (B) Two LINCL cell lines and one carrier cell line were used with three mutations (two nonsense and one splice site) in CLN2. The structure of TPP1 is shown below CLN2. The residues that comprise the catalytic triad of TPP1 are represented by light-gray circles. (C) Eleven JNCL cell lines with six mutations (one deletion, three nonsense, one missense and one splice site) in CLN3. The proposed membrane topology of CLN3 along with the structure of the truncated CLN3 protein is shown below CLN3. The truncated CLN3 protein results from a 1.02 kb deletion causing a frameshift which leads to a 181-amino acid protein with 28 novel residues (represented by a dashed line) followed by a stop codon.
NCL gene transcripts containing PTCs are less abundant
qPCR was used to quantitate the relative abundance of endogenous mutant CLN1, CLN2 and CLN3 transcripts in patient-derived lymphoblast cell lines. Endogenous CLN1 mRNA expression levels in all INCL cell lines were significantly decreased from normal (Fig. 2 and Table 1A). Two of the INCL cell lines are compound heterozygous for a nonsense and missense mutation (p.R151X/p.H39Q and p.R151X/p.T75P), and have a 1.97- and 1.64-fold decrease in CLN1 mRNA, respectively. Interestingly, two other INCL cell lines are compound heterozygous (p.R151X/p.L10X) and homozygous (p.R151X/p.R151X) for two nonsense mutations, and have a 2.68- and 6.76-fold decrease in CLN1 mRNA abundance, respectively. This shows that the presence of even a single nonsense mutation decreases endogenous mRNA levels, and that a second nonsense mutation decreases transcript abundance even further. CLN1 mRNA levels were also measured in an LINCL cell line with mutations in CLN2, which was used as a disease control to account for the secondary effects of the disease process. CLN1 mRNA expression levels in all INCL cell lines were significantly decreased from the LINCL disease control.
Figure 2.

Endogenous expression of CLN1 in INCL lymphoblast cell lines is decreased compared with age- and sex- matched controls. Quantitative real-time PCR (qPCR) was used to measure endogenous CLN1 mRNA expression in four INCL lymphoblast cell lines grown under normal conditions. Relative expression of the CLN1 mRNA transcript was normalized to at least three of four reference genes (B2M, GAPDH, GUSB, HGPRT). The INCL samples designated GM16084, NC001, WN103 and GM16083 were compared with the age- and sex- matched controls designated AG09391, GM09659, AG14798 and AG09393, respectively. NJ001 (LINCL cell line) was used as a disease control. Four technical replicates were used for each sample. All values are represented as mean ± SE; *P < 0.05.
Table 1.
(A) Relative expression of endogenous CLN1 mRNA in infantile neuronal ceroid lipofuscinosis (INCL) lymphoblasts. (B) Relative expression of endogenous CLN2 mRNA in late-infantile neuronal ceroid lipofuscinosis (LINCL) lymphoblasts. (C) Relative expression of endogenous CLN3 mRNA in juvenile neuronal ceroid lipofuscinosis (JNCL) lymphoblasts
| Sample | Gene | Mutation genotype | Fold-regulation | % of normal | P-value | |
|---|---|---|---|---|---|---|
| A | NJ001 | CLN1 | Disease control | −1.12 | 89.29 | 0.090 |
| GM16084 | CLN1 | p.R151X/p.H39Q | −1.97 | 50.76 | 0.021 | |
| NC001 | CLN1 | p.R151X/p.T75P | −1.64 | 60.98 | 0.017 | |
| WN103 | CLN1 | p.R151X/p.L10X | −2.68 | 37.31 | 0.017 | |
| GM16083 | CLN1 | p.R151X/p.R151X | −6.76 | 14.79 | 0.011 | |
| B | NL001 | CLN2 | Disease control | −1.07 | 93.46 | 0.453 |
| DT14 | CLN2 | WT/p.R208X | −1.86 | 53.76 | 0.025 | |
| NJ001 | CLN2 | p.R208X/g.G2308C | −1.62 | 61.73 | 0.027 | |
| KS212 | CLN2 | p.R208X/p.L104X | −8.00 | 12.50 | 0.027 | |
| C | DT14 | CLN3 | Disease control | −1.09 | 91.74 | 0.095 |
| NJ001 | CLN3 | Disease control | 1.07 | 93.46 | 0.150 | |
| KS212 | CLN3 | Disease control | −1.05 | 95.24 | 0.353 | |
| DT38 | CLN3 | p.R334H/p.R334H | 1.01 | 99.01 | 0.713 | |
| DT23 | CLN3 | WT/1.02kb | −3.84 | 26.04 | 0.009 | |
| DT3 | CLN3 | 1.02kb/p.R334H | −1.86 | 53.76 | 0.016 | |
| DT12 | CLN3 | 1.02kb/p.W35X | −12.35 | 8.10 | <0.001 | |
| CO011 | CLN3 | 1.02kb/p.V180X | −2.39 | 41.84 | 0.016 | |
| NL003 | CLN3 | p.Q352X/p.Q352X | −2.38 | 42.02 | 0.013 | |
| DT10 | CLN3 | 1.02kb/g.G9831A | −3.72 | 26.88 | 0.004 | |
| DT8 | CLN3 | 1.02kb/1.02kb | −1.91 | 52.36 | 0.026 | |
| DT25 | CLN3 | 1.02kb/1.02kb | −2.27 | 44.05 | 0.008 | |
| DT27 | CLN3 | 1.02kb/1.02kb | −1.68 | 59.52 | 0.026 | |
| GM08820 | CLN3 | 1.02kb/1.02kb | −2.14 | 46.73 | 0.002 |
Endogenous CLN2 mRNA expression levels in two LINCL cell lines were significantly decreased from normal (Fig. 3 and Table 1B). One of the LINCL cell lines is compound heterozygous for a single nonsense mutation and a splice-site mutation (p.R208X/g.G2308C) and had a 1.62-fold decrease in CLN2 mRNA expression. Another cell line is heterozygous for this same nonsense mutation (WT/p.R208X) and exhibited a similar 1.86-fold decrease in expression. These expression levels are similar to CLN1 mRNA expression levels in two INCL cell lines with only one nonsense mutation (Table 1). The LINCL cell line that is compound heterozygous for two nonsense mutations (p.R208X/p.L104X) had an 8.00-fold decrease in CLN2 mRNA expression, once again showing that two nonsense mutations leads to a much greater decrease in transcript abundance. There was no significant difference in CLN2 mRNA expression in a JNCL disease control cell line that has mutations in CLN3.
Figure 3.

Endogenous expression of CLN2 in LINCL lymphoblast cell lines is decreased compared with age- and sex-matched controls. Quantitative real-time PCR (qPCR) was used to measure endogenous CLN2 mRNA expression in LINCL lymphoblast cell lines grown under normal conditions. Relative expression of the CLN2 mRNA transcript was normalized to at least three of four reference genes (B2M, GAPDH, GUSB, HGPRT). The samples designated DT14, KS212 and NJ001 were compared with the age- and sex-matched controls designated GM03798, AG09393 and AG14798, respectively. NL003 (JNCL cell line) was used as a disease control. Four technical replicates were used for each sample. All values are represented as mean ± SE; *P < 0.05.
Endogenous CLN3 mRNA expression levels in all JNCL cell lines followed a similar pattern to that seen in INCL and LINCL cell line mRNA expression (Fig. 4 and Table 1C). CLN3 transcript levels in three LINCL disease controls as well as a JNCL cell line that is homozygous for a missense mutation (p.R334H/p.R334H) were not significantly decreased from normal. All four JNCL cell lines that are homozygous for the most common 1.02 kb deletion (which introduces a PTC) showed a significant decrease in CLN3 transcript abundance (Fig. 4A). All other JNCL cell lines with at least one nonsense mutation exhibited significantly decreased CLN3 mRNA levels (1.68- to 12.35-fold) compared with normal (Fig. 4B).
Figure 4.
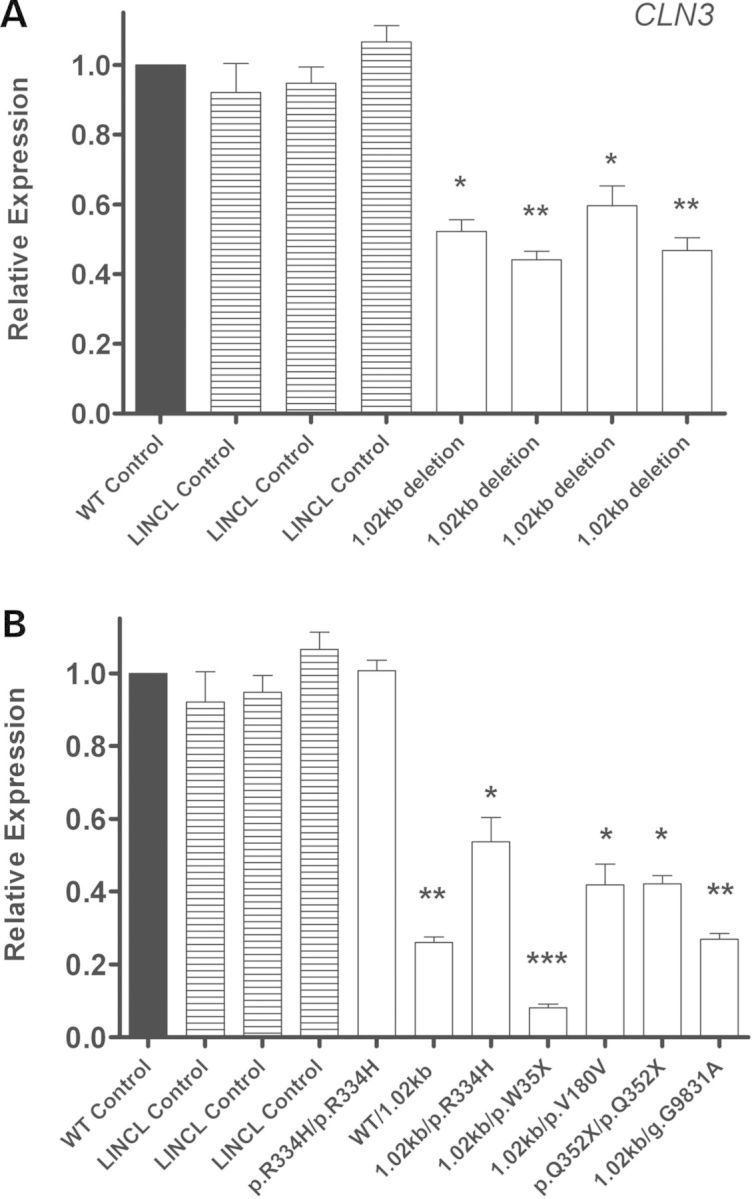
Endogenous expression of CLN3 in JNCL lymphoblast cell lines is decreased compared with age- and sex-matched controls. Quantitative real-time PCR (qPCR) was used to measure endogenous CLN3 mRNA expression in several JNCL lymphoblast cell lines grown under normal conditions. (A) The JNCL cell lines designated DT8, DT25, DT27 and GM08820 are homozygous for the 1.02 kb deletion, and were compared with the age- and sex-matched controls designated GM07535, AG09390 and AG15792. DT14, KS212 and NJ001 (LINCL cell lines) were used as disease controls. (B) The JNCL cell lines designated DT38, DT23, DT3, DT12, CO011, NL003 and DT10 have various nonsense mutations and were compared with the age- and sex-matched controls designated AG05979, GM03798, GM06160, GM07535, AG14812 and AG9390. DT14, KS212 and NJ001 (LINCL cell lines) were used as disease controls. Relative expression of the CLN3 mRNA transcript was normalized to at least three of four reference genes (B2M, GAPDH, GUSB and HGPRT). Four technical replicates were used for each sample. All values are represented as mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001.
In summary, a relative decrease in every NCL transcript containing at least one nonsense codon in three NCL genetic variants was observed. The presence of two nonsense mutations leads to an even greater decrease in transcript abundance. This provides a distinct correlation between the presence of a nonsense mutation and abundance of the respective mRNA.
NCL gene transcripts containing PTCs encode proteins with decreased enzyme activity
The function of PPT1 and TPP1 is known, allowing us to directly measure their activity within cell lines and tissue samples from patients with mutations in CLN1 and CLN2. Compromised enzyme activity is the pathological hallmark of these two genetic subtypes, and represents one of the most important methods for diagnosis. To determine the effect of different mutations on protein function, two fluorogenic assays were used to measure PPT1 (41–43) and TPP1 (44, 45) enzyme activity in INCL and LINCL cell lines, respectively. PPT1 enzyme activity in all INCL cell lines was significantly decreased from normal (Fig. 5 and Table 2A). Three of the INCL cell lines (p.R151X/p.H39Q, p.R151X/p.L10X and p.R151X/p.R151X) had 2.0–6.1% of normal PPT1 enzyme activity, and presented clinically with infantile NCL. Whereas the p.R151X/p.T75P cell line had 10.9% of normal PPT1 enzyme activity, and presented with juvenile onset NCL. The p.T75P mutation has been previously associated with juvenile NCL, whereas the p.H39Q mutation is associated with infantile NCL, a more severe clinical phenotype (33, 46). The LINCL disease control showed 84.2% of normal PPT1 enzyme activity, which was significantly decreased, but still well within an appropriate range and should not have any effect on biological function or pathology.
Figure 5.

PPT1 enzyme activity in INCL lymphoblast cell lines is decreased compared with controls. Relative PPT1 enzyme activity was measured in the INCL cell lines designated GM16084, NC001, WN103 and GM16083, and compared with three controls designated AG07535, AG09393 and AG15792, respectively. NJ001 (LINCL cell line) was used as a disease control. Four technical replicates were used for each sample. All values are represented as mean ± SE; *P < 0.001; **P < 0.0001 compared with wild-type controls using Student's t-test.
Table 2.
(A) Relative PPT1 enzyme activity in infantile neuronal ceroid lipofuscinosis (INCL) lymphoblasts. (B) Relative TPP1 enzyme activity in late-infantile neuronal ceroid lipofuscinosis (LINCL) lymphoblasts
| Sample | NCL protein | Mutation genotype | % of normal | P-value | |
|---|---|---|---|---|---|
| A | NJ001 | PPT1 | Disease control | 84.2 | 0.0002 |
| GM16084 | PPT1 | p.R151X/p.H39Q | 2.0 | <0.0001 | |
| NC001 | PPT1 | p.R151X/p.T75P | 10.9 | <0.0001 | |
| WN103 | PPT1 | p.R151X/p.L10X | 2.0 | <0.0001 | |
| GM16083 | PPT1 | p.R151X/p.R151X | 6.1 | <0.0001 | |
| B | DT14 | TPP1 | WT/p.R208X | 53.4 | <0.0001 |
| NJ001 | TPP1 | p.R208X/g.G2308C | 2.9 | <0.0001 | |
| KS212 | TPP1 | p.R208X/p.L104X | 3.1 | <0.0001 |
TPP1 enzyme activity in the two LINCL cell lines was significantly decreased from normal (Fig. 6 and Table 2B). The cell lines that are compound heterozygous for two nonsense mutations in CLN2 (p.R208X/g.G2308C and p.R208X/p.L104X) had 2.9 and 3.1% of normal TPP1 enzyme activity. An additional carrier cell line with only one mutated CLN2 allele (WT/p.R208X) had 53.4% normal TPP1 enzyme activity. The WT/p.R208X cell line presented clinically as a normal carrier, whereas the p.R208X/g.G2308C and p.R208X/p.V404X cell lines presented clinically with late-infantile NCL. The p.R208X/g.G2308C cell line, which showed similar CLN2 mRNA abundance to the WT/p.R208X cell line, had a much greater decrease in TPP1 enzyme activity, which is likely due to the additional g.G2308C splice site mutation. Unlike the p.R208X mutation, the g.G2308C splice-site mutation does not lead to a PTC and is more than likely not recognized by NMD. This explains how there are similar CLN2 mRNA expression levels in these two cell lines, but significant differences in TPP1 enzyme activity. The second LINCL cell line, p.R208X/p.L104X, showed similar TPP1 enzyme activity to the p.R208X/g.G2308C cell line. This p.R208X/p.L104X cell line also had the lowest TPP1 mRNA abundance, which is likely due to NMD recognizing both mutated CLN2 alleles.
Figure 6.

TPP1 enzyme activity in LINCL lymphoblast cell lines is decreased compared with controls. Relative TPP1 enzyme activity was measured in the LINCL cell lines designated NJ001 and KS212, and a carrier cell line designated DT14, which were compared with three controls designated GM03798, GM09393 and GM14798, respectively. Four technical replicates were used for each sample. All values are represented as mean ± SE; *P < 0.0001 compared with wild-type controls using Student's t-test.
These results confirm that these mutations in CLN1 and CLN2 lead to a significant compromise of PPT1 and TPP1 enzyme activity.
NMD targets NCL gene transcripts for degradation
Because the presence of nonsense mutations within NCL gene mRNA leads to a decrease in abundance, we propose that NMD is involved in their degradation. SiRNA-mediated knockdown of two critical NMD components, UPF1 and eIF4A3 was performed (Supplementary material, Fig. S1). Following siRNA knockdown, mRNA levels using qPCR were measured to determine whether blocking NMD would increase the abundance of CLN1, CLN2 and CLN3 mRNA.
An INCL cell line with a single nonsense mutation (p.R151X/p.H39Q) and an INCL cell line that is homozygous for two nonsense mutations (p.R151X/p.R151X) exhibited increased CLN1 mRNA expression when treated with UPF1 and eIF4A3 siRNA (Fig. 7A). Another INCL cell line that is compound heterozygous for two nonsense mutations (p.R151X/p.L10X) also showed increased CLN1 mRNA expression when treated with eIF4A3 siRNA. A normal CLN2 carrier cell line and an LINCL cell line with a single nonsense mutation (WT/p.R208X and p.R208X/g.G2308C) showed an increase in CLN2 mRNA expression when treated with eIF4A3 siRNA, while an LINCL cell line that is compound heterozygous for two nonsense mutations (p.R208X/p.L104X) exhibited increased CLN2 mRNA expression when treated with both UPF1 and eIF4A3 siRNA (Fig. 7B). UPF1 and eIF4A3 was knocked down in three JNCL lymphoblast cell lines which had various nonsense mutations in CLN3 (1.02kb del/p.W35X, p.Q352X/p.Q352X and 1.02 kb del/1.02 kb del), and showed that CLN3 mRNA expression increases when NMD is blocked (Fig. 7C). Interestingly, a JNCL cell line that is homozygous for the p.R334H missense mutation did not show an increase in CLN3 mRNA abundance when NMD was knocked down. This shows that CLN1, CLN2 and CLN3 mRNA transcripts harboring nonsense mutations in lymphoblast cells are targeted for degradation by NMD.
Figure 7.
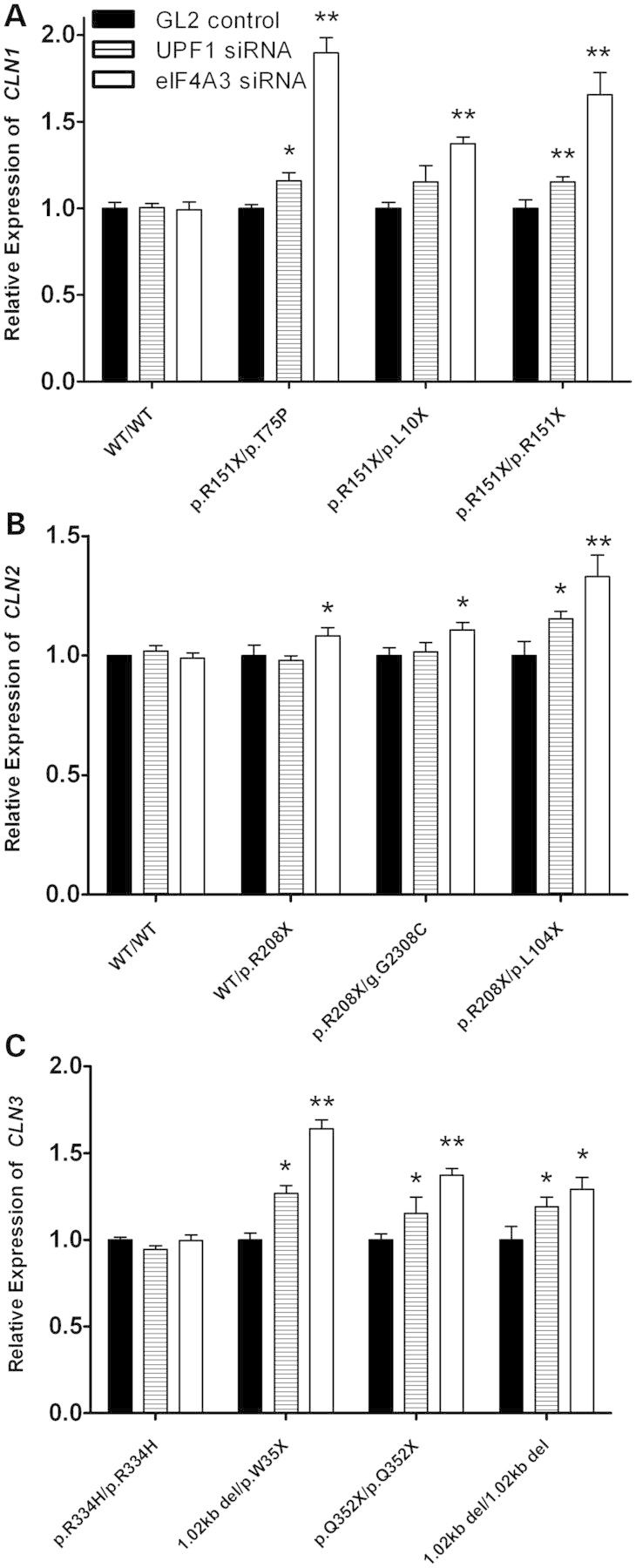
Mutated CLN1, CLN2 and CLN3 mRNA expression increases due to siRNA knockdown of nonsense-mediated decay. Lymphoblast cell lines were transiently transfected with UPF1, eIF4A3 or GL2 (luciferase) siRNA for 48 h. Quantitative real-time PCR was then used to measure CLN1, CLN2 or CLN3 mRNA in INCL, LINCL or JNCL cell lines, respectively. (A) Three INCL cell lines showed a significant increase in CLN1 mRNA abundance when UPF1 and eIF4A3 siRNA was used to knockdown NMD. No changes in CLN1 mRNA abundance were seen in a wild-type control. (B) Two LINCL cell lines showed a significant increase in CLN2 mRNA abundance when UPF1 and eIF4A3 siRNA was used to knockdown NMD. A cell line from a normal carrier also showed increased CLN2 mRNA abundance. No changes in CLN2 mRNA abundance were seen in a wild-type control. (C) Three JNCL cell lines also showed a significant increase in CLN3 mRNA abundance. UPF1 and eIF4A3 siRNA-treated samples were compared with GL2 (luciferase) controls. Lipofectamine only controls showed no change in CLN1, CLN2 and CLN3 mRNA expression. Four technical replicates were used for each sample. All values are represented as mean ± SE; *P < 0.05; **P < 0.01.
PPT1 enzyme activity was measured in INCL cell lines (p.R151X/p.T75P, p.R151X/p.L10X and p.R151X/p.R151X) and TPP1 enzyme activity was measured in LINCL cell lines (p.R208X/g.G2308C and p.R208X/p.L104X) transfected with UPF1 and eIF4A3 siRNA. All of these cell lines contained PTCs and exhibited a significant increase in enzyme activity (Fig. 8). The INCL cell line that is homozygous for the p.R151X mutation showed the greatest increase in PPT1 enzyme activity, whereas the INCL cell line that is compound heterozygous for a nonsense and missense mutation (p.R151X/p.T75P) showed the lowest increase in PPT1 enzyme activity. The LINCL cell line that is compound heterozygous for two nonsense mutations (p.R208X/p.L104X) showed the greatest increase in TPP1 enzyme activity, whereas the LINCL cell line that is compound heterozygous for a nonsense and splice site mutation (p.R208X/g.G2308C) showed the lowest increase in TPP1 enzyme activity. Currently, there is no assay to measure CLN3 protein function. In addition, there are no appropriate antibodies to quantify endogenous protein levels. The most common 1.02 kb deletion in CLN3 leads to a frameshift and subsequent nonsense mutation, most likely leading to a truncated and non-functional CLN3 protein. It is not known how other mutations will affect CLN3 function, but there are no known phenotype–genotype correlations in JNCL patients. Although the function of CLN3 is unknown, we speculate that elevating CLN3 transcript through inhibition of NMD would also result in a significant increase in functional CLN3 protein.
Figure 8.

PPT1 and TPP1 enzyme activity increases due to siRNA knockdown of nonsense-mediated decay. Lymphoblast cells were transiently transfected with UPF1, eIF4A3 or GL2 (luciferase) siRNA for 48 h. Fluorogenic enzyme assays were used to measure PPT1 or TPP1 enzyme activity in INCL or LINCL cell lines, respectively. (A) All three INCL cell lines (NC001, WN103 and GM16083) showed a significant increase in PPT1 enzyme activity after NMD was knocked down with eIF4A3 siRNA. GM16083 showed a significant increase in PPT1 activity after UPF1 was knocked down. (B) Both LINCL cell lines (NJ001 and KS212) showed a significant increase in TPP1 enzyme activity after NMD was knocked down with UPF1 and eIF4A3 siRNA. Four technical replicates were used for each sample in both PPT1 and TPP1 enzyme assays. All values are represented as mean ± SE; *P < 0.05; **P < 0.01; ***P < 0.001 compared with GL2 (luciferase) controls using Student's t-test.
Knockdown of UPF1 and eIF4A3 in INCL, LINCL and JNCL cell lines not only led to an increase in CLN1, CLN2 and CLN3 mRNA transcript abundance (Fig. 7), but also resulted in an increase in PPT1 and TPP1 enzyme activity (Fig. 8). These results show that blocking NMD in cell lines with nonsense mutations in NCL mRNA transcripts leads to increased functional protein production and a subsequent increase in enzyme activity.
Read-through drugs suppress nonsense mutations in CLN1 and CLN2 gene transcripts
An INCL cell line that is homozygous for the p.R151X mutation in CLN1 and an LINCL cell line that is compound heterozygous for the p.R208X and p.L104X mutations in CLN2 were treated with Ataluren (Fig. 9), and gentamicin for 48 h (Supplementary material, Fig. S2). Both of these cell lines contain a nonsense mutation in each allele, and read-through drug treatment was expected to increase PPT1 and TPP1 enzyme activity. Ataluren significantly increased PPT1 and TPP1 enzyme activity at 2.5 and 5.0 μg/ml. There were no significant changes in PPT1 and TPP1 activity with a DMSO-only control (data not shown). Gentamicin significantly increased PPT1 enzyme activity in the INCL cell line at 0.312, 0.625 and 1.25 mg/ml, and significantly increased TPP1 enzyme activity in the LINCL cell line at all doses. There was no significant changes in PPT1 and TPP1 activity with a phosphate buffered saline (PBS)-only control (data not shown).
Figure 9.
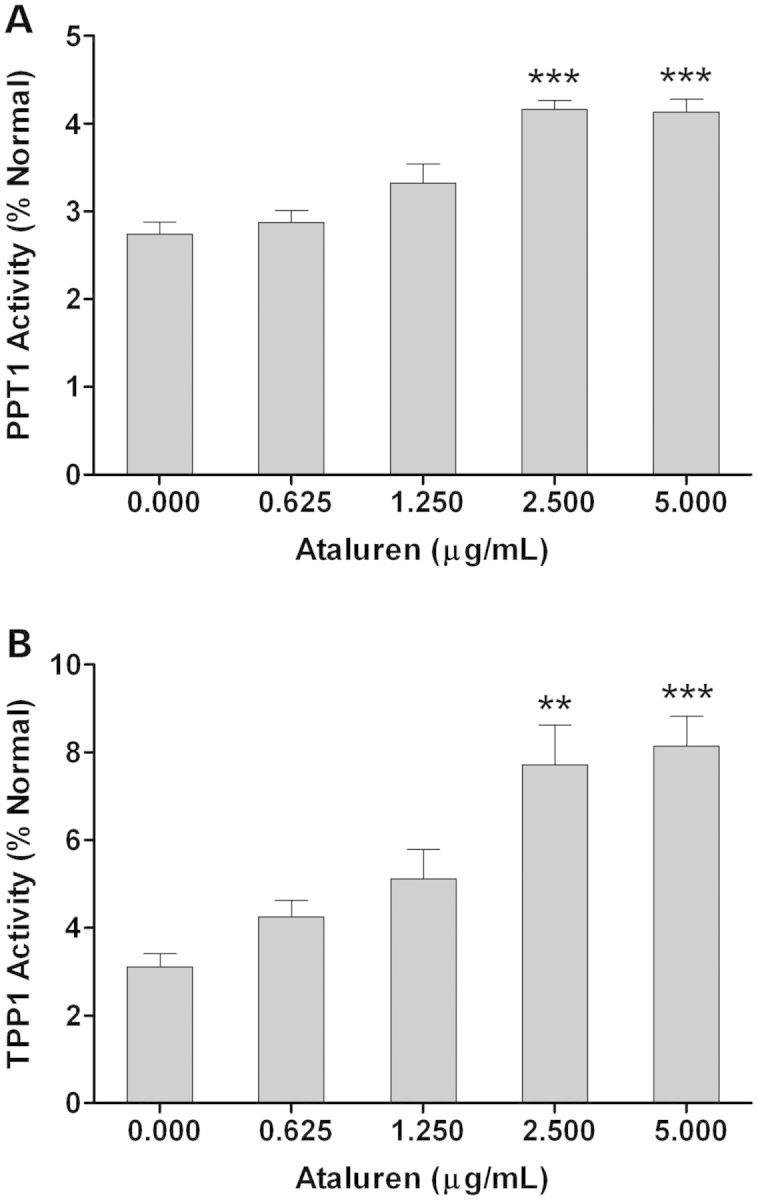
PPT1 and TPP1 enzyme activity increases following treatment with Ataluren. (A) INCL (CLN1 p.R151X/p.R151X) and (B) LINCL (CLN2 p.R208X/p.L104X) lymphoblast cell lines as well as a wild-type lymphoblast cell line were treated with Ataluren (0.625–5.0 μg/ml) for 48 h. PPT1 and TPP1 enzyme activity was assayed in the INCL and LINCL cell lines, respectively, and normalized to wild-type enzyme activity. Two biological replicates were used at each dosage. Four technical replicates were used for each biological replicate in both PPT1 and TPP1 enzyme assays. All data are plotted as a percent of normal. All values are represented as mean ± SE; **P < 0.01; ***P < 0.001 compared with wild-type controls using one-way ANOVA followed by Dunett's multiple comparison post-test.
DISCUSSION
It has been recognized that NMD is critically important in the recognition and elimination of mutant transcripts in a number of genetic disorders (14). In fact, it has been estimated that 30% of all genetic disorders are due to the presence of PTCs (24). The expression of these mutant genes could lead to the production of truncated proteins with residual or no function, as well as gain-of-function or dominant-negative effects. However, a majority of these mutant transcripts are eliminated by NMD which protects the cell from the potentially toxic side-effects of protein accumulation. Owing to its role in disease pathogenesis, NMD has emerged as an important therapeutic target in a number of genetic disorders. Current strategies that target this pathway involve the use of read-through drugs, such as the aminoglycosides, PTC124 (Ataluren) and RTC13 (17–22). Ataluren is currently being used in one ongoing phase III clinical trial for cystic fibrosis (NCT00803205) and one phase III trial currently recruiting patients for Duchenne and Becker muscular dystrophy (NCT01247207). Previously, gentamicin was shown to increase TPP1 enzyme activity in human fibroblasts from LINCL patients harboring nonsense mutations (47). It has also been recently shown that gentamicin and Ataluren were able to mildly increase PPT1 enzyme activity in INCL patient-derived fibroblast cell lines (48).
In this study, the role of NMD in neuronal ceroid lipofuscinosis was investigated using a number of cell lines with mutations in CLN1, CLN2 and CLN3. Every cell line harboring at least one nonsense mutation had a significantly lower endogenous mRNA abundance than normal cell lines. In addition, the cell lines with two nonsense mutations had an even greater decrease in endogenous mRNA abundance. This correlation between nonsense mutations and mRNA transcript abundance provides evidence of targeted degradation, for which the most likely pathway is NMD. The effects that mutations in CLN1 and CLN2 have on pathology are most easily examined through PPT1 and TPP1 enzyme activity, respectively. These enzymes are critically important for proper lysosomal function, and significant decreases in PPT1 or TPP1 enzyme activity can lead to substrate accumulation and disease pathogenesis. Every INCL and LINCL cell line in this study had a significant decrease in PPT1 and TPP1 enzyme activity, respectively. In addition, there is a distinct correlation between PPT1 and TPP1 enzyme activity, and severity of the patient's clinical phenotype from which these cell lines were derived. This supports the hypothesis that increasing enzyme activity can alleviate disease pathogenesis.
We contend that NMD-targeted degradation of NCL gene transcripts decreases enzyme activity and contributes to disease pathology. To test this hypothesis, siRNA-mediated knockdown of two proteins critically important to the NMD pathway, UPF1 and eIF4A3, was performed. Expression of CLN1, CLN2 and CLN3 increases in cell lines with nonsense mutations, but not in cell lines with missense mutations or wild-type alleles. Disrupting NMD should lead to an increase in NCL gene mRNA abundance, as well as an increase in functional protein production by read-through of the early stop codon (21). Therefore, PPT1 and TPP1 enzyme activity was measured in cell lines transfected with UPF1 and eIF4A3 siRNA. Disrupting NMD in these cell lines lead to a significant increase in PPT1 and TPP1 enzyme activity. Most interestingly, PPT1 and TPP1 enzyme activity increased the greatest in an INCL cell line (p.R151X/p.R151X) and an LINCL cell line (p.R208X/p.L104X) with two nonsense mutations.
Although the effect of NMD on CLN3 protein function cannot be adequately assessed, disrupting NMD with siRNA in JNCL cell lines leads to an increase in CLN3 mRNA abundance. This most likely leads to a significant increase in functional CLN3 protein production by read-through of PTCs, such as p.W35X, p.V180X and p.Q352X. However, read-through of the PTC that arises from the most common 1.02 kb deletion will not lead to translation of functional CLN3 due to deletion of exons 7 and 8. In this study and in previous work, we have shown that the 1.02 kb deletion leads to a significant decrease in CLN3 transcript abundance (8). In addition, cellular quality control mechanisms are likely to degrade the truncated CLN3 protein. Therefore, these data support the hypothesis that the 1.02 kb deletion in CLN3 leads to a true loss-of-function disease.
The role of NMD in NCL transcript abundance and enzyme activity was further studied using two read-through drugs gentamicin and Ataluren (PTC124). Both of these drugs were able to suppress nonsense mutations in an INCL cell line (p.R151X/p.R151X) and an LINCL cell line (p.R208X/p.L104X), leading to a significant increase in PPT1 and TPP1 enzyme activity, respectively. According to several authors, increasing enzyme activity to 1–5% (49) and 15–20% (50) of normal endogenous activity could correct a lysosomal enzyme metabolic defect. Therefore, it is possible that increasing PPT1 and TPP1 enzyme activity to ∼10% (51) of normal could sufficiently affect pathology, lessening disease severity and consequently prolonging a patient's life.
This study shows that NMD is involved in NCL mRNA degradation, and subsequent decrease in NCL protein function. Inhibiting NMD in INCL and LINCL cell lines increases PPT1 and TPP1 enzyme activity, and provides a druggable therapeutic target. NMD is involved in the molecular genetic pathology of many other genetic diseases, such as cystic fibrosis and Duchenne muscular dystrophy. As such, NMD has become an important therapeutic target and is currently the focus of a number of research studies investigating the use of read-through drugs for therapy. Read-through drugs have emerged as an important class of compounds that target NMD, leading to a significant increase in functional protein production. With this in mind, it is apparent that read-through drug treatment in NCL could potentially attenuate disease pathogenesis, and provides new treatment options for this group of fatal genetic diseases.
MATERIALS AND METHODS
Lymphoblast cell culture
Lymphoblast cell lines derived from INCL, LINCL and JNCL patients, designated NC001, WN103, DT14, KS212, NJ001, CO011, DT3, DT8, DT12, DT23, DT25, DT27, DT38, NL003 and TX008, were prepared as described previously (52). Additional patient cell lines, designated GM16083, GM16084 and GM08820, were obtained from Coriell Cell Repository. Age- and sex- matched control cell lines, designated GM03798, GM05979, GM06160, GM07535, GM09390, GM09391, GM9393, GM9659, AG14812, AG14798, AG15792 and AG15914, were also obtained from Coriell Cell Repository. All cell lines were cultured in T75 flasks using Hyclone RPMI 1640 medium (Thermo Scientific, Rockford, IL, USA) supplemented with 15% heat inactivated Hyclone fetal bovine serum (Thermo Scientific) and 10% Penicillin/Streptomycin (Thermo Scientific), at 37°C with 5% CO2.
Sample processing and handling
All lymphoblast cell lines were processed in a similar manner. Approximately 5.0 × 106 cells from each sample were centrifuged at 250g, washed in PBS, pelleted in 1.5 ml microcentrifuge tubes and frozen on dry ice. All samples were stored at −80°C no longer than 2 months before total RNA extraction or protein extraction.
Nucleic acid extraction
Total RNA was extracted from lymphoblast cell samples with a Maxwell® 16 LEV simplyRNA Cells Kit (Promega, Madison, WI, USA) using a Maxwell® 16 Instrument (Promega), according to the manufacturer's instructions. Sample purity and yield were determined using a NanoVue spectrophotometer (Denville Scientific, Metuchen, NJ, USA). All samples had A260/A280 values between 2.098 and 2.115, and yielded between 520 and 1481 ng/μl total RNA. RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) with an Agilent RNA 6000 Nano Kit (Agilent Technologies) according to the manufacturer's instructions. All relative integrity numbers were between 9.6 and 10.
Reverse transcription
Each sample was mass-normalized using ∼1000 ng of total RNA for cDNA synthesis using a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer's instructions in a 96-well plate. The reaction conditions were as follows: 25°C for 10 min, 37°C for 120 min and 85°C for 5 min. Samples were diluted with molecular grade water to 10 ng/μl following reverse transcription. Samples were assessed for DNA contamination using reactions with no reverse transcriptase added. All samples were DNA free and stored at −20°C until use.
Quantitative real-time polymerase chain reaction
qPCR was performed for target genes using TaqMan® hydrolysis assays (Life Technologies) for TPP1 (Cat.# Hs00166099_m1), CLN3 (Cat.# Hs01029229_g1), UPF1 (Cat.# Hs00161289_m1), eIF4A3 (Cat.# Hs01556773_m1) and a custom probe assay (Eurofins MWG Operon, Huntsville, Alabama, USA) for CLN1 variant 1 (CLN1v1) (NM_000310). CLN1v1 probe and primer sequences were 5′-[6-FAM]TCCTTACACA[BHA1a-Q]-3′ (probe), 5′-CAGCTTCTTCTTGAATGTC-3′ (forward primer), 5′-ATCCCATAGCATTGTAGC-3′ (reverse primer). qPCR was performed for reference genes using TaqMan® hydrolysis assays (Life Technologies) for B2M (Part.# 4333766F), GAPDH (Part.# 4333764F), GUSB (Part.# 4333767F) and HGPRT (Part.# 4333768F). Amplification was performed with 20 ng cDNA in 10 μl reaction volumes for four technical replicates using Absolute Blue qPCR Mix (Thermo Scientific) in 384-well plates (Roche Diagnostics, Indianapolis, IN, USA). Thermal cycling and fluorescence data collection were performed on a LightCycler® 480 (Roche Diagnostics) using the following reaction conditions: 95°C for 15 min, followed by 40 cycles at 95°C for 15 s, 60°C for 1 min.
qPCR data analysis
Raw fluorescence data were analyzed with the second derivative method to determine Cq values using LightCycler 480 software version 1.5 (Roche Diagnostics). The Cq values for each sample had a standard deviation <0.15. No-template controls were used for each qPCR assay and revealed no nucleic acid contamination. Calibration curves were established for each qPCR assay to determine gene amplification efficiency, which was calculated as: PCR efficiency = 10−1/slope – 1. All qPCR assays had PCR efficiencies between 0.92 and 0.99. Relative expression of all target genes was calculated compared with reference genes using REST 2009 (53, 54).
PPT1 enzyme assay
Palmitoyl-protein thioesterase 1 (PPT1) enzyme activity was measured as previously described (41–43). INCL cell lines, designated GM16083, GM16084, NC001 and WN103 as well as normal cell lines, designated AG07535 and AG09393, and an LINCL cell line, designated NJ001 were used. To summarize, live cells were pelleted, and re-suspended in PBS two to three times. One-hundred thousand cells per well were then incubated in a mixture containing 0.64 mm 4-methylumbelliferyl-6-thiopalmitoyl-β-glucoside (MU-6S-palm- βGlc, Moscerdam Substrates, Netherlands), 15 mm dithiothreitol (DTT) in 5.1 mg/ml bovine serum albumin and 0.02% Na-azide, 0.375% (w/v) Triton X-100 in 2/1 chloroform/methanol, 0.1 U β-glucosidase from almonds (Sigma-Aldrich, St Louis, MO, USA) in double-concentrated McIlvains phosphate/citric-acid buffer, pH 4.0. The live cell reaction mixture was then incubated for 1 h at 37°C. The reaction was stopped with the addition of 0.5 m NaHCO3/0.5 m Na2CO3 buffer, pH 10.7 with 0.025% Triton X-100 and fluorescence was measured using a SpectraMax M5 instrument (Molecular Devices, Sunnyvale, CA, USA). Relative enzyme activity was estimated using total fluorescence minus background, and analyzed using an unpaired Student's t-test.
TPP1 enzyme assay
TPP1 enzyme activity was measured as previously described (44, 45). LINCL cell lines, designated DT14, KS212 and NJ001, and normal cell lines, designated GM03798, GM09393 and GM14798, were used. Live cells were pelleted, and re-suspended in PBS two to three times. One-hundred thousand cells per well were then incubated with 250 μm Ala-Ala-Phe-7-amido-4-methylcoumarin (Ala-Ala-Phe-AMC, Sigma-Aldrich) in DMSO. The live cell reaction mixture was then incubated for 1 h at 37°C. The reaction was stopped with the addition of 0.1 m monochloroacetic acid/0.13 m NaOH/0.1 m acetic acid, pH 4.3 and fluorescence was measured using a SpectraMax M5 instrument (Molecular Devices). Relative enzyme activity was estimated using total fluorescence minus background, and analyzed using an unpaired Student's t-test.
Lymphoblast cell siRNA transfection
Lymphoblast cell lines were tested for transfection efficiency using 25, 50 and 100 nm BLOCK-iT™ Fluorescent Oligo (Life Technologies) (results not shown). To test for knockdown efficiency, lymphoblast cell lines were transfected at 5.0 × 105 cells per ml with 25, 50 or 100 nm ON-TARGETplus SMARTpool RENT1 (UPF1) siRNA or DDX48 (eIF4A3) siRNA (Thermo Scientific) in 30 mm dishes (4 ml total) with 10 μl Lipofectamine 2000 (Life Technologies) according to manufacturer's instructions. Luciferase GL2 Duplex siRNA (Thermo Scientific) was used as an siRNA negative control. Lipofectamine 2000 only controls showed no changes in gene expression. All additional experiments used 100 nm UPF1 or eIF4A3 siRNA and 100 nm GL2 Duplex siRNA. Cells were collected after 48 h (see above) and used for enzyme assays or stored at −80°C for further use.
Immunoblotting
Cells were collected and stored at −80°C (see above). Protein samples were prepared by treating cell pellets for 15 min with lysis buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 0.2% Triton X-100 and 300 mm NP-40, supplemented with 1:1000 protease cocktail and 1:10 000 PMSF, for extraction on ice. Following a high-speed spin (12 000 g), the supernatant was collected, and the protein concentration was measured using the Pierce 660 nm Protein Assay (Thermo Scientific). Samples were heated for 5 min in Laemmli buffer at 100°C. Proteins were resolved on 10% SDS–polyacrylamide gels at 200 V for 40 min, and transferred to PVDF membranes (Millipore Corporation, Billerica, MA, USA) at 100 V for 120 min. Membranes were then incubated in blocking buffer [5% milk in 100 mm Tris–HCl, pH 7.5, 150 mm NaCl, 0.1% Tween-20 (TBST)] for 1 h. Anti-UPF1 (1:10 000, Abcam) and anti-GAPDH (1:5000; Sigma-Aldrich) antibodies were used to probe the membranes incubated in blocking buffer overnight at 4°C. Following primary incubation, membranes were washed three times, once for 10 min and twice for 5 min with TBST, then probed with anti-rabbit antibody conjugated with horse-radish peroxidase (GE Healthcare Life Sciences, Piscataway, NJ, USA) in a blocking buffer for 1 h at room temperature. Membranes were then washed four times, once for 10 min and three times for 5 min. Signal was detected by chemiluminescence using Luminata Forte Western HRP Substrate (Millipore) and a BioSpectrum UVP Imaging system (Upland, CA, USA).
Read-through drug treatment
Lymphoblast cell lines were incubated with varying concentrations of Ataluren (0.625–5.000 µg/ml) [Selleck Chemicals, Houston, TX, USA] or Gentamicin (0.156–1.250) [Sigma-Aldrich] in 12-well plates for 48 h. Cells were collected and used for enzyme assays (see above) or stored at −80°C for further use.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by the Beat Batten Foundation, Noah's Hope, Hope for Bridget and Sanford Health.
Supplementary Material
ACKNOWLEDGEMENTS
Thanks to Andrew Cardillo and Peter Vitiello for technical guidance and useful discussions, and to Sarah Radel for technical assistance with the enzyme assays.
Conflict of Interest statement None declared.
REFERENCES
- 1.Haltia M. The neuronal ceroid-lipofuscinoses. J. Neuropathol. Exp. Neurol. 2003;62:1–13. doi: 10.1093/jnen/62.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Santavuori P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988;10:80–83. doi: 10.1016/S0387-7604(88)80075-5. [DOI] [PubMed] [Google Scholar]
- 3.Jalanko A., Braulke T. Neuronal ceroid lipofuscinoses. BBA Mol. Cell. Res. 2009;1793:697–709. doi: 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Arsov T., Smith K.R., Damiano J., Franceschetti S., Canafoglia L., Bromhead C.J., Andermann E., Vears D.F., Cossette P., Rajagopalan S., et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am. J. Hum. Genet. 2011;88:566–573. doi: 10.1016/j.ajhg.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bras J., Verloes A., Schneider S.A., Mole S.E., Guerreiro R.J. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 2012;21:2646–2650. doi: 10.1093/hmg/dds089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kousi M., Lehesjoki A.E., Mole S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012;33:42–63. doi: 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- 7.Noskova L., Stranecky V., Hartmannova H., Pristoupilova A., Baresova V., Ivanek R., Hulkova H., Jahnova H., van der Zee J., Staropoli J.F., et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011;89:241–252. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan C.H., Mitchison H.M., Pearce D.A. Transcript and in silico analysis of CLN3 in juvenile neuronal ceroid lipofuscinosis and associated mouse models. Hum. Mol. Genet. 2008;17:3332–3339. doi: 10.1093/hmg/ddn228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Y.-F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 10.Hwang J., Sato H., Tang Y., Matsuda D., Maquat L.E. UPF1 association with the cap-binding protein, CBP80, promotes nonsense-mediated mRNA decay at two distinct steps. Mol. Cell. 2010;39:396–409. doi: 10.1016/j.molcel.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang J., Maquat L.E. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr. Opin. Genet. Devel. 2011;21:422–430. doi: 10.1016/j.gde.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isken O., Kim Y.K., Hosoda N., Mayeur G.L., Hershey J.W., Maquat L.E. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133:314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H.C., Oh N., Cho H., Choe J., Kim Y.K. Nonsense-mediated translational repression involves exon junction complex downstream of premature translation termination codon. FEBS Lett. 2010;584:795–800. doi: 10.1016/j.febslet.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Muhlemann O., Eberle A., Stalder L., Zamudioorozco R. Recognition and elimination of nonsense mRNA. BBA - Gene Reg. Mech. 2008;1779:538–549. doi: 10.1016/j.bbagrm.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Silva A.L., Romao L. The mammalian nonsense-mediated mRNA decay pathway: to decay or not to decay! Which players make the decision? FEBS Lett. 2009;583:499–505. doi: 10.1016/j.febslet.2008.12.058. [DOI] [PubMed] [Google Scholar]
- 16.Kervestin S., Jacobson A. NMD: a multifaceted response to premature translational termination. Nature Rev. Mol. Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks D.A., Muller V.J., Hopwood J.J. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol. Med. 2006;12:367–373. doi: 10.1016/j.molmed.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Finkel R.S. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and ataluren (PTC124) J. Child. Neurol. 2010;25:1158–1164. doi: 10.1177/0883073810371129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldmann T., Overlack N., Wolfrum U., Nagel-Wolfrum K. PTC124-mediated translational readthrough of a nonsense mutation causing usher syndrome type 1C. Hum. Gene Ther. 2011;22:537–547. doi: 10.1089/hum.2010.067. [DOI] [PubMed] [Google Scholar]
- 20.Lee H.L., Dougherty J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012;136:227–266. doi: 10.1016/j.pharmthera.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S., et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 22.Kayali R., Ku J.-M., Khitrov G., Jung M.E., Prikhodko O., Bertoni C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012;21:4007–4020. doi: 10.1093/hmg/dds223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis B.P., Green R.E., Brenner S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl Acad. Sci. USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 25.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 26.Stehlikova K., Zapletalova E., Sedlackova J., Hermanova M., Vondracek P., Marikova T., Mazanec R., Zamecnik J., Vohanka S., Fajkus J. Quantitative analysis of CAPN3 transcripts in LGMD2A patients: involvement of nonsense-mediated mRNA decay. Neuromuscul. Disord. 2007;17:143–147. doi: 10.1016/j.nmd.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Kerr T.P., Sewry C.A., Robb S.A., Roberts R.G. Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay? Hum. Genet. 2001;109:402–407. doi: 10.1007/s004390100598. [DOI] [PubMed] [Google Scholar]
- 28.Williams R.E., Mole S.E. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79:183–191. doi: 10.1212/WNL.0b013e31825f0547. [DOI] [PubMed] [Google Scholar]
- 29.Das A.K., Becerra C.H., Yi W., Lu J.Y., Siakotos A.N., Wisniewski K.E., Hofmann S.L. Molecular genetics of palmitoyl-protein thioesterase deficiency in the U.S. J. Clin. Invest. 1998;102:361–370. doi: 10.1172/JCI3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchison H.M., Hofmann S.L., Becerra C.H., Munroe P.B., Lake B.D., Crow Y.J., Stephenson J.B., Williams R.E., Hofman I.L., Taschner P.E., et al. Mutations in the palmitoyl-protein thioesterase gene (PPT; CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular osmiophilic deposits. Hum. Mol. Genet. 1998;7:291–297. doi: 10.1093/hmg/7.2.291. [DOI] [PubMed] [Google Scholar]
- 31.Mole S.E., Zhong N.A., Sarpong A., Logan W.P., Hofmann S., Yi W., Franken P.F., van Diggelen O.P., Breuning M.H., Moroziewicz D., et al. New mutations in the neuronal ceroid lipofuscinosis genes. Euro. J. Paediatr. Neurol. 2001;5(Suppl A):7–10. doi: 10.1053/ejpn.2000.0427. [DOI] [PubMed] [Google Scholar]
- 32.Bellizzi J.J., III, Widom J., Kemp C., Lu J.Y., Das A.K., Hofmann S.L., Clardy J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc. Natl Acad. Sci. USA. 2000;97:4573–4578. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohno K., Saito S., Sugawara K., Suzuki T., Togawa T., Sakuraba H. Structural basis of neuronal ceroid lipofuscinosis 1. Brain Dev. 2010;32:524–530. doi: 10.1016/j.braindev.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Sleat D.E., Gin R.M., Sohar I., Wisniewski K., Sklower-Brooks S., Pullarkat R.K., Palmer D.N., Lerner T.J., Boustany R.M., Uldall P., et al. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am. J. Hum. Genet. 1999;64:1511–1523. doi: 10.1086/302427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pal A., Kraetzner R., Gruene T., Grapp M., Schreiber K., Gronborg M., Urlaub H., Becker S., Asif A.R., Gartner J., et al. Structure of tripeptidyl-peptidase I provides insight into the molecular basis of late infantile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2009;284:3976–3984. doi: 10.1074/jbc.M806947200. [DOI] [PubMed] [Google Scholar]
- 36.Steinfeld R., Steinke H.B., Isbrandt D., Kohlschutter A., Gartner J. Mutations in classical late infantile neuronal ceroid lipofuscinosis disrupt transport of tripeptidyl-peptidase I to lysosomes. Hum. Mol. Genet. 2004;13:2483–2491. doi: 10.1093/hmg/ddh264. [DOI] [PubMed] [Google Scholar]
- 37.Mole S.E. The genetic spectrum of human neuronal ceroid-lipofuscinoses. Brain Pathol. 2004;14:70–76. doi: 10.1111/j.1750-3639.2004.tb00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mole S.E., Williams R.E., Goebel H.H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107–126. doi: 10.1007/s10048-005-0218-3. [DOI] [PubMed] [Google Scholar]
- 39.Siintola E., Lehesjoki A.E., Mole S.E. Molecular genetics of the NCLs—status and perspectives. Biochim. Biophys. Acta. 2006;1762:857–864. doi: 10.1016/j.bbadis.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Phillips S.N., Benedict J.W., Weimer J.M., Pearce D.A. CLN3, the protein associated with batten disease: structure, function and localization. J. Neurosci. Res. 2005;79:573–583. doi: 10.1002/jnr.20367. [DOI] [PubMed] [Google Scholar]
- 41.Voznyi Y.V., Keulemans J.L., Mancini G.M., Catsman-Berrevoets C.E., Young E., Winchester B., Kleijer W.J., van Diggelen O.P. A new simple enzyme assay for pre- and postnatal diagnosis of infantile neuronal ceroid lipofuscinosis (INCL) and its variants. J. Med. Genet. 1999;36:471–474. [PMC free article] [PubMed] [Google Scholar]
- 42.van Diggelen O.P., Keulemans J.L.M., Winchester B., Hofman I.L., Vanhanen S.L., Santavuori P., Voznyi Y.V. A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol. Genet. Metab. 1999;66:240–244. doi: 10.1006/mgme.1999.2809. [DOI] [PubMed] [Google Scholar]
- 43.van Diggelen O.P., Thobois S., Tilikete C., Zabot M.-T., Keulemans J.L.M., van Bunderen P.A., Taschner P.E., Losekoot M., Voznyi Y.V. Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann. Neurol. 2001;50:269–272. doi: 10.1002/ana.1103. [DOI] [PubMed] [Google Scholar]
- 44.Sohar I., Sleat D.E., Jadot M., Lobel P. Biochemical characterization of a lysosomal protease deficient in classical late infantile neuronal ceroid lipofuscinosis (LINCL) and development of an enzyme-based assay for diagnosis and exclusion of LINCL in human specimens and animal models. J. Neurochem. 1999;73:700–711. doi: 10.1046/j.1471-4159.1999.0730700.x. [DOI] [PubMed] [Google Scholar]
- 45.Sohar I., Lin L., Lobel P. Enzyme-based diagnosis of classical late infantile neuronal ceroid lipofuscinosis: comparison of tripeptidyl peptidase i and pepstatin-insensitive protease assays. Clin. Chem. 2000;46:1005–1008. [PubMed] [Google Scholar]
- 46.Das A.K., Lu J.Y., Hofmann S.L. Biochemical analysis of mutations in palmitoyl-protein thioesterase causing infantile and late-onset forms of neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2001;10:1431–1439. doi: 10.1093/hmg/10.13.1431. [DOI] [PubMed] [Google Scholar]
- 47.Sleat D.E., Sohar I., Gin R.M., Lobel P. Aminoglycoside-mediated suppression of nonsense mutations in late infantile neuronal ceroid lipofuscinosis. Euro. J. Paediatr. Neurol. 2001;5(Suppl A):57–62. doi: 10.1053/ejpn.2000.0436. [DOI] [PubMed] [Google Scholar]
- 48.Sarkar C., Zhang Z., Mukherjee A.B. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol. Genet. Metab. 2011;3:383–345. doi: 10.1016/j.ymgme.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum. Genet. 2007;121:1–22. doi: 10.1007/s00439-006-0280-4. [DOI] [PubMed] [Google Scholar]
- 50.Sands M.S., Davidson B.L. Gene therapy for lysosomal storage diseases. Mol. Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Kohan R., Cismondi I.A., Oller-Ramirez A.M., Guelbert N., Anzolini T.V., Alonso G., Mole S.E., de Kremer D.R., de Halac N.I. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr. Pharm. Biotechnol. 2011;12:867–883. doi: 10.2174/138920111795542633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramirez-Montealegre D., Pearce D.A. Defective lysosomal arginine transport in juvenile Batten disease. Hum. Mol. Genet. 2005;14:3759–3773. doi: 10.1093/hmg/ddi406. [DOI] [PubMed] [Google Scholar]
- 53.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pfaffl M.W., Horgan G.W., Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}