

Abstract
A polymorphism located in the G6PC2 gene, which encodes an islet-specific glucose-6-phosphatase catalytic subunit, is the most important common determinant of variations in fasting blood glucose (FBG) levels in humans. Studies of G6pc2 knockout (KO) mice suggest that G6pc2 represents a negative regulator of basal glucose-stimulated insulin secretion (GSIS) that acts by hydrolyzing glucose-6-phosphate (G6P), thereby reducing glycolytic flux. However, this conclusion conflicts with the very low estimates for the rate of glucose cycling in pancreatic islets, as assessed using radioisotopes. We have reassessed the rate of glucose cycling in pancreatic islets using a novel stable isotope method. The data show much higher levels of glucose cycling than previously reported. In 5 mmol/L glucose, islets from C57BL/6J chow-fed mice cycled ∼16% of net glucose uptake. The cycling rate was further increased at 11 mmol/L glucose. Similar cycling rates were observed using islets from high fat–fed mice. Importantly, glucose cycling was abolished in G6pc2 KO mouse islets, confirming that G6pc2 opposes the action of the glucose sensor glucokinase by hydrolyzing G6P. The demonstration of high rates of glucose cycling in pancreatic islets explains why G6pc2 deletion enhances GSIS and why variants in G6PC2 affect FBG in humans.
Introduction
Glucose-6-phosphatase catalyzes the hydrolysis of glucose-6-phosphate (G6P) to glucose and inorganic phosphate (1–5). It exists as a multicomponent system located in the endoplasmic reticulum (ER) and is composed of several integral membrane proteins, namely, a catalytic subunit (G6PC), a glucose transporter, and a G6P/inorganic phosphate antiporter (1–5). Three G6PC isoforms have been identified, which are designated G6PC, G6PC2, and G6PC3 (5). Each isoform is encoded by a separate gene with a distinct pattern of tissue-specific expression (5). G6PC is predominantly expressed in the liver, where it catalyzes the final step in gluconeogenesis and glycogenolysis (1–5). G6PC3, also known as UGRP and G6Pase-β, is ubiquitously expressed (6,7). Mutations that reduce G6PC3 activity result in neutropenia; however, the physiological function of G6PC3 is unclear (8,9). G6PC2, also known as IGRP (10,11), is selectively expressed in pancreatic islet β-cells (12). G6PC2 is a major autoantigen in both mouse (13–15) and human (16,17) type 1 diabetes.
Historically, the question as to whether glucose-6-phosphatase activity is present in islets has been controversial, though it is now generally agreed that activity is detectable, but at a lower level than that found in the liver (2,10,12,18,19). While a majority of studies agree that glucose-6-phosphatase activity exists in pancreatic islets, the issue as to whether the level of activity is enough to affect glucose-stimulated insulin secretion (GSIS), and therefore whether the activity is of biological significance, is currently unresolved. Sweet et al. (19) concluded that, while glucose-6-phosphatase activity is present in rat islets, the level of activity is not enough to result in sufficient G6P hydrolysis so as to affect GSIS. However, two caveats have subsequently arisen with respect to this conclusion. First, in contrast to all other vertebrate species examined (see http://genome.ucsc.edu/), G6PC2 is a pseudogene in rats (11). Second, in various rat models associated with impaired glucose tolerance, G6PC expression is induced such that G6P hydrolysis would be elevated (20–22).
Several articles have also addressed the issue of G6P hydrolysis in mouse islets. Early studies (23) suggested that, even though glucose-6-phosphatase activity is present in mouse islets, G6P hydrolysis does not occur. While seemingly counterintuitive, such a scenario would be possible if G6P entry into the ER lumen was blocked. However, later studies (24,25) challenged this conclusion and showed that the measurement of G6P hydrolysis within islets is critically dependent on assay conditions. More recently, we have shown that G6pc2 accounts for the low glucose-6-phosphatase enzyme activity detected in mouse islets (26) and that a global knockout (KO) of G6pc2 in mice on both a mixed (27) and pure C57BL/6J genetic background (26) results in a mild metabolic phenotype characterized by an ∼15% decrease in fasting blood glucose (FBG) levels. These KO mouse data are consistent with recent genome-wide association studies (GWAS) showing that single nucleotide polymorphisms within the human G6PC2 gene are associated with variations in FBG levels (28,29). Based on these observations, we hypothesize that G6PC2 forms a futile substrate cycle with the β-cell glucose sensor glucokinase (30,31) and acts as a negative regulator of basal GSIS by hydrolyzing G6P, thereby modulating β-cell glycolytic flux (26). Consistent with this model, a reduction in G6pc2 expression results in a leftward shift in the dose-response curve for GSIS, such that under fasting conditions blood glucose levels are reduced (26).
A major caveat with this model for the function of G6PC2 is the fact that estimates of glucose cycling (GC) obtained by radiotracer studies of pancreatic islets are very low (24,25). Because the glucose-6-phosphatase activity of G6PC2 is ∼40-fold lower than that of G6PC (26,32), and because G6PC2 also possesses a phosphatidic acid phosphatase domain (6), the possibility exists that G6PC2 influences GSIS through a mechanism that is independent of its ability to hydrolyze G6P. We have revisited this issue using an alternate, novel stable isotope methodology. With this approach, we can demonstrate much higher levels of GC in islets than has previously been reported. Importantly, GC is abolished in G6pc2 KO mouse islets, suggesting that G6pc2 modulates GSIS, at least in part, through its ability to hydrolyze G6P, and thereby oppose the action of the β-cell glucose sensor glucokinase.
Research Design and Methods
Animal Care
The animal housing and surgical facilities used for this study meet the standards set by the American Association for the Accreditation of Laboratory Animal Care. The Vanderbilt University Medical Center Animal Care and Use Committee approved all the protocols that were used. Prior to islet isolation, mice were maintained either continuously on a standard rodent chow diet (calorie contributions: 28% protein, 12% fat, 60% carbohydrate [14% disaccharides]; LabDiet 5001; PMI Nutrition International) or on a high-fat diet (calorie contributions: 15% protein, 59% fat, 26% carbohydrate [42% disaccharides]; Mouse Diet F3282; BioServ) for 5–8 months. Food and water were provided ad libitum.
Generation of G6pc2 KO Mice
The generation of G6pc2 KO mice on a pure C57BL/6J genetic background has been previously described (26,27).
Islet Isolations
Islets were isolated from adult (8–12 months of age) male wild-type (WT) and G6pc2 KO mice, as previously described (26). Isolated islets were incubated overnight in petri dishes in RPMI 1640 medium containing 11 mmol/L glucose. Aliquots of ∼100 islets were then transferred to 96-well plates and incubated for 24 or 72 h in RPMI 1640 medium containing 5 or 11 mmol/L glucose in a volume of 175 μL. Islets were incubated in either naturally labeled glucose or [1,2,3,4,5,6,6-2H7]glucose (D7-glucose) (98% isotopic purity per site; Cambridge Isotope Laboratories, Andover, MA). After the 24 or 72 h of incubation, islets were resuspended by pipetting and were pelleted by centrifugation. The supernatant was retained for the analysis of insulin content and glucose isotopomers, whereas the cell pellet was washed in PBS and then solubilized in passive lysis buffer (Promega) before quantitation of protein content using the Bio-Rad colorimetric protein assay.
Preparation of Glucose Derivatives
The supernatants collected after islet incubations were divided into three aliquots, and each was derivatized separately to obtain di-O-isopropylidene propionate, aldonitrile pentapropionate, and methyloxime pentapropionate derivatives of glucose. For di-O-isopropylidene derivatization, 20 μL of supernatant was mixed with 20 μL of 5 mmol/L [U-13C6; 1,2,3,4,5,6,6-2H7]glucose (Cambridge Isotope Laboratories), which served as an internal standard. The derivatization then proceeded, as described previously (33), to produce glucose 1,2,5,6-di-isopropylidene propionate. The glucose aldonitrile pentapropionate and glucose methyloxime pentapropionate derivatives were produced as described previously (33).
Gas Chromatography–Mass Spectrometry Analysis
Gas chromatography–mass spectrometry analysis was performed using a 7890A Gas Chromatography System (Agilent) with an HP-5 ms capillary column (30 m × 0.25 mm × 0.25 μm; Agilent J&W Scientific) interfaced with a 5975C mass spectrometer (Agilent). One microliter of di-O-isopropylidene, aldonitrile, or methyloxime derivatives was injected into a 270°C injection port in splitless mode. Helium flow was maintained at 0.88 mL/min. The column temperature was held at 80°C for 1 min, ramped at 20°C/min to 280°C and held for 4 min, then ramped at 40°C/min to 325°C. After a 5-min solvent delay, the mass spectrometer collected data in scan mode from mass/charge ratio (m/z) values of 100 to 500. Each derivative peak was integrated using a custom MATLAB function (34) to obtain mass isotopomer distributions (MIDs) for specific ion ranges.
Estimation of Total Glucose Concentration in the Presence of Deuterated Glucose
Because enzymatic methods for glucose quantification are sensitive to kinetic isotope effects, we used a gas chromatography–mass spectrometry–based method for quantification. The di-O-isopropylidene glucose peak was integrated over a mass range from m/z of 301 (M + 0) to 321 (M + 20), and the ratio of the integrated ion counts of the glucose in the medium supernatant (M + 0 to M + 10) to the fully labeled [U-13C6; 1,2,3,4,5,6,6-2H7]glucose internal standard (M + 11 to M + 20) was determined. We used known glucose standards that were derivatized and analyzed alongside the experimental samples in each run to build a standard curve, as follows:
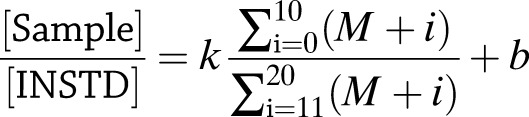 |
(1) |
where [Sample] is the concentration of glucose in the supernatant sample, [INSTD] is the known concentration of the internal standard, (M + i) is the relative abundance of the ith mass isotopomer, k is the slope of the calibration curve, and b is its y-intercept. Cell-free evaporation controls were collected at 24 and 72 h from replicate wells. The glucose concentration of these unlabeled samples was measured with a YSI 2300 STAT Plus Glucose & Lactate Analyzer (YSI Life Sciences). An evaporation correction factor was calculated by dividing the average concentration of the 72-h cell-free controls by the average concentration of the 24-h cell-free controls, and all 72-h sample concentrations were divided by the correction factor.
Calculation of Glucose Uptake and Cycling
Net glucose uptake (vnet) was calculated from the change in medium glucose concentration from 24 to 72 h and expressed in picomoles/islet/hour, as follows:
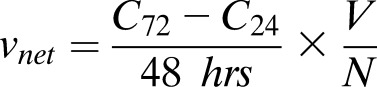 |
(2) |
where C72 is the extracellular glucose concentration at 72 h, C24 is the glucose concentration at 24 h, V is the liquid volume in each well (175 μL), and N is the number of islets in each well. To quantify GC, the rate of G6P hydrolysis (v2) was expressed as a percentage of vnet using an equation derived from the model illustrated in Fig. 1.
Figure 1.

GC is controlled by the action of glucokinase (GK) and G6PC2. Tracer glucose (D7-glucose) is converted to G6P (D7-G6P) through the action of the enzyme glucokinase at the rate v1. G6P is assumed to fully equilibrate with fructose-6-phosphate (F6P). The PGI-catalyzed isomerization of G6P to F6P results in an exchange of hydrogen at the second carbon (C2) of G6P. Any deuterium at C2 derived from the D7-glucose tracer is replaced by an unlabeled hydrogen derived from water during the reverse isomerization reaction. The action of G6PC2 counteracts the activity of GK, converting G6P to glucose at the rate v2. vnet is equal to v1 − v2. Black circles represent a carbon bound to deuterium, and white circles represent a carbon with hydrogen bound.
Using the glucose concentration and relative isotopomer abundance measurements, GC (v2/vnet) was calculated as follows:
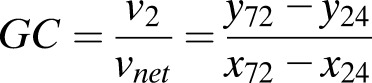 |
(3) |
where
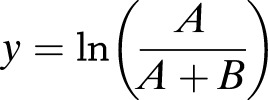 |
and
In this equation, y represents the natural logarithm of the ratio of M + 7 glucose abundance (A) to total M + 1 through M + 7 abundance (A + B), and x is the natural logarithm of the glucose concentration (C). The subscripts on x and y represent the time at which each measurement was obtained. Equation 3 was applied to estimate GC by using the measured di-O-isopropylidene glucose MIDs, corrected for natural isotope abundance (35), to calculate the relative abundances B (m/z 302–307) and A (m/z 308).
Calculation of Positional Enrichment
The MIDs of six ion fragments, with m/z values of 145, 173, 259, 284, 301, and 370, derived from all three glucose derivatives were compiled into a matrix and compared with theoretical distributions using a custom least squares regression program (33). The program was used to estimate the relative abundance of each of the 128 possible hydrogen isotopomers of glucose (Fig. 2). Previous studies (33) validated the positional enrichment methodology using glucose standards.
Figure 2.

A novel stable isotope methodology can be used to assess GC in pancreatic islets. The glucose isotopomers in the media sample are derivatized in three ways. Each derivative is distinct in structure and mass. The derivatives elute at different retention times and produce different ion fragments. The overlapping ion fragments are used to estimate the abundance of each of the possible 27 hydrogen isotopomers of glucose using custom integration and least squares (LSQ) regression programs. The changes in mass of the glucose in the medium, specifically the changes in mass at the second carbon position, are used to quantify cycling. Black circles represent a carbon bound to deuterium, and white circles represent a carbon with hydrogen bound. GC-MS, gas chromatography–mass spectrometry.
Statistical Analyses
Data were analyzed using a two-tailed Student t test, with two samples assuming equal variance. The level of significance is indicated in the figure legends.
Results
Quantification of GC in WT and G6pc2 KO Islets Isolated From Chow-Fed Mice
During incubations with isolated islets, lower mass isotopomers of glucose would be expected to accumulate in the culture medium if a D7-glucose tracer is converted to D7-G6P through the action of glucokinase but is then converted back to D6-glucose through the action of G6pc2 (Fig. 1). To address this hypothesis, we incubated WT and G6pc2 KO islets, which were isolated from chow-fed (CF) mice, in the presence of 5 mmol/L D7-glucose, and measured the MIDs of medium glucose samples collected at 24 and 72 h. We first examined the MIDs obtained from integrating the glucose 1,2,5,6-di-isopropylidene propionate derivative peak, which preserves all seven carbon-bound hydrogen atoms from the parent glucose molecule. After correcting for natural isotope abundance, we summed the relative abundances of the lower mass isotopomers from m/z values of 302 (M + 1) to 307 (M + 6) and normalized them to the total labeled glucose signal from M + 1 to M + 7. The results show a significant accumulation of total M + 1 to M + 6 isotopomers in the incubation media from WT islets but no increase in the media from KO islets (Fig. 3A).
Figure 3.

GC in WT or G6pc2 KO islets isolated from CF mice and incubated in 5 mmol/L D7-glucose. A: The average relative abundance of lower mass isotopomers (M + 1 to M + 6) of the 1,2,5,6-di-isopropylidene propionate derivative fragment in the media from WT (black squares) or KO (white circles) islet incubations at 24 and 72 h. The relative abundances are corrected for natural stable isotope labeling (35) and normalized to the total labeled glucose signal from M + 1 to M + 7. The average relative abundance of the 24-h KO samples was subtracted from baseline values to correct for the isotopic purity of the D7-glucose tracer. In all experiments where tracer control samples were analyzed, the MID of the 24-h KO samples was nearly identical to the MID of the pure D7-glucose tracer (Supplementary Table 1). B: The average estimated abundance ± SEM of the [1,3,4,5,6,6-2H6]glucose isotopomer with deuterium loss at C2 in the media from WT (black bar) or KO (white bar; all experiments reported zero abundance) islet incubations at 72 h. C: Average GC ± SEM, reported as a percentage of vnet. WT islets at 24 h, n = 4; WT islets at 72 h, n = 4; KO islets at 24 h, n = 4; KO islets at 72 h, n = 4 incubations of 100 islets. *P < 0.05 vs. WT islets. hr, hours.
Next, we examined the positional 2H enrichment. Our model suggests that cycling of D7-glucose should result in accumulation of a specific M + 6 mass isotopomer of glucose with deuterium replaced by hydrogen at the C2 position (i.e., [1,3,4,5,6,6-2H6]glucose). Least squares regression analysis using mass isotopomer data obtained from three glucose derivatives (Fig. 2) enabled us to calculate the 2H enrichment at each carbon position of glucose in the culture media. This calculation confirmed that the loss of deuterium at the C2 position of glucose from the fully labeled D7-glucose tracer accounted for the majority of the lower mass isotopomers that were formed in the media from WT islet incubations (Fig. 3B). Accumulation of the [1,3,4,5,6,6-2H6]glucose mass isotopomer was not observed in the media from KO islet incubations (Fig. 3B).
Finally, using equations described in research design and methods, with derivations provided in the Supplementary Material, we calculated v2/vnet (Table 1 and Supplementary Fig. 1A) and found that WT islets exhibited cycling rates that were ∼16% of vnet (Fig. 3C). This is much higher than the 3% rate of GC previously reported by Khan et al. (36), who used a radiotracer method to quantify GC in islets isolated from CF mice and incubated in 5.5 mmol/L glucose. In contrast, GC in KO islets was negligible (Fig. 3C). Taken together, these findings are consistent with our hypothesis that G6PC2 acts by hydrolyzing G6P, thereby opposing the action of glucokinase.
Table 1.
Comparison of GC and uptake among experimental groups
| Diet | Glucose concentration (mmol/L) |
v2 (pmol/islet/h) |
vnet (pmol/islet/h) |
v2/vnet (%) |
|||
|---|---|---|---|---|---|---|---|
| WT | G6pc2 KO | WT | G6pc2 KO | WT | G6pc2 KO | ||
| CF | 5 | 2.7 ± 0.4 (4)A | 0.1 ± 0.3 (4) | 18 ± 1 (4)A | 14 ± 3 (4)A | 16 ± 4 (4)A | 2 ± 2 (4) |
| HFF | 5 | 3.1 ± 0.1 (5)A | 0.1 ± 0.3 (5) | 14 ± 2 (5)A | 14 ± 3 (5)A | 25 ± 3 (5)A/B | 0.5 ± 2 (5) |
| CF | 11 | 11 ± 1 (5)B | 0.9 ± 0.3 (4) | 28 ± 4 (5)A/B | 31 ± 5 (4)A/B | 40 ± 6 (5)B | 3 ± 1 (4) |
| HFF | 11 | 14 ± 1 (7)B | 1.2 ± 0.4 (7) | 45 ± 8 (7)B | 46 ± 9 (7)B | 35 ± 5 (7)B | 3 ± 1 (7) |
Rate of v2, vnet, and v2/vnet results are reported as the mean ± SEM (number of incubations). In each column, groups were compared by ANOVA. If at least one mean value was significantly different, pairwise comparisons were performed by Tukey-Kramer test. Statistically separated groups are indicated by different letters (A and B).
Quantification of GC in WT and G6pc2 KO Islets Isolated From High Fat–Fed Mice
Both WT and G6pc2 KO mice fed a high-fat diet (i.e., high fat–fed [HFF] mice) have a ∼100 mg/dL elevation in FBG levels, though, as in CF mice, FBG levels remain ∼20 mg/dL lower in G6pc2 KO mice compared with WT mice (26). Based on this observation, we hypothesized that v2 would be similar in CF and HFF mouse islets (Supplementary Fig. 2). To address this hypothesis, we applied our stable isotope method to islets isolated from HFF mice.
Consistent with our model for the action of G6PC2 on GC, M + 1 to M + 6 glucose isotopomers accumulated in the media between 24 and 72 h in WT islet incubations only (Fig. 4A). Further validation of the cycling model was provided by the positional 2H enrichment results, which showed that the relative abundance of the [1,3,4,5,6,6-2H6]glucose isotopomer with deuterium loss at the second carbon was greater in the WT islet incubations compared with the KO islet incubations (Fig. 4B). We again calculated the rate of GC relative to vnet. As expected, and consistent with the similar accumulation of M + 1 to M + 6 isotopomers observed in the HFF incubations (Fig. 4A), v2 was nearly identical between CF and HFF islets in 5 mmol/L glucose (Table 1). vnet (Table 1 and Supplementary Fig. 1B) and GC (Table 1 and Fig. 4C) were not statistically different between CF and HFF islets. As in CF islets, GC was completely abolished in KO islets from HFF mice (Fig. 4C).
Figure 4.

GC in WT or G6pc2 KO islets isolated from HFF mice and incubated in 5 mmol/L D7-glucose. The same data are shown as described in Fig. 3 but using islets obtained from HFF mice. WT islets at 24 h, n = 5; WT islets at 72 h, n = 5; KO islets at 24 h, n = 5; KO islets at 72 h, n = 5 incubations of 100 islets. *P < 0.05 vs. WT islets. hr, hours.
Quantification of the Influence of Extracellular Glucose Concentration on GC in WT and G6pc2 KO Islets
An analysis of insulin secretion from perfused WT and G6pc2 KO mouse pancreata suggests a parallel shift in the dose-response curve for GSIS to the left in the KO pancreata when glucose increased from 5 to 11 mmol/L (26). This parallel shift is indicative of a fixed v2, since the rate of glucose phosphorylation by glucokinase (v1) should be the same in both WT and KO pancreata at the same glucose concentration, with the difference in GSIS between the two curves being solely attributable to v2 (Supplementary Fig. 2). This situation could arise if either the transport of G6P into the ER lumen or the hydrolysis of G6P to glucose by G6pc2 was operating at Vmax, with the latter being supported by enzymatic studies (32). Based on this observation, we hypothesized that v2 would be similar at 5 and 11 mmol/L, and that v2/vnet would be reduced in response to the expected increase in vnet at elevated glucose concentrations. To address this hypothesis, GC was assessed at 11 mmol/L glucose using islets isolated from CF and HFF mice.
If v2 was similar in the 5 and 11 mmol/L incubations, as hypothesized, the accumulation of M + 1 to M + 6 isotopomers of glucose in the medium would be similar. However, a significant increase in the accumulation of M + 1 to M + 6 isotopomers, consistent with an increase in v2, was observed in CF (Fig. 5A) and HFF (Fig. 6A) islets. Even at the early 24-h time point, the accumulation of M + 1 to M + 6 isotopomers in the media is significant in CF (Fig. 5A) and HFF (Fig. 6A) WT incubations, while the abundance of M + 1 to M + 6 isotopomers in KO incubations does not change from the baseline. The accumulated M + 1 to M + 6 isotopomers in the WT media contain a significant abundance of the model-predicted [1,3,4,5,6,6-2H6]glucose isotopomer with deuterium loss at the second carbon position, while this isotopomer is not present in the KO incubations (Figs. 5B and 6B). Unexpectedly, both CF and HFF WT islets exhibited increased cycling, relative to CF islets in 5 mmol/L glucose, driven by the significant increase in v2, with rates quantified as ∼35–40% of net uptake (Figs. 5C and 6C). Once again, the cycling in KO islets was negligible (Figs. 5C and 6C).
Figure 5.

GC in WT or G6pc2 KO islets isolated from CF mice and incubated in 11 mmol/L D7-glucose. The same data are shown as described in Fig. 3 but using islets incubated in 11 mmol/L D7-glucose. WT islets at 24 h, n = 5; WT islets at 72 h, n = 5; KO islets at 24 h, n = 4; KO islets at 72 h, n = 4 incubations of 100 islets. *P < 0.05 vs. WT islets. hr, hours.
Figure 6.

GC in WT or G6pc2 KO islets isolated from HFF mice and incubated in 11 mmol/L D7-glucose. The same data are shown as described in Fig. 3 but using islets obtained from HFF mice and incubated at 11 mmol/L glucose. WT islets at 24 h, n = 7; WT islets at 72 h, n = 7; KO islets at 24 h, n = 7; KO islets at 72 h, n = 7 incubations of 100 islets. *P < 0.05 vs. WT islets. hr, hours.
Discussion
This study describes a novel method for quantifying GC in isolated pancreatic islets that is based on the use of a stable D7-glucose tracer and the model presented in Fig. 1. We applied this method to islets isolated from WT and G6pc2 KO mice to investigate the role of G6pc2 in GC. In all WT islet incubations, a significant accumulation of M + 1 to M + 6 glucose isotopomers was observed over 72 h (Figs. 3–6), and a specific, model-predicted D6-glucose isotopomer, [1,3,4,5,6,6-2H6]glucose, accounted for most of the cycled glucose, as determined by a custom least squares regression algorithm (Fig. 2). GC in WT islets varied with the assay conditions and was estimated to be 16–40% of vnet (Table 1). G6pc2 KO islets took up glucose from the media at rates similar to those of WT islets (Table 1 and Supplementary Fig. 1), but no accumulation of M + 1 to M + 6 glucose isotopomers was observed (Figs. 3–6), and GC was negligible (Table 1). These observations are consistent with our hypothesis that G6PC2 controls GC by opposing the action of glucokinase, thereby regulating glycolytic flux, and ultimately GSIS. In this manner, G6PC2 may play an important role in the regulation of FBG levels, a hypothesis supported by GWAS data linking G6CP2 to FBG in humans (28,29,37).
WT islets isolated from CF mice and incubated in 5 mmol/L glucose cycled at a rate that was ∼16% of vnet (Fig. 3). Previous studies (20,36) using tritiated water quantified GC in isolated islets by measuring 3H incorporation at carbon 2 of derivatized glucose. This method was not sensitive enough to quantify cycling in 3-h, 5.5 mmol/L glucose incubations using islets isolated from lean WT mice but, based on the extrapolation of data derived from ob/ob mouse islets, was used to estimate WT cycling rates at 3–4% of total uptake (36). While our novel method for measuring GC using stable isotopes requires longer incubation times, the simplicity and sensitivity are distinct advantages. Both the tritiated water method (36) and our method for measuring GC rely on the assumption that G6P and fructose-6-phosphate are in a rapid and complete equilibrium. However, hydrogen isotopes alter the kinetics of the phosphoglucoisomerase (PGI) reaction such that equilibrium may not be complete. Nevertheless, previous work (38) suggests that the kinetic isotope effect of tritium reduces the Vmax of the PGI reaction more than deuterium, which potentially makes our stable isotope method more accurate in applying this assumption. We expect that the rates of other reactions in our model, including the reactions catalyzed by glucokinase (v1) and G6pc2 (v2), would be minimally affected by the presence of either 2H or 3H tracers because they do not involve hydrogen transfer. Both methods account only for G6P molecules that are completely cycled out of the cell and subsequently accumulate in the culture medium. Therefore, the calculated rates of GC will underestimate the true cycling rate catalyzed by G6pc2 if some of the resulting glucose molecules are redirected toward an alternative intracellular fate and do not reappear in the extracellular medium.
To investigate the effect of diet on GC, islets were isolated from HFF mice. Similar increased rates of G6P hydrolysis were observed in CF and HFF islets when glucose was increased from 5 to 11 mmol/L glucose (Table 1), which is consistent with our previous observation that the difference in FBG levels between WT and G6pc2 KO mice is similar in mice receiving both diets, despite elevated FBG levels in the HFF groups (26). Interestingly, previous studies (24,25,36,39,40) reported a significant increase in GC in islets isolated from obese ob/ob mice relative to WT mice. Kahn and Efendic (39) showed that this increase was not due to hyperglycemia, which is characteristic of ob/ob mice. However, whether hyperlipidemia or the absence of leptin signaling influences GC in ob/ob mice is unclear. Furthermore, the magnitude of this previously reported enhancement of GC in ob/ob mouse islets could, in part, be due to the underestimation of cycling in lean WT mice.
Our method requires 72-h islet incubations in order to enrich the medium in lower mass isotopomers of glucose for quantification. Over this time period in nonphysiological conditions, the data suggest that the islets are losing some of their in vivo characteristics. G6pc2 KO mice have a significantly lower FBG level than their WT littermates (26), and, consistent with this finding, isolated KO islets exhibited increased glycolytic flux (data not shown) and insulin secretion (26) in short-term assays. However, after 72 h in culture, insulin secretion between the WT and KO islets normalizes such that the concentration of insulin in the medium is not significantly different between WT and KO islets (Supplementary Fig. 3). In addition, the estimated vnet between 24 and 72 h in WT and G6pc2 KO islets is equivalent (Table 1 and Supplementary Fig. 1). While these glucose uptake data are consistent with the insulin secretion data (Supplementary Fig. 3), the acute insulin secretion assays predict increased glucose uptake in G6pc2 KO islets relative to WT islets. Similarly, a surprising elevation in the rate of G6P hydrolysis was observed in CF and HFF islets incubated in 11 mmol/L glucose (Figs. 5A and 6A and Table 1). This observation is at odds with perfused pancreas data showing a parallel, leftward shift in the dose-response curve for GSIS in CF G6pc2 KO mice (26). In combination, these observations suggest that the islets are undergoing adaptation during the long assay incubation. We hypothesize that the KO islets adapt to the absence of G6pc2 by reducing glucose uptake, which would make biological sense in that elevated glucose metabolism has been shown to be toxic to β-cells (41). We also hypothesize that the ability of glucose to stimulate G6pc2 expression (32) may explain the increase in v2. This induction would not have been a factor in short-term insulin secretion experiments, given their short time course. Addressing these hypotheses and the molecular mechanisms behind islet adaptation to prolonged culture are clearly of interest and will be the subject of future experiments. Given these apparent effects of the long incubation time and any limitation in gas exchange in the 96-well plates used for the assay, future work will focus on using microfluidic devices specifically designed for islet culture (42,43), allowing the media volume, and thus the time in culture, to be reduced. Such experiments would also reveal whether the significant increase in glucose uptake at 11 mmol/L in HFF islets, but not in CF islets (Table 1), is also an artifact of the long incubation times.
In conclusion, the data presented in this study suggest that GC in WT islets is greater than previously reported and that G6pc2 is responsible for this cycling. These findings are consistent with the GWAS linking G6PC2 to the regulation of FBG levels (28,29,37).
Article Information
Acknowledgments. The authors thank P. Jacobson (Abbott Pharmaceuticals) for assistance with the initiation of this project. The authors also thank M. Brissova and A. Coldren (Vanderbilt University) for performing islet isolations.
Funding. Research in the laboratory of R.M.O. was supported by National Institutes of Health (NIH) grant DK-92589. Research in the laboratory of J.D.Y. was supported by National Science Foundation grant CBET 0955251. The Vanderbilt Islet Procurement and Analysis Core is supported by NIH grant P60-DK-20593 to the Vanderbilt Diabetes Research Training Center and NIH grant DK-59637 to the Vanderbilt Mouse Metabolic Phenotyping Center. M.L.W. and L.D.P. were both supported by Vanderbilt Molecular Endocrinology Training Program grant 5T32 DK07563.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.L.W. took the primary lead on glucose cycling studies and helped to write the manuscript. L.D.P. and I.T. helped to perform some of the glucose cycling experiments. R.M.O. and J.D.Y. helped to perform some of the glucose cycling experiments and write the manuscript. J.D.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 74th Scientific Sessions of the American Diabetes Association, San Francisco, CA, 13–17 June 2014.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0745/-/DC1.
References
- 1.Mithieux G. New knowledge regarding glucose-6 phosphatase gene and protein and their roles in the regulation of glucose metabolism. Eur J Endocrinol 1997;136:137–145 [DOI] [PubMed] [Google Scholar]
- 2.Foster JD, Pederson BA, Nordlie RC. Glucose-6-phosphatase structure, regulation, and function: an update. Proc Soc Exp Biol Med 1997;215:314–332 [DOI] [PubMed] [Google Scholar]
- 3.van de Werve G, Lange A, Newgard C, Méchin MC, Li Y, Berteloot A. New lessons in the regulation of glucose metabolism taught by the glucose 6-phosphatase system. Eur J Biochem 2000;267:1533–1549 [DOI] [PubMed] [Google Scholar]
- 4.van Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J 2002;362:513–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem 2009;284:29241–29245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin CC, Oeser JK, Svitek CA, Hunter SI, Hutton JC, O’Brien RM. Identification and characterization of a human cDNA and gene encoding a ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol 2002;29:205–222 [DOI] [PubMed] [Google Scholar]
- 7.Boustead JN, Martin CC, Oeser JK, et al. Identification and characterization of a cDNA and the gene encoding the mouse ubiquitously expressed glucose-6-phosphatase catalytic subunit-related protein. J Mol Endocrinol 2004;32:33–53 [DOI] [PubMed] [Google Scholar]
- 8.Cheung YY, Kim SY, Yiu WH, et al. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J Clin Invest 2007;117:784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boztug K, Appaswamy G, Ashikov A, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med 2009;360:32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arden SD, Zahn T, Steegers S, et al. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic subunit-related protein. Diabetes 1999;48:531–542 [DOI] [PubMed] [Google Scholar]
- 11.Martin CC, Bischof LJ, Bergman B, et al. Cloning and characterization of the human and rat islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) genes. J Biol Chem 2001;276:25197–25207 [DOI] [PubMed] [Google Scholar]
- 12.O’Brien RM. Moving on from GWAS: functional studies on the G6PC2 gene implicated in the regulation of fasting blood glucose. Curr Diab Rep 2013;13:768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lieberman SM, Evans AM, Han B, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A 2003;100:8384–8388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han B, Serra P, Amrani A, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med 2005;11:645–652 [DOI] [PubMed] [Google Scholar]
- 15.Mukherjee R, Wagar D, Stephens TA, Lee-Chan E, Singh B. Identification of CD4+ T cell-specific epitopes of islet-specific glucose-6-phosphatase catalytic subunit-related protein: a novel beta cell autoantigen in type 1 diabetes. J Immunol 2005;174:5306–5315 [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Danke NA, Berger D, et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J Immunol 2006;176:2781–2789 [DOI] [PubMed] [Google Scholar]
- 17.Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin Immunol 2008;127:359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perales MA, Sener A, Malaisse WJ. Hexose metabolism in pancreatic islets: the glucose-6-phosphatase riddle. Mol Cell Biochem 1991;101:67–71 [DOI] [PubMed] [Google Scholar]
- 19.Sweet IR, Najafi H, Li G, Grodberg J, Matschinsky FM. Measurement and modeling of glucose-6-phosphatase in pancreatic islets. Am J Physiol 1997;272:E696–E711 [DOI] [PubMed] [Google Scholar]
- 20.Khan A, Chandramouli V, Ostenson CG, Löw H, Landau BR, Efendić S. Glucose cycling in islets from healthy and diabetic rats. Diabetes 1990;39:456–459 [DOI] [PubMed] [Google Scholar]
- 21.Laybutt DR, Glandt M, Xu G, et al. Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J Biol Chem 2003;278:2997–3005 [DOI] [PubMed] [Google Scholar]
- 22.Tokuyama Y, Sturis J, DePaoli AM, et al. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes 1995;44:1447–1457 [DOI] [PubMed] [Google Scholar]
- 23.Ashcroft SJ, Randle PJ. Glucose-6-phosphatase activity of mouse pancreatic islets. Nature 1968;219:857–858 [DOI] [PubMed] [Google Scholar]
- 24.Khan A, Chandramouli V, Ostenson CG, et al. Evidence for the presence of glucose cycling in pancreatic islets of the ob/ob mouse. J Biol Chem 1989;264:9732–9733 [PubMed] [Google Scholar]
- 25.Chandramouli V, Khan A, Ostenson CG, et al. Quantification of glucose cycling and the extent of equilibration of glucose 6-phosphate with fructose 6-phosphate in islets from ob/ob mice. Biochem J 1991;278:353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pound LD, Oeser JK, O’Brien TP, et al. G6PC2: a negative regulator of basal glucose-stimulated insulin secretion. Diabetes 2013;62:1547–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Martin CC, Oeser JK, et al. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia 2007;50:774–778 [DOI] [PubMed] [Google Scholar]
- 28.Bouatia-Naji N, Rocheleau G, Van Lommel L, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 2008;320:1085–1088 [DOI] [PubMed] [Google Scholar]
- 29.Chen WM, Erdos MR, Jackson AU, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest 2008;118:2620–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matschinsky FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 1996;45:223–241 [DOI] [PubMed] [Google Scholar]
- 31.Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 2009;66:27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrolonis AJ, Yang Q, Tummino PJ, et al. Enzymatic characterization of the pancreatic islet-specific glucose-6-phosphatase-related protein (IGRP). J Biol Chem 2004;279:13976–13983 [DOI] [PubMed] [Google Scholar]
- 33.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Measuring deuterium enrichment of glucose hydrogen atoms by gas chromatography/mass spectrometry. Anal Chem 2011;83:3211–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antoniewicz MR, Kelleher JK, Stephanopoulos G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal Chem 2007;79:7554–7559 [DOI] [PubMed] [Google Scholar]
- 35.Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom 1996;31:255–262 [DOI] [PubMed] [Google Scholar]
- 36.Khan A, Chandramouli V, Ostenson CG, et al. Glucose cycling is markedly enhanced in pancreatic islets of obese hyperglycemic mice. Endocrinology 1990;126:2413–2416 [DOI] [PubMed] [Google Scholar]
- 37.Bouatia-Naji N, Bonnefond A, Baerenwald DA, et al. Genetic and functional assessment of the role of the rs13431652-A and rs573225-A alleles in the G6PC2 promoter that are strongly associated with elevated fasting glucose levels. Diabetes 2010;59:2662–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malaisse WJ, Malaisse-Lagae F, Liemans V, Ottinger R, Willem R. Phosphoglucoisomerase-catalyzed interconversion of hexose phosphates: isotopic discrimination between hydrogen and deuterium. Mol Cell Biochem 1990;93:153–165 [DOI] [PubMed] [Google Scholar]
- 39.Khan A, Efendic S. Evidence that increased glucose cycling in islets of diabetic ob/ob mice is a primary feature of the disease. Am J Physiol 1995;269:E623–E626 [DOI] [PubMed] [Google Scholar]
- 40.Khan A, Narangoda S, Ahren B, Holm C, Sundler F, Efendic S. Long-term leptin treatment of ob/ob mice improves glucose-induced insulin secretion. Int J Obes Relat Metab Disord 2001;25:816–821 [DOI] [PubMed] [Google Scholar]
- 41.Dadon D, Tornovsky-Babaey S, Furth-Lavi J, et al. Glucose metabolism: key endogenous regulator of β-cell replication and survival. Diabetes Obes Metab 2012;14(Suppl. 3):101–108 [DOI] [PubMed] [Google Scholar]
- 42.Easley CJ, Rocheleau JV, Head WS, Piston DW. Quantitative measurement of zinc secretion from pancreatic islets with high temporal resolution using droplet-based microfluidics. Anal Chem 2009;81:9086–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rocheleau JV, Walker GM, Head WS, McGuinness OP, Piston DW. Microfluidic glucose stimulation reveals limited coordination of intracellular Ca2+ activity oscillations in pancreatic islets. Proc Natl Acad Sci U S A 2004;101:12899–12903 [DOI] [PMC free article] [PubMed] [Google Scholar]