

Abstract
Epigenome refers to “epi” meaning outside the “genome.” Epigenetics is the field of study of the epigenome. Epigenetic modifications include changes in the promoter CpG Islands, modifications of histone protein structure, posttranslational repression by micro-RNA which contributes to the alteration of gene expression. Epigenetics provides an understanding of the role of gene-environment interactions on disease phenotype especially in complex multifactorial diseases. Periodontitis is a chronic inflammatory disorder that affects the supporting structures of the tooth. The role of the genome (in terms of genetic polymorphisms) in periodontitis pathogenesis has been examined in numerous studies, and chronic periodontitis has been established as a polygenic disorder. The potential role of epigenetic modifications in the various facets of pathogenesis of periodontitis is discussed in this paper based on the available literature.
Keywords: Chronic periodontitis, epigenetic modifications, micro-RNA, pathogenesis
INTRODUCTION
Epigenetics is defined as the study of mitotically and meiotically heritable changes in gene function that are not dependent on DNA sequence.[1] The major difference between epigenetics and genetics is that epigenetic changes occur more frequently than genetic changes. The epigenetic changes are reversible by treatment with pharmacological agents.[2] Epigenome, the overall epigenetic state of an organism, is just as important as the genome for normal development and environmental factors can also have profound effects on the epigenetic changes and induce susceptibility to disease.[3]
In mammalian cells, there are three types of epigenetic regulation on gene expression- the DNA methylation, histone modification, and RNA-associated silencing (micro-RNA)[4] [Figures 1–3]. The enzymatic DNA methylation of the C-5 position of cytosine residues in the CpG islands of the promoter region of a gene is considered as the most important epigenetic mechanisms in mammals.[5] More and more evidences have shown that epigenetic modifications play a crucial role in cancer and other diseases.[6] The epigenetic modifications have been further discussed and summarized in Table 1.
Figure 1.
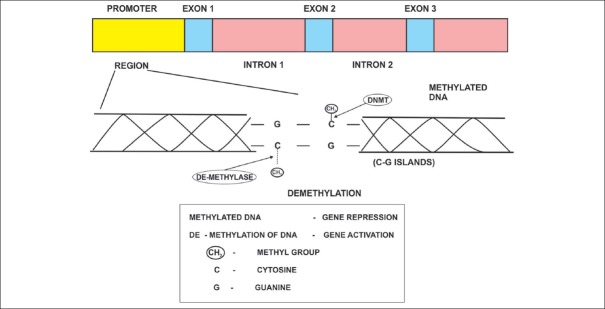
DNA methylation and de-methylation mediated by DNA methyl transferase (DNMT) and de-methylase enzymes and their role in gene expression
Figure 3.
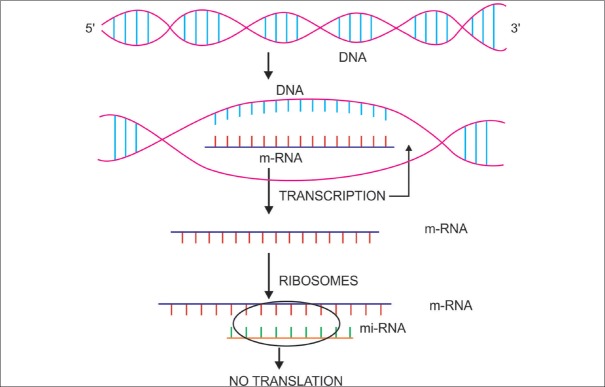
Micro RNA's and their role in translational repression
Table 1.
Summary of epigenetic modifications and their influence on gene expression

Figure 2.
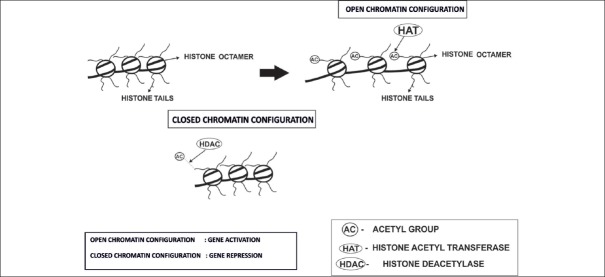
Histone modifications and its import on chromatin configuration and gene expression
ROLE OF EPIGENETIC CHANGES IN PERIODONTAL DISEASE
Epigenetic events determine gene expression through the remodeling of chromatin and selective activation or inactivation of genes. These events induce changes in cytokine profile, immune mechanisms, and thereby contribute to the pathogenesis of various infectious and inflammatory diseases.[12] Chronic periodontitis is a multifactorial disease characterized by inflammation and destruction of the tooth supporting structures.[13] Although periodontitis has a microbial etiology, its progression can be influenced by several factors such as systemic diseases, environmental factors, and genetic factors.[13] The exact role of epigenetic changes on bacteria-induced host inflammatory response has not been studied extensively. Patients with the same clinical features respond differently to the same treatment, suggesting inter-individual variability influenced by genetic,[14] as well as epigenetic factors.[15] This review focuses on such epigenetic alterations which could help in understanding the mechanisms relating to periodontal disease activity.
EPIGENETIC IMPRINT OF BIOFILM ON PERIODONTITIS PATHOGENESIS
The oral environment has a diverse microbial profile. The tooth provides a hard nonshedding surface that facilitates the formation of dental plaque. The plaque bacteria evoke host immune response in the gingival epithelium. Gingival epithelia utilize multiple signaling pathways to regulate innate immune responses to various oral bacteria, but little is understood about how these bacteria alter the epithelial epigenetic status. Recent evidence has shown, bacteria belonging to the orange and red complex can cause epigenetic changes in the periodontal tissues.[16]
A recent study by Yin and Chung[17] provides a new insight into the bacteria-specific innate immune responses via epigenetic regulation. The authors reported that the presence of bacteria results in epigenetic modifications in gingival epithelia and exposure to different oral bacteria results in differential methylation profile. In the study, stimulation of gingival epithelial cells (GECs) with Fusobacterium nucleatum resulted in hypermethylation of Mucosa associated lymphoid tissue lymphoma translocation gene 1 (MALT1) and thereby a lack of Nuclear factor kappa B production by GEC. On the other hand, stimulation of GEC with Porphyromonas gingivalis resulted in hypomethylation of ZNF287 a DNA binding protein believed to be involved in transcriptional regulation. In addition, P. gingivalis significantly decreased the tri-methylation of histone H3 K4 protein expression, but F. nucleatum did not, indicating that P. gingivalis could suppress the activation of transcription, which was found to be consistent with the “stealth-like” properties of P. gingivalis.
Chung and Dale,[18] reported a differential induction of downstream innate immune markers following exposure of GECs to different oral bacteria. The authors reported that different signaling pathways were involved for different bacteria. Similar studies by Yin et al.[19] and Chung et al.,[20] have also demonstrated a lower level of cytokines and chemokines secretion in P. gingivalis – stimulated dendritic cells and GEC's.
Earlier reports had demonstrated that P. gingivalis produces a broad array of virulence factors that could trigger GECs through pathogen-associated molecular pattern receptors such as toll-like receptor (TLR) TLR2/TLR4[21] or protease-activated receptor[22] which lead to the activation of transcription signaling pathways NF-κB, MAPK (mitogen-activated protein kinase), and MEK/ERK (mitogen-activated protein kinase/extracellular regulated kinase). Huang et al.[23] reported that both recruitments of NF-κB to target promoters and NF-κB-induced transcriptional genes are modulated through chromatin modification. It is, therefore, likely that connections exist between these transduction signaling pathways and DNA methyltransferase and histone deacetylase.
From the above-mentioned studies, it can be inferred that perio-pathogens like P. gingivalis can modulate the host defense through inducing epigenetic changes in transcription factor expression in GECs.
EPIGENETIC CHANGES IN CYTOKINE GENES IN CHRONIC PERIODONTITIS
Several studies have been performed in recent years on epigenetic changes in the cytokine genes implicated in periodontal disease pathogenesis. One of the earliest studies on epigenetic changes in subjects with chronic periodontitis,[24] evaluated the methylation status of DNA in the promoter region of interleukin-8 (IL-8; a chemokine) in gingival and oral mucosal cells, leucocytes in blood from healthy individuals, smokers and nonsmoker subjects with chronic periodontitis. The authors also co-related the methylation status with m-RNA levels of IL-8 and reported a higher percentage of hypomethylation of IL-8 gene in chronic periodontitis subjects (independent of smoking) in DNA of oral mucosal cells, no significant differences were observed in gingival and blood cells between different groups with regard to the methylation status.
Evaluation of epigenetic changes in the promoter region of Prostaglandin synthase 2, the gene coding for cyclooxygenase (COX-2) in chronic periodontitis subjects, revealed a hyper-methylation status of the gene and lower levels of COX-2 transcription in inflamed gingival biopsy.[25] Another study in the subjects with aggressive periodontitis evaluated the DNA methylation status in the promoter region of IL-8 gene in oral and GECs also. The authors reported a hypomethylated status in oral and GECs of subjects who presented with generalized aggressive periodontitis.[26]
Zhang et al.,[27] evaluated the presence of epigenetic modifications in the promoter region of interferon gamma (IFNG) gene in different stages of periodontal disease and the authors reported a significant hypomethylation and increased IFNG transcription in gingival biopsies from chronic periodontitis sites.
Epigenetic changes in E-Cadherin and COX-2 genes have been examined in chronic periodontitis and nonperiodontitis subjects and breast cancer biopsies.[28] A hypermethylation of E-Cadherin and COX-2 was observed in similar proportions in breast cancer (E Cadherin - 38% and COX-2: 35%) and chronic periodontitis subjects (E Cadherin - 25%, COX - 2:19%). The authors concluded that epigenetics changes observed in the two genes in periodontitis subjects might be related to the irreversible destruction in the tissues similar to that observed in cancer.
The epigenetic alteration of cytokine levels assumes significance in that, it can influence the type of Immune (Th) response and also regulate DNA methyltransferase (DNMT) expression in T cells and, therefore, play a significant role in regulation of immune response.
EPIGENETIC BASIS FOR IMMUNE-REGULATION IN PERIODONTITIS PATHOGENESIS
Chronic periodontitis is characterized by a host inflammatory response to the specific periodontal pathogens. The host immune response is characterized by an innate and adaptive phase. One of the most important immune mechanisms through which the innate and adaptive pathways are interconnected is the TLR pathway. Micro-RNAs have been implicated in controlling TLR responses to the bacteria. A recent study[29] reported that polymicrobial infection with three periodontal pathogens (P. gingivalis, Treponema denticola, Tannerella forsythia) enhanced the levels of the miR-146a in ApoE-/- mice with experimental periodontitis. In the same study, THP 1 monocyte cell line cultures were stimulated with a combination of the periodontal pathogens, and an increase in mi-RNA levels was shown in a time-dependent manner and this co-related with the down-regulation of adaptor kinases IL-1 receptor-associated kinase 1 (IRAK-1), tumor necrosis factor (TNF) receptor-associated factor (TRAF-6), and TNF-alpha production by these cells. The authors concluded that the elevated levels of miR-146a contributed to endotoxin tolerance by negatively influencing IRAK-1 and TRAF-6 levels at a post transcriptional level and that miR-146a may represent a target for therapeutic intervention in periodontal disease.
A similar observation was made by Ceppi et al.,[30] who examined the levels of miR-155 in monocyte-derived dendritic cells exposed to endotoxin lipopolysaccharide (LPS). An elevated level of miR-155 was demonstrated, and it was shown to down-regulate TAB2 (TAK1 binding protein 2) which plays an important role in IL-1 signaling pathways. The authors concluded that the miR-155 mediated the negative feedback loop and helped modulate the inflammatory responses of dendritic cells.
The micro RNA expression profile in chronic periodontitis as compared to healthy gingiva was evaluated in a recent study using the microarray technique,[31] and miR-181b, mi-R19b, miR-30a, miR-let 7a, and miR-301a expression were shown to be significantly higher in periodontitis group as compared to the healthy group. The significance of these findings, however, remains to be yet determined.
In the pathogenesis of periodontitis, the progression from gingivitis to periodontitis is characterized by a transition from Th1 to Th2 subsets.[32] The naïve T cells undergo differentiation into various lineages and during the differentiation process, changes in the chromatin structure occur through epigenetic mechanisms such as histone modification, DNA methylation, generation of DNAse I hypersensitive sites.[33] The primary cytokine genes which define the lineage specificity include IFN Gamma gene for Th1 cell lineage, IL-4 gene for Th2 cell lineage, and IL-17 gene for Th17 cells. Difference in methylation patterns has been observed in the various T cell subsets. T cells belonging to the Th1 lineage were marked H3K4 me3 (active IFN-gamma gene), whereas Th2 cells have H3 K27 me3 in their IFN-gamma gene (repression of gene expression).[34] Induced Treg cells have been marked with H3K4 me3 in the FoxP3 gene, which was, however, not observed in Th17 subset and n Tregs.[35] Taking into consideration the above literature, and periodontitis being an inflammatory condition whose pathogenesis involves the different T cell subsets, further studies can be performed to analyze the role of the above-mentioned epigenetic marks in disease pathogenesis.
The role of autoimmunity in the pathogenesis of periodontitis has been explored since 1965.[36] A later study by Hirsch et al.,[37] demonstrated the presence of a high number of cells that secreted antibody against Type I and to a lesser extent type III collagen in gingival tissue biopsies of subjects with adult (chronic) periodontitis. The results of this study suggested the possibility of autoimmunity contributing to the pathogenesis of adult (chronic) periodontitis. Autoreactivity by T cells has been demonstrated following the inhibition of DNA methylation by methylation inhibitors, 5’ Azacytidine in mature T cells. Anti-DNA antibody production may be augmented by overstimulation of B cells due to overexpression of cytokines, cell surface co-stimulatory molecules by de-methylated T cells.[38] A similar overstimulation has been observed in periodontitis with polyclonal activation of B cells triggered by the periodontal pathogen F. nucleatum.[39,40] Natural T regulatory cells represent a subset of immune cells that have a suppressive function and also play a role in control of immune responses and maintain peripheral tolerance.[41] Nakajima et al.,[42] have demonstrated elevated levels of T regulatory cells in gingival biopsies from periodontitis subjects suggesting a role for this T cell subset in periodontitis pathogenesis. Fox P3, a transcription factor is very important for development and function of T regulatory cells. Epigenetic alterations at the Foxp3 locus by DNA methylation/histone modifications contribute to the establishment of a committed lineage of phenotypically stable regulatory T cells.[43] Whether epigenetic pathways are involved in autoimmune mechanisms in periodontitis pathogenesis remains to be determined.
It is interesting to note that not all cases of gingivitis progress to periodontitis and that there are different models for progression of periodontitis. Epigenetic pathways by the virtue of their action on epithelial cells, pathogen-associated molecular pattern signaling pathways, immune regulatory mechanisms, and cytokine production variability may contribute significantly to periodontitis pathogenesis. [Figure 4]
Figure 4.
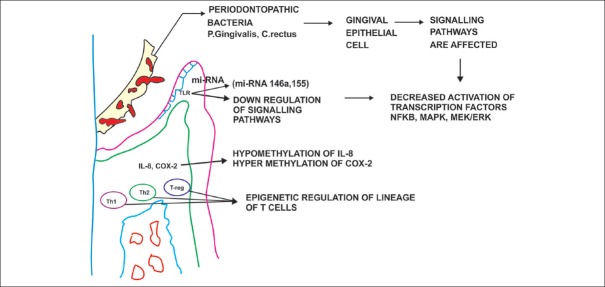
The potential epigenetic pathways involved in periodontitis pathogenesis
ROLE OF ENVIRONMENT ON PERIODONTITIS PATHOGENESIS: THE EPIGENETIC PATHWAY
Environmental influences have been proposed to contribute to the progression of periodontitis, and one of the important environmental risk factors for progression of periodontitis is smoking. Recent studies have demonstrated a global de-methylation induced by smoking increases the risk for development of tumors.[44] A recent study by Oliveira et al.,[24] evaluated the methylation status of Il-8 promoter in smokers and nonsmoker subjects with periodontitis and the authors concluded there was no significant difference between the groups with regard to the methylation status. However, no global methylation status studies have been done in smokers with periodontitis to assess the impact of smoking at an epigenetic level in subjects with periodontitis.
The role of micronutrients in periodontitis pathogenesis has been elicited in previous studies.[45,46] Nutrition has been shown to influence gene expression through epigenetic modifications.[47] A well-documented example of this influence is folate deficiency during pregnancy leads to a lack of S-Adenosylmethionine a substrate required for the enzyme DNMT to methylate CpG residues during embryonic development.[48]
Epigenetic mechanisms represent a pathway through which the gene expression can be modified by environmental factors such as smoking and nutritional factors.
EPIGENETIC LINK FOR PERIODONTITIS AS A RISK FOR SYSTEMIC DISEASES
Periodontitis has been proposed to be a co-morbidity factor in several systemic conditions. Few studies have evaluated the epigenetic changes in systemically compromised individuals with periodontitis, and the significance of these changes remains unknown.
The expression profile of micro-RNA in obese individuals with and without periodontitis was studied by Perri et al.[49] The micro RNA's miR-18a, miR-30e were up-regulated among obese individuals with healthy periodontium and increased expression of miR-30e, miR-106b was observed in nonobese individuals with periodontal disease. However, in obese individuals with periodontal disease, several different mi-RNAs (mi R15a, mi R 18a, miR-22, mi R30 d, e, mi R-103, mi R 106b, miR-130 a, mi R-142–3p, miR-185, miR-210) were up-regulated. These micro-RNAs are involved in the regulation of genes coding for cytokines, collagen, chemokines, and several important lipid mediators. The significance of variation in these levels needs to be evaluated further in order to get a clearer understanding of the significance of these findings and their import on periodontitis as a risk for systemic diseases.
Recent studies have shown that the fetal environment can cause changes in the epigenome, with long-term consequences for gene regulation and age-related diseases.[50] Bobetsis et al.,[51] showed that periodontal infection can lead to placental-fetal exposure and when coupled with a fetal inflammatory response, leads to preterm delivery. Bobetsis et al.,[52] established that infection with Campylobacter rectus induced hypermethylation in the promoter region of the Insulin-like growth factor (Igf2) gene in a murine placenta. The authors concluded that epigenetic alterations could be induced in the placenta by oral bacteria which could lead to alteration of the placental phenotype that influences the development of the fetus.
Epigenetic modifications represent a fertile field of research for establishing a link between focal infections such as periodontitis and systemic conditions.
EPIGENETIC MARKS IN PROGENITOR CELL LINEAGE DIFFERENTIATION AND IMPLICATION IN PERIODONTAL REGENERATION
A study by Dangaria et al.,[53] demonstrated that dental follicle progenitor population were distinguished from periodontal ligament cells, alveolar bone osteoblasts, cementoblast population by a switch from the euchromatin marker H3K4 me3 to the transcriptional repressor H3 K9 me3. The switch of epigenetic marks following dental follicle progenitor differentiation into different lineages was suggested to be an important event that was mediated by methyltransferases.
A recent study[54] reported on the potential use of histone deacetylase inhibitor (sodium butyrate) in inducing the differentiation of periodontal ligament fibroblasts into osteoblasts. The authors observed an increased expression of osteoblast phenotype characteristics and inhibition of LPS induced reactive oxygen species, pro-inflammatory cytokine production in the periodontal ligament fibroblasts following exposure to the histone deacetylase inhibitor. The authors concluded that sodium butyrate was a potential therapeutic agent for periodontal regeneration.
The findings of the above two studies assume significance in that the switching of the epigenetic marks by means of growth factors/use of enzyme inhibitors can be applied in tissue engineering for achieving more predictable outcomes for periodontal regeneration.
EPIGENETIC THERAPY AS A PART OF PERIODONTAL DISEASE MANAGEMENT
Histone deacetylase inhibitors have been used for management of chronic inflammatory diseases involving bone. The deacetylase inhibitors help in promoting osteoblast maturation[55] and suppressing resorption of bone by osteoclasts.[56] Cantley et al.,[57] examined the effectiveness of using 1179.4b (Class I, II HDAC inhibitor) and MS-275 (Class I HDAC inhibitor) on bone volume changes in P. gingivalis induced experimental periodontitis in mice. The authors concluded that 1179.4b reduced the P. gingivalis induced bone loss, whereas MS-275 had no significant effect. The above studies represent preliminary work for epigenetics as part of host modulation therapy for the management of periodontitis. The potential for further research is wide open, and epigenetic therapy may represent a new avenue for inflammatory disease management.
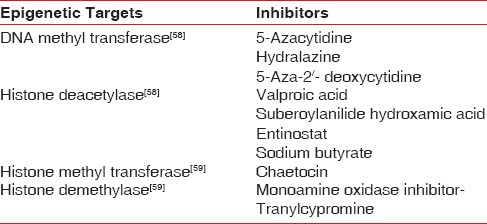
The various epigenetic marks that have been targeted with inhibitors to modulate epigenetic influences are summarized in Table 2.
Table 2.
Epigenetic marks for therapeutic intervention and their inhibitors
CONCLUSION
Periodontitis is a chronic inflammatory disease, and its persistent nature could also exert a significant systemic impact on health, by serving a risk factor for atherosclerosis, chronic obstructive pulmonary disease, diabetes, adverse pregnancy outcomes, and rheumatoid arthritis. The traditional method for periodontal disease management involves techniques targeting the bacteria/pathogens. These have limitations, such as recurrence of the disease and bacterial resistance. Thus, developing new therapeutic strategies for chronic inflammation based on regulation of the host innate immune response is highly desirable. Knowledge about alterations in histone modifications, DNA methylation, and microRNA regulation will provide a better understanding of the molecular basis for various chronic inflammatory diseases. Progress in studies of epigenetic alterations during the inflammatory response opens opportunities for the development of effective medications for specific targets. Among the drugs currently proposed for epigenetic therapy are histone deacetylase inhibitors and demethylating agents, which target chromatin in rapidly dividing tumor cells and restore normal cell functions.[60] The integration of the latest technological achievements in whole-genome microarray expression profiling and chromatin immunoprecipitation-based sequencing (ChIP-seq) methods will be instrumental in the development of epigenetic drugs with greater specificity.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462–82. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 2.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 3.Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res. 2011;90:9–17. doi: 10.1177/0022034510378683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 5.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 6.Patai AV, Molnár B, Kalmár A, Schöller A, Tóth K, Tulassay Z. Role of DNA methylation in colorectal carcinogenesis. Dig Dis. 2012;30:310–5. doi: 10.1159/000337004. [DOI] [PubMed] [Google Scholar]
- 7.Esteller M, Almouzni G. How epigenetics integrates nuclear functions. Workshop on epigenetics and chromatin: Transcriptional regulation and beyond. EMBO Rep. 2005;6:624–8. doi: 10.1038/sj.embor.7400456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campos EI, Reinberg D. Histones: Annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 9.Wysocka J. Identifying novel proteins recognizing histone modifications using peptide pull-down assay. Methods. 2006;40:339–43. doi: 10.1016/j.ymeth.2006.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonkoly E, Pivarcsi A. MicroRNAs in inflammation. Int Rev Immunol. 2009;28:535–61. doi: 10.3109/08830180903208303. [DOI] [PubMed] [Google Scholar]
- 11.Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–9. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 12.Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008;58:2686–93. doi: 10.1002/art.23758. [DOI] [PubMed] [Google Scholar]
- 13.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–44. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 14.Michalowicz BS, Diehl SR, Gunsolley JC, Sparks BS, Brooks CN, Koertge TE, et al. Evidence of a substantial genetic basis for risk of adult periodontitis. J Periodontol. 2000;71:1699–707. doi: 10.1902/jop.2000.71.11.1699. [DOI] [PubMed] [Google Scholar]
- 15.Offenbacher S, Barros SP, Beck JD. Rethinking periodontal inflammation. J Periodontol. 2008;79:1577–84. doi: 10.1902/jop.2008.080220. [DOI] [PubMed] [Google Scholar]
- 16.Iacopino AM. Epigenetics: New explanations for old problems? J Can Dent Assoc. 2010;76:a76. [PubMed] [Google Scholar]
- 17.Yin L, Chung WO. Epigenetic regulation of human ß-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 2011;4:409–19. doi: 10.1038/mi.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung WO, Dale BA. Differential utilization of nuclear factor-kappaB signaling pathways for gingival epithelial cell responses to oral commensal and pathogenic bacteria. Oral Microbiol Immunol. 2008;23:119–26. doi: 10.1111/j.1399-302X.2007.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin L, Sanson B, An J, Hacker BM, Silverman GA, Dale BA, et al. Differential effects of peripopathogens on host protease inhibitors SLP1, elafin, SCCA1, SCCA 2. J Oral Microbiol. 2010;2:1–12. doi: 10.3402/jom.v2i0.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung WO, An JY, Yin L, Hacker BM, Rohani MG, Dommisch H, et al. Interplay of protease-activated receptors and NOD pattern recognition receptors in epithelial innate immune responses to bacteria. Immunol Lett. 2010;131:113–9. doi: 10.1016/j.imlet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–51. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung WO, Hansen SR, Rao D, Dale BA. Protease-activated receptor signaling increases epithelial antimicrobial peptide expression. J Immunol. 2004;173:5165–70. doi: 10.4049/jimmunol.173.8.5165. [DOI] [PubMed] [Google Scholar]
- 23.Huang GT, Zhang HB, Dang HN, Haake SK. Differential regulation of cytokine genes in gingival epithelial cells challenged by Fusobacterium nucleatum and Porphyromonas gingivalis. Microb Pathog. 2004;37:303–12. doi: 10.1016/j.micpath.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Oliveira NF, Damm GR, Andia DC, Salmon C, Nociti FH, Jr, Line SR, et al. DNA methylation status of the IL8 gene promoter in oral cells of smokers and non-smokers with chronic periodontitis. J Clin Periodontol. 2009;36:719–25. doi: 10.1111/j.1600-051X.2009.01446.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, Barros SP, Niculescu MD, Moretti AJ, Preisser JS, Offenbacher S. Alteration of PTGS2 promoter methylation in chronic periodontitis. J Dent Res. 2010;89:133–7. doi: 10.1177/0022034509356512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andia DC, de Oliveira NF, Casarin RC, Casati MZ, Line SR, de Souza AP. DNA methylation status of the IL8 gene promoter in aggressive periodontitis. J Periodontol. 2010;81:1336–41. doi: 10.1902/jop.2010.100082. [DOI] [PubMed] [Google Scholar]
- 27.Zhang S, Crivello A, Offenbacher S, Moretti A, Paquette DW, Barros SP. Interferon-gamma promoter hypomethylation and increased expression in chronic periodontitis. J Clin Periodontol. 2010;37:953–61. doi: 10.1111/j.1600-051X.2010.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loo WT, Jin L, Cheung MN, Wang M, Chow LW. Epigenetic change in E-cadherin and COX-2 to predict chronic periodontitis. J Transl Med. 2010;8:110. doi: 10.1186/1479-5876-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nahid MA, Rivera M, Lucas A, Chan EK, Kesavalu L. Polymicrobial infection with periodontal pathogens specifically enhances microRNA miR-146a in ApoE-/- mice during experimental periodontal disease. Infect Immun. 2011;79:1597–605. doi: 10.1128/IAI.01062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A. 2009;106:2735–40. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YH, Na HS, Jeong SY, Jeong SH, Park HR, Chung J. Comparison of inflammatory microRNA expression in healthy and periodontitis tissues. Biocell. 2011;35:43–9. [PubMed] [Google Scholar]
- 32.Gemmell E, Yamazaki K, Seymour GJ. Destructive periodontitis lesions are determined by the nature of the lymphocytic response. Crit Rev Oral Biol Med. 2002;13:17–34. doi: 10.1177/154411130201300104. [DOI] [PubMed] [Google Scholar]
- 33.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 34.Cuddapah S, Barski A, Zhao K. Epigenomics of T cell activation, differentiation, and memory. Curr Opin Immunol. 2010;22:341–7. doi: 10.1016/j.coi.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–73. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandtzaeg P, Kraus FW. Autoimmunity and periodontal disease. Odontol Tidskr. 1965;73:281–393. [PubMed] [Google Scholar]
- 37.Hirsch HZ, Tarkowski A, Miller EJ, Gay S, Koopman WJ, Mestecky J. Autoimmunity to collagen in adult periodontal disease. J Oral Pathol. 1988;17:456–9. doi: 10.1111/j.1600-0714.1988.tb01315.x. [DOI] [PubMed] [Google Scholar]
- 38.Richardson BC, Liebling MR, Hudson JL. CD4+ cells treated with DNA methylation inhibitors induce autologous B cell differentiation. Clin Immunol Immunopathol. 1990;55:368–81. doi: 10.1016/0090-1229(90)90125-a. [DOI] [PubMed] [Google Scholar]
- 39.Mangan DF, Lopatin DE. Polyclonal activation of human peripheral blood B lymphocytes by Fusobacterium nucleatum. Infect Immun. 1983;40:1104–11. doi: 10.1128/iai.40.3.1104-1111.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mangan DF, Won T, Lopatin DE. Nonspecific induction of immunoglobulin M antibodies to periodontal disease-associated microorganisms after polyclonal human B-lymphocyte activation by Fusobacterium nucleatum. Infect Immun. 1983;41:1038–45. doi: 10.1128/iai.41.3.1038-1045.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson C, Powrie F. Regulatory T cells. Curr Opin Pharmacol. 2004;4:408–14. doi: 10.1016/j.coph.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Nakajima T, Ueki-Maruyama K, Oda T, Ohsawa Y, Ito H, Seymour GJ, et al. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. 2005;84:639–43. doi: 10.1177/154405910508400711. [DOI] [PubMed] [Google Scholar]
- 43.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–7. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Velden U, Kuzmanova D, Chapple IL. Micronutritional approaches to periodontal therapy. J Clin Periodontol. 2011;38(Suppl 11):142–58. doi: 10.1111/j.1600-051X.2010.01663.x. [DOI] [PubMed] [Google Scholar]
- 46.Milward MR, Chapple IL. Dental health the role of diet in periodontal disease. Dent Health. 2013;52:18–21. [Google Scholar]
- 47.Wilson AG. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol. 2008;79:1514–9. doi: 10.1902/jop.2008.080172. [DOI] [PubMed] [Google Scholar]
- 48.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 49.Perri R, Nares S, Zhang S, Barros SP, Offenbacher S. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012;91:33–8. doi: 10.1177/0022034511425045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson RF, Einstein FH. Epigenetic basis for fetal origins of age-related disease. J Womens Health (Larchmt) 2010;19:581–7. doi: 10.1089/jwh.2009.1408. [DOI] [PubMed] [Google Scholar]
- 51.Bobetsis YA, Barros SP, Offenbacher S. Exploring the relationship between periodontal disease and pregnancy complications. J Am Dent Assoc. 2006;137(Suppl):7S–13. doi: 10.14219/jada.archive.2006.0403. [DOI] [PubMed] [Google Scholar]
- 52.Bobetsis YA, Barros SP, Lin DM, Weidman JR, Dolinoy DC, Jirtle RL, et al. Bacterial infection promotes DNA hypermethylation. J Dent Res. 2007;86:169–74. doi: 10.1177/154405910708600212. [DOI] [PubMed] [Google Scholar]
- 53.Dangaria SJ, Ito Y, Luan X, Diekwisch TG. Differentiation of neural-crest-derived intermediate pluripotent progenitors into committed periodontal populations involves unique molecular signature changes, cohort shifts, and epigenetic modifications. Stem Cells Dev. 2011;20:39–52. doi: 10.1089/scd.2010.0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim TI, Han JE, Jung HM, Oh JH, Woo KM. Analysis of histone deacetylase inhibitor-induced responses in human periodontal ligament fibroblasts. Biotechnol Lett. 2013;35:129–33. doi: 10.1007/s10529-012-0992-6. [DOI] [PubMed] [Google Scholar]
- 55.Schroeder TM, Westendorf JJ. Histone deacetylase inhibitors promote osteoblast maturation. J Bone Miner Res. 2005;20:2254–63. doi: 10.1359/JBMR.050813. [DOI] [PubMed] [Google Scholar]
- 56.Cantley MD, Bartold PM, Marino V, Fairlie DP, Le GT, Lucke AJ, et al. Histone deacetylase inhibitors and periodontal bone loss. J Periodontal Res. 2011;46:697–703. doi: 10.1111/j.1600-0765.2011.01392.x. [DOI] [PubMed] [Google Scholar]
- 57.Cantley MD, Bartold PM, Fairlie DP, Rainsford KD, Haynes DR. Histone deacetylase inhibitors as suppressors of bone destruction in inflammatory diseases. J Pharm Pharmacol. 2012;64:763–74. doi: 10.1111/j.2042-7158.2011.01421.x. [DOI] [PubMed] [Google Scholar]
- 58.Ivanov M, Barragan I, Ingelman-Sundberg M. Epigenetic mechanisms of importance for drug treatment. Trends Pharmacol Sci. 2014;35:384–96. doi: 10.1016/j.tips.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009;49:243–63. doi: 10.1146/annurev-pharmtox-061008-103102. [DOI] [PubMed] [Google Scholar]
- 60.Karberg S. Switching on epigenetic therapy. Cell. 2009;139:1029–31. doi: 10.1016/j.cell.2009.11.038. [DOI] [PubMed] [Google Scholar]