

 This work is licensed under a
This work is licensed under a Summary
A de novo heterozygous inactivating mutation of calcium-sensing receptor (CASR) gene typically causes neonatal hyperparathyroidism (NHPT) with moderate hypercalcemia and hyperparathyroid bone disease. We present a case of asymptomatic hypocalciuric hypercalcemia with a de novo heterozygous mutation in CASR, S591C, which is primarily reported to be responsible for NHPT. A 54-year-old female was referred for investigation of asymptomatic hypercalcemia that was initially found in the 1980s but without a history of bone disease during the perinatal period. She had moderate hypercalcemia (12.4 mg/dl) and relative hypocalciuria (fractional extraction of calcium 1.07%) but normal intact parathyroid hormone and serum 1,25-dihydroxyvitamin D3. Pedigree analysis revealed that she carried a de novo heterozygous mutation of S591C, which she transmitted to an affected child with moderate hypercalcemia but not to other children, who had normal serum calcium levels. A de novo heterozygous CASR mutation that is responsible for NHPT may also present in individuals with asymptomatic hypocalciuric hypercalcemia. Caution is required when predicting course and outcome in a pedigree with CASR mutation, as well as incidental hypercalcemia, because of its variable phenotypes.
Learning points
The phenotype and severity of CASR mutations are thought to be dependent on genotypes.
We report an asymptomatic case of the de novo heterozygous S591C mutation in CASR, which has previously been reported as a responsible mutation of NHPT with bone diseases.
Variable phenotypes of CASR raise a cautionary note about predicting outcome by genotyping in a pedigree with CASR mutation.
Background
Calcium-sensing receptor (CASR), which is mainly expressed in parathyroid chief cells and renal tubular cells, regulates phospho-calcic homeostasis by regulating the secretion of parathyroid hormone (PTH) as well as renal calcium reabsorption. Germ-line inactivating mutations of CASR cause persistent hypercalcemia with inappropriately normal or elevated PTH along with low urinary calcium extraction, which results in three phenotypes that are usually dependent on an apparent gene dosage effect. Homozygous or compound heterozygous mutations typically cause neonatal severe hyperparathyroidism (NSHPT), an autosomal-recessive disorder with life-threatening hypercalcemia and multiple fractures. Heterozygous mutations of CASR cause familial hypocalciuric hypercalcemia (FHH), an autosomal-dominant asymptomatic disorder with mild or moderate hypercalcemia. Infants that are heterozygous for CASR mutations may present neonatal hyperparathyroidism (NHPT) with moderate hypercalcemia and a marked elevation of PTH levels, which leads to apparent hyperparathyroid bone disease (1) (2) (3). They usually have a de novo heterozygous mutation or a paternally inherited mutation.
The phenotypes of mutations in CASR, however, do not always correspond to genotypes. Some homozygous mutations in CASR unexpectedly do not confer any severe clinical manifestations of NSHPT; instead, they just confer the mild phenotype of FHH (4) (5). A de novo heterozygous mutation may cause asymptomatic FHH but not NHPT (4). We herein present a case of asymptomatic hypocalciuric hypercalcemia with a de novo heterozygous mutation, S591C, which has, however, been previously reported to be responsible for NHPT with multiple fractures and bone erosion (6). The mutation was transmitted to the next generation and led to asymptomatic FHH in the pedigree we identified.
Case presentation
The proband (II-1, Fig. 1), a 54-year-old female referred for investigation of hypercalcemia, was the first child of non-consanguineous parents. Her medical record indicated no history of bone disease during the peripartum period. She had experienced no clinical symptoms attributable to hypercalcemia, such as frequent urination, muscle weakness, or delirium, although her hypercalcemia had been pointed out when she was 24 years old.
Figure 1.
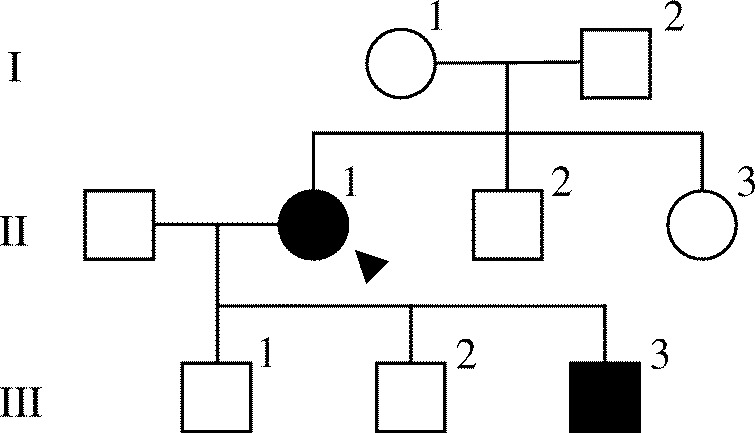
Pedigree of the studied family. Squares represent male family members, and circles represent female family members. Clinical status is indicated by open symbol (unaffected) and solid symbol (affected). The proband is indicated by an arrowhead.
Investigation
Laboratory evaluation (Table 1) revealed hypercalcemia (serum calcium 12.4 mg/dl, when corrected for a mild hypoalbuminemia; reference values 8.8–10.8), mild hypophosphatemia (serum phosphate 2.6 mg/dl; reference values 2.7–4.4), normal intact PTH levels (34 pg/ml; reference values 10–65), and relative hypocalciuria (fractional extraction of calcium (FECa) 1.07%). Serum concentration of 1,25-dihydroxyvitamin D3 (44.7 pg/ml; reference values 20–60) was not elevated. A 99mTc-methoxyisobutylisonitrile scintigraphy, as well as an ultrasound examination, revealed no parathyroid tumor (data not shown). Evaluation of bone mineral density of the lumbar spine showed no evidence of low bone mass or bone loss (T-score −0.9, Z-score 0.1). The combination of hypercalcemia, inappropriately unsuppressed PTH, and low FECa suggested a diagnosis of FHH, although her parents (I-1 and I-2, Fig. 1) and two siblings (II-2 and II-3, Fig. 1) had normal serum calcium levels (Table 2).
Table 1.
Biochemical characteristics of the proband
| Biochemical tests | Result | Reference range |
|---|---|---|
| Serum albumin (g/dl) | 3.9 | 3.9–4.9 |
| Serum calcium (mg/dl) | 12.2 | 8.8–10.8 |
| Serum phosphate (mg/dl) | 2.6 | 2.7–4.4 |
| Serum magnesium (mg/dl) | 2.5 | 1.9–2.5 |
| Serum creatinine (mg/dl) | 0.58 | 0.4–0.8 |
| Urinary calcium (mg/gCre) | 225 | |
| FECa (%) | 1.07 | |
| % TRP (%) | 81.72 | |
| Intact PTH (pg/ml) | 34 | 10–65 |
| 1,25-(OH)2vitamin D (pg/ml) | 44.7 | 20–60 |
Table 2.
Biochemical characteristics of proband's family
| Pedigree | Serum albumin | Serum calcium | Serum phosphate | Serum creatinine | Intact PTH |
|---|---|---|---|---|---|
| I-1 | 4.6 | 9.7 | NA | 0.88 | NA |
| I-2 | 4.2 | 9.2 | 2.8 | 0.85 | NA |
| II-1 | 3.9 | 12.2 | 2.6 | 0.58 | 34 |
| II-2 | 4.7 | 9.8 | 3.4 | 0.9 | 33 |
| II-3 | 4.3 | 9.2 | 2.7 | 0.62 | 71 |
| III-1 | 4.8 | 10.0 | 3.3 | 0.79 | 22 |
| III-2 | 4.5 | 9.3 | 4.4 | 0.88 | 29 |
| III-3 | 4.6 | 12.0 | 2.8 | 0.77 | 38 |
| Reference range | 3.9–4.9 (g/dl) | 8.8–10.8 (mg/dl) | 2.7–4.4 (mg/dl) | M: 0.5–1.1 F: 0.4–0.8 (mg/dl) |
10–65 (pg/ml) |
NA, not available.
To identify whether the hypocalciuric hypercalcemia in the proband was inherited, laboratory evaluations of all of her children (III-1, III-2, and III-3, Fig. 1) were performed. The evaluations revealed hypercalcemia (12.0 mg/dl) with normal PTH (38 pg/ml) and low FECa (0.63%) in one of them (III-3, Table 2), which was consistent with the proband (II-1).
Genetic analysis
To confirm whether the kindred had a unique mutation, germ-line sequences of CASR were analyzed in the proband (II-1) and two of the three children (III-1 and III-3). All of the coding exons of CASR were amplified by PCR with genomic DNA from peripheral blood leukocytes. The amplicons were directly sequenced with BigDye Terminator Cycle Sequencing kits (v1.1) and an ABI Prism 3130XL genetic analyzer (Applied Biosystems). The proband was heterozygous for a cytosine to guanine transition at the nucleotide position 1772 in exon 7 (c.1772 C>G), which led to the replacement of serine (TCC) by cysteine (TGC) at codon 591 (S591C) (Fig. 2). Sequence analysis in the affected child (III-3) revealed the heterozygous mutation of c.1772 C>G, whereas the unaffected child (III-1) did not carry the mutation (Fig. 2). These results indicated that the FHH in the kindred was most likely associated with the S591C in CASR. Because the parents and all of the siblings of the proband had normal serum calcium levels, the mutation was likely de novo in the proband.
Figure 2.
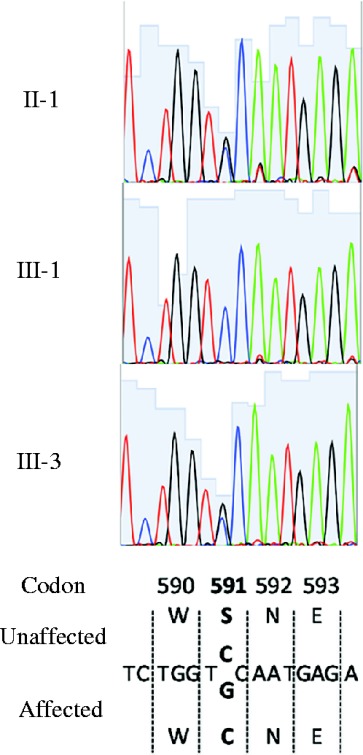
The sequencing chromatograms for germ-line sequence analysis of amplicon of exon 7 of CASR. Unaffected and affected sequences of part of exon 7.
Treatment, outcome, and follow-up
The proband (II-1) and the affected child (III-3) have been followed up every 6 months, so far without apparent symptoms or related abnormal laboratory findings. Therefore, no treatment for hypercalcemia has been administered.
Discussion
We identified a heterozygous missense mutation, S591C, in CASR in two affected family members with asymptomatic hypocalciuric hypercalcemia. A case of NHPT carrying the heterozygous S591C has previously been described with multiple fracture and bone erosion (6). The affected family members in the present study, however, were diagnosed with benign FHH, because they did not have any severe clinical manifestations.
Increasing evidence has demonstrated clinical variability of heterozygous CASR mutants, although the mutants generally cause asymptomatic FHH. A search of the CASR database (www.casrdb.mcgll.ca) revealed that six mutations of CASR have thus far been reported in NSHPT with de novo heterozygous mutations, which is referred as NHPT (1) (2) (3). Among the six mutations, only one mutation, R220W, has been described as being responsible for both NHPT (7) and FHH (8), which is similar to the S591C we identified. In the pedigree of FHH with R220W, however, no de novo cases have been described (8). We report for the first time here that the de novo CASR mutation responsible for NHPT, S591C, can also cause just asymptomatic hypocalciuric hypercalcemia.
Another de novo heterozygous mutation, G459R, in CASR has been reported to cause asymptomatic FHH (4). In the pedigree, however, apparent hypercalcemia has been observed only in homozygous carriers because of low potency of the mutation, as verified by in vitro experiments (4). In the pedigree we identified, the heterozygous S591C caused moderate hypercalcemia and/or the development of NHPT.
It remains unclear what factor determines the differential phenotypes of the de novo heterozygous mutation of S591C, NHPT with hyperparathyroid bone disease or asymptomatic FHH without hyperparathyroidism. In NHPT, the affected CASR in the fetus senses maternal normal Ca2 + levels as ‘low’ and thus stimulates the fetal parathyroid to release PTH. Maternal vitamin D deficiency and/or low calcium intake could be other stimuli of the fetal parathyroid glands. Variable maternal environment and/or exposure in utero, as well as genetic background, including polymorphisms of other FHH-related genes, G protein subunit α11 (GNA11), and adaptor-related protein complex2 σ subunit (AP2S1), could bring about the differential phenotypes. It would be of interest if the paternal transmission of S591C caused NHPT in the pedigree we identified.
In the CASR mutant described in the present study, the serine 591 was replaced with cysteine containing a free thiol group (-SH), which easily forms a disulphide bridge with another cysteine. The residue 591 is located in the extracellular cysteine-rich domain and transmits the signal from the Ca2 +-binding domain to intracellular signaling pathways. Studies of site-directed mutagenesis have indicated the functional importance of the seven cysteine residues in the cysteine-rich domain (9). Those native cysteines likely contribute to stabilizing the conformational structure of CASR by forming intramolecular disulphide bridges (10). The replacement by cysteine at residue 591 might affect the formation of disulphide bridges and the three-dimensional structure of the extracellular domain of CASR.
In conclusion, the present report is the first case of FHH with the de novo heterozygous S591C mutation in CASR, which was previously reported as being a responsible mutation of NHPT. Variable phenotypes observed with S591C suggest the need for caution in the prediction of course and outcome in a pedigree with CASR mutation. The accumulation of data not just from genotyping but also from the evaluation of the maternal environment of carrier fetuses, as well as in vitro studies of molecular functionality, would define the factors that contribute differential phenotypes in heterozygous CASR mutations.
Patient consent
Written informed consent was obtained from the patient for publication of this report.
Author contribution statement
K Taki, T Kogai, and A Hishinuma analyzed the data and drafted the manuscript. K Taki was the endocrinologist responsible for the patients. T Kogai, J Sakumoto, and T Namatame performed the gene analysis.
Acknowledgements
We thank Hiroko Suda for assistance with the sequence data analysis.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1. Pearce S & Steinmann B. 1999. Casting new light on the clinical spectrum of neonatal severe hyperparathyroidism. Clinical Endocrinology 50 691–693. 10.1046/j.1365-2265.1999.00788.x [DOI] [PubMed] [Google Scholar]
- 2. Brown EM. 2005. Editorial: mutant extracellular calcium-sensing receptors and severity of disease. Journal of Clinical Endocrinology and Metabolism 90 1246–1248. 10.1210/jc.2004-2483 [DOI] [PubMed] [Google Scholar]
- 3. Reh CM, Hendy GN, Cole DE & Jeandron DD. 2011. Neonatal hyperparathyroidism with a heterozygous calcium-sensing receptor (CASR) R185Q mutation: clinical benefit from cinacalcet. Journal of Clinical Endocrinology and Metabolism 96 E707–E712. 10.1210/jc.2010-1306 [DOI] [PubMed] [Google Scholar]
- 4. Lietman SA, Tenenbaum-Rakover Y, Jap TS, Yi-Chi W, De-Ming Y, Ding C, Kussiny N & Levine MA. 2009. A novel loss-of-function mutation, Gln459Arg, of the calcium-sensing receptor gene associated with apparent autosomal recessive inheritance of familial hypocalciuric hypercalcemia. Journal of Clinical Endocrinology and Metabolism 94 4372–4379. 10.1210/jc.2008-2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szczawinska D, Schnabel D, Letz S & Schöfl C. 2014. A homozygous CaSR mutation causing a FHH phenotype completely masked by vitamin D deficiency presenting as rickets. Journal of Clinical Endocrinology and Metabolism 99 E1146–E1153. 10.1210/jc.2013-3593 [DOI] [PubMed] [Google Scholar]
- 6. Nyweide K, Feldman KW, Gunther DF, Done S, Lewis C & Van Eenwyk C. 2006. Hypocalciuric hypercalcemia presenting as neonatal rib fractures: a newly described mutation of the calcium-sensing receptor gene. Pediatric Emergency Care 22 722–724. 10.1097/01.pec.0000238747.19477.d3 [DOI] [PubMed] [Google Scholar]
- 7. Fox L, Sadowsky J, Pringle KP, Kidd A, Murdoch J, Cole DE & Wiltshire E. 2007. Neonatal hyperparathyroidism and pamidronate therapy in an extremely premature infant. Pediatrics 120 e1350–e1354. 10.1542/peds.2006-3209 [DOI] [PubMed] [Google Scholar]
- 8. D'Souza-Li L, Yang B, Canaff L, Bai M, Hanley DA, Bastepe M, Salisbury SR, Brown EM, Cole DE & Hendy GN. 2002. Identification and functional characterization of novel calcium-sensing receptor mutations in familial hypocalciuric hypercalcemia and autosomal dominant hypocalcemia. Journal of Clinical Endocrinology and Metabolism 87 1309–1318. 10.1210/jc.87.3.1309 [DOI] [PubMed] [Google Scholar]
- 9. Fan GF, Ray K, Zhao XM, Goldsmith PK & Spiegel AM. 1998. Mutational analysis of the cysteines in the extracellular domain of the human Ca2+ receptor: effects on cell surface expression, dimerization and signal transduction. FEBS Letters 436 353–356. 10.1016/S0014-5793(98)01165-X [DOI] [PubMed] [Google Scholar]
- 10. Hu J & Spiegel AM. 2007. Structure and function of the human calcium-sensing receptor: insights from natural and engineered mutations and allosteric modulators. Journal of Cellular and Molecular Medicine 11 908–922. 10.1111/j.1582-4934.2007.00096.x [DOI] [PMC free article] [PubMed] [Google Scholar]