

Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder characterized in man by parathyroid, pancreatic, pituitary and adrenal tumours. The MEN1 gene encodes a 610-amino acid protein (menin) which is a tumour suppressor. To investigate the in vivo role of menin, we developed a mouse model, by deleting Men1 exons 1 and 2 and investigated this for MEN1-associated tumours and serum abnormalities. Men1+/− mice were viable and fertile, and 220 Men1+/− and 94 Men1+/+ mice were studied between the ages of 3 and 21 months. Survival in Men1+/− mice was significantly lower than in Men1+/+ mice (<68% vs >85%, P<0.01). Men1+/− mice developed, by 9 months of age, parathyroid hyperplasia, pancreatic tumours which were mostly insulinomas, by 12 months of age, pituitary tumours which were mostly prolactinomas, and by 15 months parathyroid adenomas and adrenal cortical tumours. Loss of heterozygosity and menin expression was demonstrated in the tumours, consistent with a tumour suppressor role for the Men1 gene. Men1+/− mice with parathyroid neoplasms were hypercalcaemic and hypophosphataemic, with inappropriately normal serum parathyroid hormone concentrations. Pancreatic and pituitary tumours expressed chromogranin A (CgA), somatostatin receptor type 2 and vascular endothelial growth factor-A. Serum CgA concentrations in Men1+/− mice were not elevated. Adrenocortical tumours, which immunostained for 3-β-hydroxysteroid dehydrogenase, developed in seven Men1+/− mice, but resulted in hypercorticosteronaemia in one out of the four mice that were investigated. Thus, these Men1+/− mice are representative of MEN1 in man, and will help in investigating molecular mechanisms and treatments for endocrine tumours.
Introduction
Multiple endocrine neoplasia type 1 (MEN1), in man, is characterised by the combined occurrence of tumours of parathyroids, pancreatic islets and anterior pituitary (Marx 2005, Thakker 2006). Some patients may also develop adrenal cortical tumours, carcinoid tumours, facial angiofibromas, collagenomas and lipomas. MEN1 is inherited as an autosomal dominant disorder with a high degree of penetrance, such that >95% of patients develop clinical manifestations of the disorder by the fifth decade (Calender et al. 1995, Trump et al. 1996, Marx et al. 1998). Parathyroid tumours, which lead to hypercalcaemia, are the most common feature of MEN1 and occur in ∼95% of patients (Calender et al. 1995, Trump et al. 1996, Marx et al. 1998). Pancreatic islet cell tumours, which consist of gastrinomas, insulinomas, pancreatic polypeptidomas, glucagonomas and vasoactive intestinal polypeptidomas occur in ∼40% of patients; anterior pituitary tumours, which consist of prolactinomas, somatotrophinomas, corticotrophinomas or non-functioning adenomas, occur in ∼30% of patients; and adrenal cortical tumours, which are usually non-secreting but may occasionally be associated with hypercortisolaemia or hyperaldosteronism, may occur in 35% of patients (Calender et al. 1995, Trump et al. 1996, Marx et al. 1998, Vierimaa et al. 2007).
The MEN1 gene, which is a tumour-suppressor gene located in human chromosome 11q13, consists of 10 exons (Fig. 1) that span approximately >9 kb of genomic DNA and encodes a 610-amino acid protein referred to as menin (Chandrasekharappa et al. 1997, The European Consortium on MEN1 1997). The main transcript of the MEN1 gene is a 2.8 kb mRNA. However, at least six alternative transcripts, which utilise alternative exons 1, have been reported with variations in their content of the 5′-untranslated region (Khodaei-O'Brien et al. 2000), and another rare variant that would result in an elongation of the reading frame by 15 bases at the exon 2/intron 2 junction has also been reported (Chandrasekharappa & Teh 2003). In order to study the in vivo role of menin, we embarked on generating a mouse model for MEN1 through homologous recombination (i.e. knockout) of the mouse Men1 gene. During the course of our studies eight mouse models (four conventional and four conditional knockouts) for MEN1 have been reported (Table 1; Crabtree et al. 2001, 2003, Scacheri et al. 2001, Bertolino et al. 2003a, b, Libutti et al. 2003, Biondi et al. 2004, Loffler et al. 2007). Each one of these Men1 mouse models has been generated by disrupting different parts of the Men1 gene and by different strategies, e.g. conventional or conditional knockout (Table 1). The resulting phenotypes in Men1+/− mice have been variable and ranged from embryonic lethality (Scacheri et al. 2001) to full viability and fertility into adult life with the development of a range of different MEN1-associated and non-MEN1-associated tumours, e.g. phaeochromocytomas, thyroid tumours and gonadal tumours (Table 1). The basis for these differences remain to be elucidated; however, it is interesting to note that in two of the conventional Men1+/− models, exon 2 which contains the ATG translational start signal had not been deleted (Crabtree et al. 2001, Bertolino et al. 2003a), and that in the other conventional Men1+/− model exon 1 and the alternative exons 1 remained intact (Table 1). This raises the possibility that there may be potentially other alternative partial transcripts that may be able to compensate for the absence of the full normal Men1 transcript, and the situation may be analogous to that recently reported for the extracellular calcium-sensing receptor which has tissue-specific alternative spliced transcripts (Chang et al. 2008). Although, an important role for the alternative Men1 transcripts is unlikely, as six out of the seven do not normally affect the coding region (Khodaei-O'Brien et al. 2000, Lemos & Thakker 2008), we nevertheless decided to generate a targeting vector that lacked exons 1 and 2, and a >1.5 kb region of the 5′ sequence that contained the untranslated region (UTR), promoter and alternative exons 1, and ∼1 kb of the 5′ region of intron 2 (Fig. 1).
Figure 1.

Targeted disruption of the Men1 gene. (A) Restriction maps of the Men1 gene representing the wild-type (+) and mutant-recombinant (−) alleles. The exons are represented by boxes (filled boxes depict translated regions), and the locations of restriction enzymes sites (BamHI (B), EcoRV (Ev), EcoRI (E), HpaI (H) and SmaI (S)) together with genomic fragments used as 5′, 3′ and neo-cassette (Neo) probes for Southern blot analysis are shown. PCR primers (1R, 1F and NeoF) were designed to facilitate detection of the wild-type (+) and mutant (−) alleles. Exons 1 and 2 together with ∼1.9 kb of the 5′ adjacent region and ∼1.0 kb of intron 2 were disrupted by introduction of the neomycin transferase gene linked to the phosphoglycerate (PGK) promoter (PGK-Neo), in the opposite transcriptional orientation to Men1; the PGK-Neo cassette was flanked by LoxP recognition sequences (open triangles). (B) BamHI Southern blot analysis of genomic DNA extracted from untransfected ES cells (wild-type, Men1+/+) and transfected with Men1 targeting construct (Men1+/−), using the 5′ or Neo probe. Hybridisation with the 5′ probe yielded a 17.4 kb band for the wild-type allele and a 9.7 kb band for the mutant allele, while hybridisation with the Neo probe yielded a 6.8 kb band for the mutant allele, and no signal from wild-type DNA. (C) Genotyping by PCR, utilising primers 1F, 1R and NeoF revealed the presence of the wild-type (735 bp) and mutant (499 bp) alleles from the targeted Men1+/− ES cells, but only the 735 bp from the normal (Men1+/+) ES cells. (D) Southern blot analysis of genomic DNA extracted from mouse tails or pituitary tumours, digested with BamHI and hybridised with the 3′ probe. DNA from Men1+/+ mice showed the expected 17.4 kb band, and the Men1+/− mice showed both the 17.4 and 6.8 kb bands; DNA from the pituitary tumours that developed in Men1+/− mice showed only the mutant 6.8 kb band, consistent with a loss of heterozygosity (LOH) in the tumour. (E) Genotyping by PCR utilising primers 1R, 1F and NeoF revealed the presence of the wild-type (735 bp) and mutant (499 bp) alleles in the normal pituitary of Men1+/− mice, but only the mutant (499 bp) allele from pituitary tumours of these mice, consistent with LOH in the tumours. (F) RT-PCR analysis of normal kidney extracts from Men1+/+ and Men1+/− mice revealed the presence of a Men1 transcript, which was absent in pituitary tumours from Men1+/− mice. Control calmodulin 2 (Calm2) expression is shown. (G) Western blot analysis of normal pituitary extracts from Men1+/+ and Men1+/− mice revealed the expression of menin; however, extracts of pituitary tumours that developed in Men1+/− mice revealed a loss of menin expression. Control immunoblotting for α-tubulin is shown.
Table 1.
Multiple endocrine neoplasia type 1 (MEN1) mouse models and their main phenotypic features
| Model | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Typea | Cv | Cv | Cv | Cn | Cn | Cn | Cn | Cv | Cv |
| Men1 exons deleted | 2–4 | 3–8 | 3 | 3–8 | 3 | 3–8 | 2 | 2 | 1–2 |
| Replacement cassetteb | PGK-Neo | PGK-Neo | Neo-TK | PGK-Neo/Rip-Cre | PGK-Neo/Rip-Cre | PGK-Neo/Pth-Cre | PGK-Neo/Rip-Cre | PGK-Neo | PGK-Neo |
| Strainc | − | Mixed NIH Black Swiss/129 | 129 | Mixed C57/FVB/129 | Mixed C57/129 | Mixed FVB/129 | Mixed C57/129 | Mixed C57/129 | Mixed C57/129 |
| Outcome | Embryonic lethality | Viable | Viable | Viable | Viable | Viable | Viable | Viable | Viable |
| Tumour developmentd | |||||||||
| Parathyroid | − | + | + | − | − | + (↑Ca) | − | + | + (↑Ca) |
| Pancreas | − | + (INS) | + (INS, GLU) | + (INS) | + (INS) | − | + (INS) | + (INS, GLU) | + (INS, GLU, SSTR2, CgA) |
| Pituitary | − | + (PRL) | + (PRL, GH) | − | − | − | + (PRL) | + (PRL, NF) | + (PRL, GH, ACTH, SSTR2, CgA) |
| Adrenal cortex | − | + | + | − | − | − | − | + | +(3β-HSD), ↑Cort |
| Phaeochromocytoma | − | + | − | − | − | − | − | + | + (TH) |
| Gastric NET | − | + | − | − | − | − | − | − | − |
| Thyroid | − | + | + | − | − | − | − | + | + |
| Testicular | − | − | + (LD) | − | − | − | − | + (LD) | + (LD) |
| Extra pancreatic gastrinoma | − | − | + | − | − | − | − | − | − |
| Ovarian | − | − | + (SC) | − | − | − | − | + (SC) | + (SC) |
| Year published | 2001 | 2001 | 2003 | 2003 | 2003 | 2003 | 2004 | 2007 | 2009 |
| References | Scacheri et al. (2001) | Crabtree et al. (2001) | Bertolino et al. (2003a) | Crabtree et al. (2003) | Bertolino et al. (2003b) | Libutti et al. (2003) | Biondi et al. (2004) | Loffler et al. (2007) | (This report) |
Cv, conventional; Cn, conditional.
PKG-Neo, phosphoglycerate kinase-neomycin; Neo-TK, neomycin-thymidine kinase; Rip-Cre, rat insulin promoter-causes recombination; Pth-Cre, parathyroid hormone-causes recombination.
129-129S6/SvEv; C57-C57BL/6.
↑Ca, hypercalcaemia; INS, insulin; GLU, glucagon; SSTR2, somatostatin receptor type 2; CgA, chromogranin A; PRL, prolactin; GH, growth hormone; NF, non-functioning; ACTH, adrenocorticotrophin; 3β-HSD, 3-beta-hydroxy steroid dehydrogenase; TH, tyrosine hydroxylase; NET, neuroendocrine tumours; LD, Leydig cell tumour; SC, sex cord stromal cell tumours; Cort, corticosterone.
Materials and methods
Construction of the gene targeting vector and generation of mouse model for MEN1
Genomic clones containing overlapping portions of the murine Men1 gene were isolated, using PCR and Men1-specific primers, from a 129/Sv BAC library (Incyte Genomics, Inc., St Louis, MO, USA). A 13.5 kb ApaI fragment containing exons 1–2 and the adjacent 5′ region, and a 5.1 kb HpaI fragment containing exons 3–10 and the adjacent 3′ region were selected for generating the targeting construct. The 13.5 kb ApaI fragment was digested with EcoRI to generate a 4.8 kb fragment that contained 1.9 kb of the UTR adjacent to the coding region as well as the promoter and alternative exons 1 (5′ homology arm), and the 5.1 kb HpaI fragment was digested with HpaI and EcoRV to generate a 4.4 kb fragment that contained part of intron 2, exons 3–10 and the 3′ adjacent region (3′ homology arm; Fig. 1A). The targeting vector was constructed by ligating the 5′ arm into the EcoRI site and the 3′ arm into the SmaI site of the 38LoxPNeo vector (Incyte Genomics, Inc). The resulting targeting vector lacked a 3 kb sequence that comprised 1.9 kb of the 5′ sequence, exons 1–2 (including the ATG initiation codon), and ∼1 kb of the 5′ region of intron 2 of the Men1 gene; this 3 kb sequence was replaced by a 2.1 kb selectable phosphoglycerate kinase (PGK)-neomycin resistance (Neo) cassette that was flanked with LoxP sites, in the opposite transcriptional orientation to Men1 (Fig. 1A). The 38LoxPNeo/Men1 vector was linearised and electroporated into murine 129/SvJ embryonic stem (ES) cells. ES cells that had a correct homologous recombination were identified by Southern blot (Fig. 1B) and PCR analysis (Fig. 1C), and expanded for injection into blastocysts. Between five and seven ES cells were used for each injection into 3.5-day-old C57BL/6 blastocysts that were then transferred to pseudo-pregnant female recipients. The resulting male chimaeras were bred with C57BL/6 females to obtain germline transmission of the targeted allele.
Phenotype studies
Mice were kept in accordance with UK Home Office welfare guidelines and project license restrictions. Mice were maintained on a mixed C57BL/6×129S6/SvEv background and fed a standard diet (RM1 expanded diet, Special Diet Services Ltd, Witham, UK). Wild-type (Men1+/+) and heterozygous (Men1+/−) mice were killed every 3 months, beginning at 3 months and continuing until 21 months, and a blood sample was immediately taken by cardiac puncture and the serum stored at −20 °C.
Genotype studies
Genotypes were determined by Southern blot (Fig. 1D) and PCR (Fig. 1E) analysis, as previously described (Thakker et al. 1989, Pannett & Thakker 2001). PCR analysis was performed using primers (1R (5′-CCA AAC TCC ATG TTC CAA TAT GAC AGC-3′); 1F (5′-CAC GAA GTC TGT AAT GAC CCT GTT TCC-3′); and NeoF (5′-CTC TCG TGG GAT CAT TGT TTT TCT C-3′)). Primers 1R and 1F yielded a 735 bp wild-type band and primers 1R and NeoF yielded a 499 bp band (Fig. 1C and E).
Reverse transcriptase-PCR (RT-PCR) analysis
RT-PCR, using total RNA extracted from Men1+/+ and Men1+/− kidneys, as controls, and pituitary tumours obtained from Men1+/− mice (Fig. 1F), was performed using either Men1-specific primers (forward 5′-GCT TCG TGG AGC ATT TCC T-3′; and reverse 5′-TCC AGT CCC TCT TCA GCT TC-3′) or control calmodulin-2 primers (forward 5′-AT GGC TGA CCA ACT GAC TGA A-3′; and reverse 5′-C ATT CTG TAC AAT GTC TTC ACT T-3′), as described (Nesbit et al. 2004).
Western blot analysis
Western blot analysis using total protein from normal pituitaries and pituitary tumours from Men1+/− mice was performed (Fig. 1G), as previously described (Nesbit et al. 2004).
Histology and immunohistochemistry
Tissues were fixed in neutral buffered 4% formalin, except the testes, which were fixed in Bouin's solution. Detailed histology was initially undertaken on the majority of mice from the older cohort after which progressively younger age groups were studied until a very low tumour occurrence had been identified, after which histology studies were performed on smaller numbers of mice from subsequently younger age groups. In mice ≤12 months, survey sections of thyroid/parathyroids were obtained, and in mice ≥15 months, serial sections were performed whenever possible. However, this approach often failed to locate the parathyroids because of their high occurrence at extra-thyroidal sites (Capen et al. 1996). For immunohistochemical analysis, commercially available antibodies were obtained (details available upon request) and used according to the manufacturer's instructions. The slides were counterstained with haematoxylin.
Clinical chemistry
Serum was analysed for calcium, phosphate, creatinine and albumin, as previously described (Hough et al. 2002). Total serum calcium (Ca) was adjusted (ACa) for albumin (Alb) using the formula: ACa=Ca (mmol/l)−((Alb(g/l)−30)×0.017). Serum parathyroid hormone (PTH) concentration was measured using an ELISA-specific for mouse intact PTH (Immutopics, San Clemente, CA, USA). Serum chromogranin A (CgA) was measured using a direct ELISA (Dako, Glostrup, Denmark). Corticosterone was measured using an enzyme immunoassay (IDS Ltd, Boldon, Tyne & Wear, UK) following prior dilution of the mouse sera.
Statistical analysis
Comparisons of tumour occurrence were made by appropriate use of Fisher's exact test and 2×2 contingency tables. Two-tailed P values were calculated and statistical significance set at P<0.05. A two-tailed Student's t-test and χ2 test were appropriately used to analyse the results of serum measurements. Kaplan–Meier analysis was performed and a two-tailed log rank test used.
Results
Generation of the Men1+/− mice
Men1+/− mice were generated by targeted deletion of exons 1 and 2, a 1.9 kb 5′ sequence that encompassed the UTR, promoter and alternative exons 1, and an ∼1 kb sequence of the 5′ region of intron 2; these sequences were replaced by a neomycin resistance gene in ES cells (Fig. 1A–C). PCR analysis using primers 1F, 1R and NeoF showed that ES cells transfected with the targeting construct had the wild-type (735 bp) and mutant (499 bp) bands (Fig. 1C). Similarly, BamHI-digested DNA from the transfected ES cells used for Southern blot analysis that utilised a 5′ probe, which consisted of a 1.4 kb XbaI–EcoRI fragment, yielded a wild-type allele of 17.4 kb and a mutant allele of 9.7 kb (Fig. 1B). The additional use of a 0.6 kb PCR product from the neomycin resistance gene as a probe yielded a hybridisation signal of 6.8 kb, thereby confirming the corrected incorporation of the PGK-Neo cassette in transfected ES cells. ES cell clones with a correct homologous recombination of the construct were injected into blastocysts, which were then transferred to pseudo-pregnant female mice. The resulting male chimaeras were bred with C57BL/6 females to obtain germline transmission, which was confirmed by Southern blot analysis of genomic DNA (Fig. 1D). Men1+/− mice were viable and fertile but Men1−/− mice were embryonically lethal (Lemos et al. 2007).
Development of tumours and pathological analysis
Men1+/− mice and their wild-type (Men1+/+) littermates were studied for the development of tumours at 3 monthly intervals over a 21 month period, commencing at 3 months of age. A total of 314 mice (94 control Men1+/+ (40 males and 54 females) and 220 Men1+/− (90 males and 130 females)) were studied. Premature unexpected death occurred in 85 mice (14 Men1+/+ (5 males and 9 females) and 71 Men1+/− (13 males and 58 females)) and tumour development was therefore studied in the remaining 229 live mice (80 control Men1+/+ (35 males and 45 females) and 149 Men1+/− mice (77 males and 72 females)). The weights of the Men1+/+ and Men1+/− mice, at the time of necropsy were not significantly different (data not shown). In total, 211 tumours were identified from 71 Men1+/− mice (37 males and 34 females). Tumours were found to have loss of heterozygosity (LOH) and loss of menin expression (Figs 1D–G, and 2; see below).
Figure 2.

Proliferative endocrine lesions in Men1+/− mice. (A–D) Parathyroids, (E–H) pancreas islets, (I–M) pituitary, (N–Q) adrenals and (R–U) thyroids. (A) Nodular parathyroid hyperplasia (black arrow), normal parathyroid (white arrow); (B) parathyroid adenoma (black arrow) and normal parathyroid (white arrow); (C) papillary parathyroid adenoma (black arrow) and adjacent parathyroid (white arrow); (D) parathyroid adenomas showing loss of menin expression (indicated by asterisks); (E) pancreatic islet tumour (black arrow) and a normal sized islet (white arrow); (F) insulin staining of the same tumour showing a non-staining peripheral nodule (black arrow); (G) glucagon-positive nodule of the same tumour (black arrow); (H) multinodular neoplasia of pancreas with loss of menin expression (asterisks); (I) pituitary macroadenoma compressing the overlying cerebrum; (J) anterior pituitary macroadenoma containing prolactin; (K) anterior pituitary macroadenoma containing GH; (L) cystic ACTH containing pars intermedia tumour (arrow); (M) nuclear menin expression was lost in the pituitary adenoma cells, but preserved in adjacent non-neoplastic pituitary cells; (N) unilateral adrenal cortex adenoma (black arrow) and contralateral adrenal cortex hyperplasia (white arrow); (O) this adrenocortical tumour immunostained for 3β-HSD (black arrow) and has compressed residual cortex at the margin (white arrow); (P) phaeochromocytoma showing immunostaining for tyrosine hydroxylase (TH) (black arrow) with compressed cortex unstained with TH at the margin (white arrow); (Q) loss of menin expression in adrenal adenoma (asterisk); (R) thyroid follicular adenoma; (S) serial section showing loss of menin expression in the follicular adenoma (asterisk); (T) thyroid C-cell adenoma; (U) serial section showing loss of menin expression in the C-cell adenoma (asterisk). Scale bars =1000 μm (N), 500 μm (E, F, H, I and R– U), 200 μm (A, L, O and P), 100 μm (D, G and M), 50 μm (B, C and Q).
Overall, only 5% of Men1+/− mice ≤9 months of age developed tumours, whereas 84% of the Men1+/− mice aged ≥12 months developed tumours, consistent with an age-related penetrance for the condition (Fig. 3A). The higher occurrence of tumours in Men1+/− mice aged ≥12 months was coincident with the onset of an increased mortality in these mice (Fig. 3B); the survival in the Men1+/− mice was significantly lower at <68% while that in the Men1+/+ mice was >85% (P<0.01, Fig. 3B). The ≥12 months old Men1+/− mice developed tumours of the parathyroid, pancreatic islet, anterior pituitary, adrenal cortex, thyroid, testis and ovary (Fig. 3C). In addition, parathyroid and pancreatic islet cell tumours occurred in >25% of Men1+/− mice, pancreatic islet tumours and anterior pituitary tumours occurred in >15% of Men1+/− mice, anterior pituitary and parathyroid tumours were found in ∼1% of Men1+/− mice; and 6% of Men1+/− mice were found to have parathyroid, pancreatic islet cell and anterior pituitary tumours (Fig. 3D). The occurrence of parathyroid and pancreatic islet cell tumours was not significantly different in male and female Men1+/− mice, but the occurrence of anterior pituitary and adrenal cortical tumours showed significant gender differences (Fig. 3E). Thus, anterior pituitary tumours occurred more frequently in female Men1+/− mice, while adrenal cortical tumours occurred solely in male Men1+/− mice (Fig. 3E). Detailed analysis of the tumours in the Men1+/− mice revealed the following.
Figure 3.
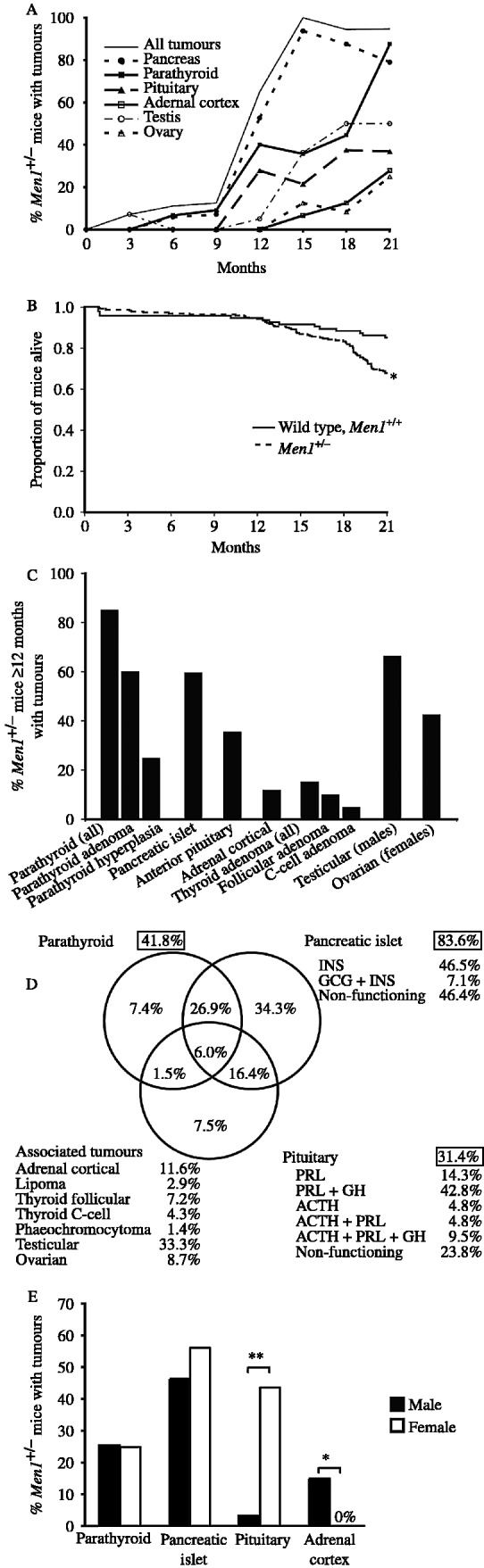
Tumour occurrence in Men1+/− mice with tumours. (A) Percentage of Men1+/− mice at different ages with tumours. The numbers of Men1+/− mice studied at 3, 6, 9, 12, 15, 18 and 21 months of age were 20, 20, 21, 23, 22, 24 and 19, respectively. The results for the testicular and ovarian tumours are shown as a percentage of the combined total of male and female Men1+/− mice. (B) Kaplan–Meier analysis revealed that survival among the Men1+/− mice (149 out of 220, i.e. <68%) was significantly lower than that in Men1+/+ mice (80 out of 94, i.e. >85%) (*P<0.01, log rank test). Survival in the Men1+/+ and Men1+/− mice was similar up to the age of 12 months, after which survival in the Men1+/− mice began to decrease; this was coincident with the higher frequency of tumour development in the Men1+/− mice that were ≥12 months of age (Fig. 3A and B). (C) Tumour development in Men1+/− mice ≥12 months age, revealed that >85% had parathyroid proliferative abnormalities, which consisted of hyperplasia (∼60%) and adenomas (>25%); >60% had pancreatic islet cell tumours, >35% had anterior pituitary tumours, >10% had adrenal cortical tumours, and >15% had thyroid tumours which consisted of follicular adenomas (>10%) and C-cell adenomas (>5%). Two Men1+/− mice had lipomas, and one had a phaeochromocytoma. Testicular tumours developed in >65% of male Men1+/− mice, and ovarian tumours occurred in >40% of Men1+/− female mice. (D) Schematic representation of 211 tumours that were found in 71 Men1+/− mice. The proportions of Men1+/− mice in which parathyroid, pancreatic or pituitary tumours occurred are shown in the respective boxes. For example, 41.8% of the Men1+/− mice had parathyroid hyperplasia or adenomas. The Venn diagram indicates the proportion of Men1+/− mice with each combination of tumours. For example, 26.9% of Men1+/− mice had both a parathyroid and pancreatic tumour, whereas 34.3% of the Men1+/− mice had a pancreatic tumour only. A similar analysis on the Men1+/− mice aged ≥12 months revealed that >85% had parathyroid abnormalities, >60% had pancreatic islet cell tumours, and >35% had pituitary tumours. In addition, >60% had combined parathyroid and pancreatic, >25% had pancreatic and pituitary tumours; and ∼10% had combined parathyroid, pancreatic islet cell and pituitary tumours; whilst none had combined parathyroid and pituitary tumours. The hormones contained within each of these tumours are indicated: INS, insulin; GCG, glucagon; PRL, prolactin; GH, growth hormone and ACTH, adrenocorticotrophin. (E) Percentage of Men1+/− male and female mice with tumours. P values, *<0.001, **<0.0001.
The parathyroid abnormalities consisted of adenomas or focal hyperplasia. The foci of hyperplasia that were characterised by clusters of pale atypical hypertrophic cells that did not compress the surrounding tissue or enlarge the parathyroid (Fig. 2A), were observed in Men1+/− mice aged ≥9 months. These parathyroid abnormalities are equivalent to those previously reported as focal dysplasia (Crabtree et al. 2001, Bertolino et al. 2003a) or hyperplasia (Loffler et al. 2007). The adenomas (Fig. 2B and C) that either had solid or papillary patterns with compression of adjacent normal tissue at the margin, occurred in Men1+/− mice aged ≥15 months. The Men1+/− mice with parathyroid hyperplasia had a significantly lower mean age (mean±s.d.=16.95±4.05 months) than those with adenomas (19.88±2.23 months, P=0.02). Both parathyroid hyperplasia and adenomas in man, cause primary hyperparathyroidism (Eastell et al. 2009) and are found in patients with MEN1 (Calender et al. 1995, Trump et al. 1996). The clinical and serum biochemical features of primary hyperparathyroidism, in man, arising from these two types of parathyroid proliferative abnormalities are indistinguishable, and the data from the Men1+/− mice with these parathyroid abnormalities were therefore pooled for further analysis (see below).
Investigation of the pancreatic islets, stomach and proximal duodenum revealed abnormalities only of the pancreatic islets. These consisted of islet cell hyperplasia and adenomas; there was a continuum in islet size between hyperplasia and adenoma, and an islet with a width greater than one ×100 objective field was chosen as an arbitrary cut-off to designate an adenoma. The islet cell hyperplasia occurred in Men1+/− mice aged ≥6 months while the earliest islet cell adenoma occurred in Men1+/− mice aged 9 months. Moreover, ∼60% of Men1+/− mice aged ≥12 months were found to have islet cell adenomas. The majority of these islet cell adenomas contained insulin (Fig. 2E and F), but ∼5% contained both insulin and glucagon (Fig. 2F and G). Forty percent of these pancreatic islet cell adenomas were also immunostained for pancreatic polypeptide and gastrin, and this revealed an absence of these peptides. Moreover, gastrin containing cells were not detected in the pancreatic islets of Men1+/+ or Men1+/− mice (data not shown), and as duodenal gastrinomas are often found in MEN1 patients (Gibril et al. 2004), a detailed analysis of the stomach and proximal duodenum from 36 Men1+/− mice aged 18–21 months was undertaken. This did not reveal the occurrence of any gastric or proximal duodenal neuroendocrine tumours.
Pituitary proliferative changes, which consisted of focal hyperplasia without compression or gland enlargement, occurred in Men1+/+ and Men1+/− mice aged ≥6 months but only the Men1+/− mice developed tumours. Some pituitary macroadenomas were large enough to compress the overlying hypothalamus (Fig. 2I). The majority (∼90%) of the pituitary tumours, originated from the pars distalis, and contained both prolactin and GH, while the remainder (∼10%) originated from the pars intermedia and contained ACTH (Fig. 2I–L); >10% of Men1+/− mice had both pars distalis and pars intermedia tumours. These pituitary tumours in the Men1+/− mice which involved the pars distalis or pars intermedia are equivalent to the anterior pituitary tumours that arise in MEN1 patients. Pituitary tumours involving the pars nervosa, which is equivalent to the posterior pituitary (neurohypophysis) in man, were not found in the Men1+/− mice. More than 75% of the pituitary tumours contained prolactin, GH and ACTH, and the remainder were considered to be non-functioning tumours.
Age-related incidental adrenal hyperplastic conditions consisting of cortical or subcapsular hyperplasia, and accessory adrenocortical nodules (Nyska & Maronpot 1999) occurred in Men1+/+ and Men1+/− mice aged ≥6 months, but only Men1+/− mice developed adrenal tumours, seven of which consisted of cortical adenomas (Fig. 2N) that immunostained for hydroxysteroid dehydrogenase (3β-HSD; Fig. 2O), and one was an adrenal medullary tumour, which immunostained for tyrosine hydroxylase (Fig. 2P) and hence was consistent with a phaeochromocytoma.
Gonadal tumours, which are not associated with MEN1 in man, consisted of either Leydig cell tumours or teratoma in Men1+/− male mice, or tubulostromal or sex cord/stromal cell tumours in Men1+/− female mice (data not shown). Approximately 45% of Men1+/− males ≥9 months of age had Leydig cell hyperplasia, but this was not observed in any of the Men1+/+ mice. Unilateral or bilateral Leydig cell tumours, which developed in Men1+/− male mice aged ≥12 months, occurred in ∼60% of Men1+/− mice and were sometimes associated with Leydig cell hyperplasia in the contralateral testis. Moreover, these tumours were malignant with invasion of the capsule in ∼10% of mice. Only one Men1+/− male mouse, aged 3 months, had a testicular teratoma. In six Men1+/− female mice aged >15 months, there were seven ovarian tumours; three were granulosa cell tumours, two were dysgerminomas and there were single cases of luteoma and tubulostromal tumours. One female Men1+/− mouse had a unilateral granulosa cell tumour and a contralateral dysgerminoma.
Macroscopic abnormalities were not detected in any of the Men1+/+ littermates below 18 months of age. In those Men1+/+ control mice over the age of 18 months, macroscopic lesions were observed in five mice. Histology revealed that these lesions consisted of: a mesenteric anaplastic spindle cell sarcoma; a lung adenoma; a focus of mesenteric fat necrosis; and two mice had cystic endometrial hyperplasia of the uterus and ovarian cysts. Men1+/+ mice had common age-related pituitary and adrenal hyperplasia, but importantly, none of the Men1+/+ control mice, including those at 21 months of age, were found to have macroscopic or microscopic tumours of the pancreas, pituitary, parathyroids, adrenals, thyroids or gonads, thereby indicating that the development of tumours in Men1+/− mice was not due to aging or the background of the mouse strain, but instead specific to loss of functioning Men1 alleles and menin expression.
LOH and absence of menin expression in tumours
Tumour development in Men1+/− mice was shown by Southern blot (Fig. 1D), PCR (Fig. 1E), RT-PCR (Fig. 1F), and western blot (Fig. 1G) analyses, to be associated with LOH in which there was a loss of the wild-type Men1 allele. LOH was detected in 100% of anterior pituitary tumours (n=20), 83% of pancreatic islet cell tumours (n=12) and in 67% of adrenal cortical tumours (n=6). In addition, loss of menin expression was shown by immunostaining to have occurred in parathyroid, pancreatic islet cell, anterior pituitary, adrenal cortical, thyroid (Fig. 2D, H, M, Q, S and U) and testicular tumours (data not shown). These results demonstrate that tumour development in the Men1+/− mice is not a sporadic event in this strain, but instead it is specific and dependent on LOH of the wild-type Men1 allele and a loss of menin expression. The loss of menin expression in the thyroid follicular and C-cell adenomas is of particular interest (Fig. 2R–U). Thyroid tumours consisting of adenomas, colloid goitres and carcinomas have been reported to occur in 25% of patients with MEN1 (Thakker 2006). However, the prevalence of thyroid disorders in the general population is high, and it has been suggested that the association of thyroid abnormalities in patients with MEN1 may be incidental and not specific for MEN1. Our results showing loss of menin expression in two types of thyroid tumours (Fig. 2R–U) that occurred in Men1+/− mice, indicate that their development is specific to loss of Men1 expression; this suggests the possibility that the occurrence of thyroid tumours may be part of the MEN1 syndrome in man and indicates that similar studies in human thyroid tumours from MEN1 patients are warranted.
Serum analysis
Serum total calcium (adjusted for albumin), phosphate, PTH, creatinine and CgA in Men1+/− mice with tumours, and also control Men1+/+ mice were measured (Fig. 4). The serum creatinine and albumin were similar in Men1+/+ male and female mice, and in Men1+/− male and female mice with and without tumours (data not shown). In addition, these values were similar in the different age groups (data not shown) and total and adjusted serum calcium (ACa) in the Men1+/+ mice was also similar in both sexes and in the different age groups (data not shown). The data from both sexes and the different age groups were therefore pooled for analysis.
Figure 4.
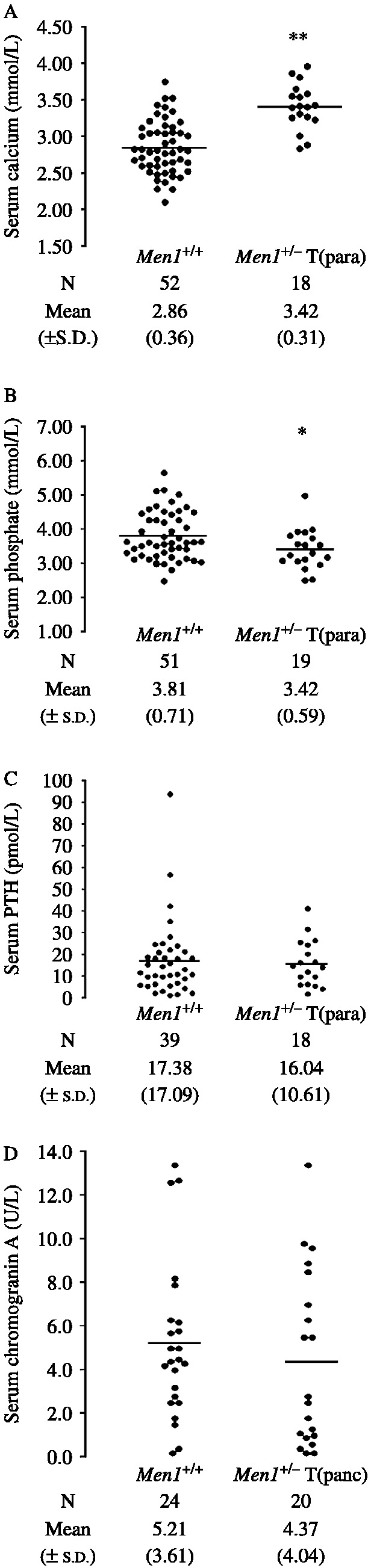
Serum calcium, phosphate, PTH and chromogranin A concentrations. (A) Serum calcium adjusted for albumin, (B) serum phosphate, and (C) serum PTH concentrations in Men1+/+ and Men1+/− mice with parathyroid hyperplasia or adenomas (Men1+/−T(para)). (D) Serum chromogranin A concentrations in Men1+/+ and Men1+/− mice with histologically proven pancreatic tumours (Men1+/−T(panc)). The results for individual mice are shown. The bars show the mean for each group. P values, *<0.05, **<0.001. The Men1+/−T(para) mice were significantly hypercalcaemic and hypophosphataemic when compared to the Men1+/+ mice. The serum PTH concentrations were similar in both groups, but the occurrence of a serum PTH concentration in the normal range in association with hypercalcaemia is considered to be inappropriate, and consistent with primary hyperparathyroidism (Eastell et al. 2009).
Men1+/− mice with parathyroid proliferative abnormalities were found to have significant hypercalcaemia and hypophosphataemia when compared to Men1+/+ mice, consistent with PTH overactivity (Fig. 4A and B). However, the serum PTH concentrations in these Men1+/− mice were similar to that in the normocalcaemic Men1+/+ mice (Fig. 4C). These findings of serum PTH concentrations within the normal range, in association with hypercalcaemia are indicative of inappropriate elevations in serum PTH concentrations for the prevailing ACa and are consistent with primary hyperparathyroidism (Eastell et al. 2009). Men1+/− mice (n=60) in whom parathyroid abnormalities were not detected by histology, were also found to have significant hypercalcaemia (mean ACa±s.d.=3.31±0.30 mmol/l) when compared to Men1+/+ mice (2.86±0.36 mmol/l, n=52, P<0.01), but not hypophosphataemia (Men1+/− mean phosphate±s.d.=3.71±0.82 mmol/l versus Men1+/+ 3.81±0.71 mmol/l). These Men1+/− mice likely represent a heterogeneous group in which some have no parathyroid abnormalities, while others have parathyroid adenomas or hyperplasia that were not detected by histology owing to the sampling difficulties outlined above. The mean (±s.d.) serum PTH in these Men1+/− mice (17.61±14.09 pmol/l, n=59) without parathyroid proliferative abnormalities was not significantly different from that in Men1+/+ mice (17.38±17.09 pmol/l, n=39).
The serum CgA was not significantly different in Men1+/− mice with pancreatic islet cell tumours when compared with Men1+/+ mice (Fig. 4D). In addition, Men1+/− mice (n=40) without pancreatic islet cell tumours when compared to Men1+/+ mice (n=24) had similar mean serum CgA concentrations (Men1+/− mean±s.d., 4.42±2.93 U/l versus Men1+/+, 5.21±3.61 U/l). These findings indicate that serum CgA is unlikely to be a useful marker for pancreatic neuroendocrine tumours in Men1+/− mice.
The adrenal cortical tumours, which occurred in the Men1+/− male mice, showed immunostaining for 3β-HSD (Fig. 2O). Serum corticosterone which is the main glucocorticoid hormone in rodents and precursor of aldosterone (Malisch et al. 2009) was measured. The mean serum corticosterone concentrations in the Men1+/− male mice with adrenal cortical hyperplasia (53±9 nmol/l, n=14) were similar to those Men1+/− male mice without adrenal cortical abnormalities (74±14 nmol/l, n=54) and Men1+/+ male mice (56±13 nmol/l, n=21). However, among the seven Men1+/− male mice with adrenal cortical tumours, serum samples were available for analysis from only four mice; one of these was found to have a serum corticosterone that was markedly elevated at 655 nmol/l (i.e. >10 s.d. above the normal Men1+/+ male mean) while the other three had serum corticosterones of 52, 87 and 90 nmol/l which were in the normal range. The Men1+/− male mouse with hypercorticosteronaemia and the adrenal cortical tumour did not have an anterior pituitary tumour, indicating that the hypercorticosteronaemia is not secondary to ACTH stimulation but instead due to the adrenal tumour. The mean serum corticosterone concentrations in the Men1+/− female mice with adrenal hyperplasia (70±17 nmol/l, n=10), Men1+/− female mice without adrenal cortical abnormalities (94±15 nmol/l, n=49), and Men1+/+ female mice (81±25 nmol/l, n=19) were not significantly different. The finding of a hormone secreting adrenal tumour in 1 out of 4 (∼25%) of the Men1+/− male mice, is consistent with reports that such secreting adrenal tumours occur in 35% of MEN1 patients (Calender et al. 1995, Trump et al. 1996, Marx et al. 1998, Vierimaa et al. 2007). However, corticosterone-producing adrenal tumours, in man, are usually carcinomas and cause sodium retention with a suppression of the renin–angiotensin system and decreased plasma aldosterone concentrations (Edwards & Stowasser 2006). The adrenal cortical tumour in the Men1+/− male mouse with excess corticosterone did not have features of an adrenal carcinoma, and additional sera were not available to further study the effects of the corticosterone excess on glucocorticoid and mineralocorticoid homeostasis.
Expression of CgA, somatostatin receptor type 2, and vascular endothelial growth factor-A
Pancreatic islet cell and pituitary tumours of Men1+/− mice were investigated for expression of CgA, somatostatin receptor type 2 (SSTR2) and vascular endothelial growth factor-A (VEGF-A). CgA was expressed abundantly in the pancreatic islet cell tumours of Men1+/− mice, but mainly on the periphery of pancreatic islets in Men1+/+ mice (Fig. 5A). CgA expression also occurred abundantly in the anterior pituitary tumours of Men1+/− mice, but sparsely in the anterior pituitaries of Men1+/+ mice. The differences in CgA expression in the tumours did not correlate with serum CgA concentrations (Fig. 4D). SSTR2 expression occurred in the pancreatic islets and anterior pituitaries of Men1+/+ mice as well as the pancreatic islet cell and anterior pituitary tumours of Men1+/− mice (Fig. 5B), suggesting that these tumours may be amenable to treatment with somatostatin analogues. VEGF-A expression occurred abundantly in the pancreatic islets of Men1+/+ mice and pancreatic islet cell tumours of Men1+/− mice (Fig. 5C). VEGF-A expression was also observed in the anterior pituitaries of Men1+/+ mice and the anterior pituitary tumours of Men1+/− mice (Fig. 5C). These results indicate that the pancreatic islet cell and anterior pituitary tumours of Men1+/− represent neuroendocrine tumours that express CgA, that may be amenable to treatment with somatostatin analogues and inhibitors of angiogenesis, as they express SSTR2 and VEGF-A, respectively.
Figure 5.
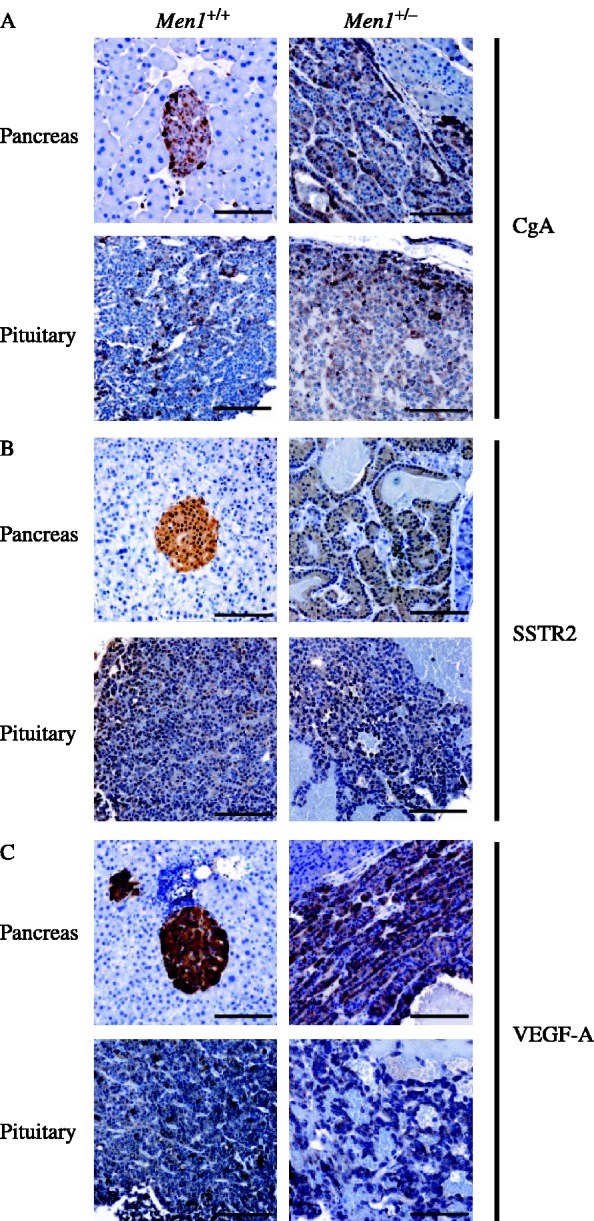
Immunohistochemistry revealing expression of chromogranin A (CgA), somatostatin receptor type 2 (SSTR2), and vascular endothelial growth factor-A (VEGF-A) in pancreatic islets and anterior pituitary tumours of Men1+/− mice. (A) Chromogranin A was present in normal pancreatic islets and anterior pituitary cells of Men1+/+ mice, and in the pancreatic islet cell tumours and anterior pituitary tumours of Men1+/− mice. (B) Somatostatin receptor type 2 was present in Men1+/+ pancreatic islets and anterior pituitary, as well as pancreatic islet and anterior pituitary tumours of Men1+/− mice; (C) VEGF-A expression was observed in Men1+/+ pancreatic islets and anterior pituitary as well as pancreatic islet cell tumours and anterior pituitary tumours of Men1+/− mice.
Discussion
The results of our study, which have established Men1+/− mice that are deleted for exons 1 and 2, together with >1.5 kb of the 5′ sequence that contains the UTR, promoter and alternative exons 1, and ∼1 kb of the 3′ region of intron 2 of the Men1 gene (Fig. 1), reveal that these Men1+/− mice are representative of MEN1 in man. Thus, Men1+/− mice develop parathyroid, pancreatic islet, anterior pituitary, adrenal cortical and lipomatous tumours (Figs 2 and 3). Moreover, the greater occurrence of anterior pituitary tumours in female Men1+/− mice is similar to the gender difference observed in the occurrence of these tumours in MEN1 patients (Verges et al. 2002). The causes of this gender difference are not certain, and further studies of this MEN1 mouse model for gender-specific gene modifiers may help to elucidate these. Furthermore, the age-related onset of tumour development in the Men1+/− mice is similar to the age-related penetrance of MEN1 in man (Trump et al. 1996). Finally, LOH and a lack of menin expression was observed in the majority of the tumours from the Men1+/− mice consistent with a tumour suppressor role for Men1, and similar to the findings reported in MEN1 tumours in man (Larsson et al. 1988, Lubensky et al. 1996). However, there are some interesting differences in the frequencies of tumours that develop in the Men1+/− mice and in patients with MEN1, and five of these are as follows: 1) parathyroid tumours are the commonest manifestation, occurring in ∼95%, of MEN1 patients from <10 to >75 years (Benson et al. 1987, Calender et al. 1995, Trump et al. 1996, Marx et al. 1998), whereas they occurred in only ∼40% of Men1+/− mice aged 3–21 months (Fig. 3), although amongst the ≥12 months cohort they occurred in >85% of Men1+/− mice, 2) gastrinomas are the commonest pancreatic islet cell tumours in MEN1 patients (Trump et al. 1996, Marx et al. 1998), whereas they did not occur in any of the Men1+/− mice, 3) prolactinomas are the commonest anterior pituitary tumours in MEN1 patients (Trump et al. 1996, Marx et al. 1998, Verges et al. 2002), whereas somatolactotrophinomas are the commonest anterior pituitary tumour in the Men1+/− mice, 4) adrenal cortical tumours occurred only in Men1+/− male mice, and this gender difference has not been reported to occur in man, and 5) gonadal tumours, which are rarely found in MEN1 patients, occurred commonly in Men1+/− mice. The basis of these inter-species differences remain to be defined but they may partly be due to: the methods of detection used, e.g. autopsy in the mice which may identify more tumours as opposed to biochemical and radiological investigations in man; the effects of species-specific genetic modifiers that might alter the phenotypic expression of the Men1 mutation in a species; and the absence of a particular lineage, e.g. gastrin producing cells, in the pancreatic islets of Men1+/− mice and hence the absence of gastrinomas in these mice.
The Men1+/− mice described in this report develop tumours that are similar to those reported in other conventional MEN1 mouse models (Table 1; Crabtree et al. 2001, Bertolino et al. 2003a, Loffler et al. 2007). Thus, all the Men1+/− mice develop parathyroid, pancreatic islet cell, anterior pituitary, adrenal cortical and gonadal tumours. However, there are important differences that follow. First, gastrinomas which were extrapancreatic were reported in only one MEN1 model (Bertolino et al. 2003a). Second, gastric neuroendocrine tumours were reported in only one other model (Crabtree et al. 2001). Third, invasive carcinomas were reported in one model (Bertolino et al. 2003a), but not in the other two models (Crabtree et al. 2001, Loffler et al. 2007) or this MEN1 mouse model. The reasons underlying these differences in observed phenotype are not clear, but may be strain dependent or due to differences in the extent to which regional lymph nodes and lungs were sampled to detect carcinomas and metastases. Fourth, the occurrence of hypercalcaemia (Fig. 4A) and hypophosphataemia (Fig. 4B) in association with parathyroid hyperplasia and adenomas were observed only in the Men1+/− mice of this report. The hypercalcaemia was associated with inappropriately normal serum PTH concentrations for the prevailing ACa in these Men1+/− mice with parathyroid abnormalities (Fig. 4C); this is consistent with primary hyperparathyroidism (Eastell et al. 2009). Moreover, the finding of hypercalcaemia in association with hypophosphataemia is consistent with the presence of elevations in biologically active PTH concentrations. The absence of hypercalcaemia in the other MEN1 models (Table 1) is difficult to reconcile, and possible explanations may be the different responses of the mouse strains, the calcium and vitamin D content of the diets, or the method used to measure the serum calcium. Conditional and specific deletion of both of the Men1 alleles in the parathyroids results in hypercalcaemia and parathyroid tumours (Libutti et al. 2003), in agreement with our observations (Table 1).
To further investigate the possible utility of the established MEN1 model for treatments, we investigated the Men1+/− mice for alterations in serum CgA (Fig. 4D) and for tumour expression of CgA, SSTR2 and VEGF-A (Fig. 5). Pancreatic islet cell and pituitary tumours represent neuroendocrine tumours that are highly vascularised, and previous studies in man, have reported that these tumours, as assessed by immunohistochemistry, express CgA, SSTR2 and VEGF-A (Moller et al. 2003, Turner et al. 2003, Helle et al. 2007). In addition, serum CgA has been reported to correlate with pancreatic neuroendocrine tumour burden in MEN1 patients (Janson et al. 1997, Granberg et al. 1999) although the variation in serum CgA can be marked due to tumour instability and liability to release hormones (Granberg et al. 1999). The pancreatic islet and anterior pituitary tumours in the Men1+/− mice were found to contain CgA (Fig. 5A), but serum CgA was not found to be elevated in these mice (Fig. 4D), indicating that CgA may not be easily released from these tumour cells and that serum CgA is unlikely to be a useful marker for monitoring response to treatments in these Men1+/− mice. However, the finding of CgA expression within the Men1+/− mice anterior pituitary tumours that also contain prolactin, is unusual, as in man it has been reported that prolactinomas, in contrast to all other pituitary adenomas, do not express CgA (Lloyd et al. 1989). Moreover, normal endocrine cells of the pituitary, in man, usually express CgA, and the observation of abundant CgA expression in the prolactin containing anterior pituitary tumour cells of Men1+/− mice implies that CgA expression may be upregulated upon neoplastic transformation. In order to facilitate investigation of future therapies for the tumours in Men1+/− mice, we investigated them for SSTR2 (Fig. 5B) and VEGF-A (Fig. 5C) expression. The expression of SSTR2 and VEGF-A by the pancreatic islet and anterior pituitary tumours of these Men1+/− mice, indicates that they are likely to respond to treatment with somatostatin analogues and inhibitors, e.g. monoclonal antibodies, of angiogenesis, respectively.
In summary, we have established a Men1+/− mouse model that is representative of MEN1 in man, and our demonstration of SSTR2 and VEGF-A expression in the pancreatic islet cell tumours and anterior pituitary tumours, indicates that these Men1+/− mice will be of use in evaluating therapies with established and emerging somatostatin analogues, and inhibitors of angiogenesis.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported in this article.
Funding
This work was supported by the Medical Research Council (G9825289, 2004), UK (B Harding, M C Lemos, A A C Reed, M R Bowl, G V Walls, J Jeyabalan, H Tateossian, T Hough, M T Cheeseman and R V Thakker), and the Portuguese Foundation for Science and Technology (BD/12415/2003) (M C Lemos).
Acknowledgements
We are grateful to T Hacker and J Humphreys (MRC Mary Lyon Centre, Harwell) for assistance in preparation of histology sections, S Thomas (MRC, Mary Lyon Centre, Harwell) for preparing Fig. 2, Prof I Mason, Reproductive and Developmental Sciences, University of Edinburgh, for the gift of the anti-3β-HSD antibody, Dr A F Parlow, National Institute of Diabetes and Digestive and Kidney Diseases, Torrance, CA, USA for the anti-prolactin antibody and to Mrs Tracey Walker for typing the manuscript.
Footnotes
(B Harding and M C Lemos contributed equally to this work)
References
- Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. American Journal of Medicine. 1987;82:731–737. doi: 10.1016/0002-9343(87)90008-8. [DOI] [PubMed] [Google Scholar]
- Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Molecular Endocrinology. 2003a;17:1880–1892. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Research. 2003b;63:4836–4841. [PubMed] [Google Scholar]
- Biondi CA, Gartside MG, Waring P, Loffler KA, Stark MS, Magnuson MA, Kay GF, Hayward NK. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Molecular and Cellular Biology. 2004;24:3125–3131. doi: 10.1128/MCB.24.8.3125-3131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calender A, Giraud S, Cougard P, Chanson P, Lenoir G, Murat A, Hamon P, Proye C. Multiple endocrine neoplasia type 1 in France: clinical and genetic studies. Journal of Internal Medicine. 1995;238:263–268. doi: 10.1111/j.1365-2796.1995.tb00933.x. [DOI] [PubMed] [Google Scholar]
- Capen CC, Gröne A, Bucci TJ, Rosol TJ. Changes in structure and function of the parathyroid gland. In: Mohru U, Dungworth DL, Capen CC, Carlton WW, Sundberg JP, Ward JM, editors. Pathobiology of the Aging Mouse. International Life Sciences Institute Press; Washington DC: 1996. pp. 109–123. [Google Scholar]
- Chandrasekharappa SC, Teh BT. Functional studies of the MEN1 gene. Journal of Internal Medicine. 2003;253:606–615. doi: 10.1046/j.1365-2796.2003.01165.x. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chang W, Tu C, Chen TH, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Science Signaling. 2008;1:ra1. doi: 10.1126/scisignal.1159945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. PNAS. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, McNally SR, Swain GP, Montagna C, Hager JH, Hanahan D, Edlund H, Magnuson MA, et al. Of mice and MEN1: insulinomas in a conditional mouse knockout. Molecular and Cellular Biology. 2003;23:6075–6085. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastell R, Arnold A, Brandi ML, Brown EM, D'Amour P, Hanley DA, Rao DS, Rubin MR, Goltzman D, Silverberg SJ, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. Journal of Clinical Endocrinology and Metabolism. 2009;94:340–350. doi: 10.1210/jc.2008-1758. [DOI] [PubMed] [Google Scholar]
- Edwards CRW & Stowasser M 2006 Primary mineralocorticoid excess syndromes. In Endocrinology, edn 5, pp 2461–2490. Eds LJ DeGroot & JL Jameson. Philadelphia: Elsevier Saunders.
- Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger–Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine. 2004;83:43–83. doi: 10.1097/01.md.0000112297.72510.32. [DOI] [PubMed] [Google Scholar]
- Granberg D, Stridsberg M, Seensalu R, Eriksson B, Lundqvist G, Oberg K, Skogseid B. Plasma chromogranin A in patients with multiple endocrine neoplasia type 1. Journal of Clinical Endocrinology and Metabolism. 1999;84:2712–2717. doi: 10.1210/jcem.84.8.5938. [DOI] [PubMed] [Google Scholar]
- Helle KB, Corti A, Metz-Boutigue MH, Tota B. The endocrine role for chromogranin A: a prohormone for peptides with regulatory properties. Cellular and Molecular Life Sciences. 2007;64:2863–2886. doi: 10.1007/s00018-007-7254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough TA, Nolan PM, Tsipouri V, Toye AA, Gray IC, Goldsworthy M, Moir L, Cox RD, Clements S, Glenister PH, et al. Novel phenotypes identified by plasma biochemical screening in the mouse. Mammalian Genome. 2002;13:595–602. doi: 10.1007/s00335-002-2188-1. [DOI] [PubMed] [Google Scholar]
- Janson ET, Holmberg L, Stridsberg M, Eriksson B, Theodorsson E, Wilander E, Oberg K. Carcinoid tumors: analysis of prognostic factors and survival in 301 patients from a referral center. Annals of Oncology. 1997;8:685–690. doi: 10.1023/a:1008215730767. [DOI] [PubMed] [Google Scholar]
- Khodaei-O'Brien S, Zablewska B, Fromaget M, Bylund L, Weber G, Gaudray P. Heterogeneity at the 5′-end of MEN1 transcripts. Biochemical and Biophysical Research Communications. 2000;276:508–514. doi: 10.1006/bbrc.2000.3471. [DOI] [PubMed] [Google Scholar]
- Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Human Mutation. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- Lemos MC, Harding B, Thakker RV. Genetic background influences expression of multiple endocrine neoplasia type 1 (MEN1) mutation, implicating a role for genetic modifiers. Endocrine Abstracts. 2007;13:P19. [Google Scholar]
- Libutti SK, Crabtree JS, Lorang D, Burns AL, Mazzanti C, Hewitt SM, O'Connor S, Ward JM, Emmert-Buck MR, Remaley A, et al. Parathyroid gland-specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Research. 2003;63:8022–8028. [PubMed] [Google Scholar]
- Lloyd RV, Iacangelo A, Eiden LE, Cano M, Jin L, Grimes M. Chromogranin A and B messenger ribonucleic acids in pituitary and other normal and neoplastic human endocrine tissues. Laboratory Investigation. 1989;60:548–556. [PubMed] [Google Scholar]
- Loffler KA, Biondi CA, Gartside M, Waring P, Stark M, Serewko-Auret MM, Muller HK, Hayward NK, Kay GF. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. International Journal of Cancer. 2007;120:259–267. doi: 10.1002/ijc.22288. [DOI] [PubMed] [Google Scholar]
- Lubensky IA, Debelenko LV, Zhuang Z, Emmert-Buck MR, Dong Q, Chandrasekharappa S, Guru SC, Manickam P, Olufemi SE, Marx SJ, et al. Allelic deletions on chromosome 11q13 in multiple tumors from individual MEN1 patients. Cancer Research. 1996;56:5272–5278. [PubMed] [Google Scholar]
- Malisch JL, Kelly SA, Bhanvadia A, Blank KM, Marsik RL, Platzer EG, Garland T. Lines of mice with chronically elevated baseline corticosterone levels are more susceptible to a parasitic nematode infection. Zoology. 2009;112:316–324. doi: 10.1016/j.zool.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nature Reviews. Cancer. 2005;5:367–375. doi: 10.1038/nrc1610. [DOI] [PubMed] [Google Scholar]
- Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: clinical and genetic topics. Annals of Internal Medicine. 1998;129:484–494. doi: 10.7326/0003-4819-129-6-199809150-00011. [DOI] [PubMed] [Google Scholar]
- Moller LN, Stidsen CE, Hartmann B, Holst JJ. Somatostatin receptors. Biochimica et Biophysica Acta. 2003;1616:1–84. doi: 10.1016/s0005-2736(03)00235-9. [DOI] [PubMed] [Google Scholar]
- Nesbit MA, Bowl MR, Harding B, Ali A, Ayala A, Crowe C, Dobbie A, Hampson G, Holdaway I, Levine MA, et al. Characterization of GATA3 mutations in the hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome. Journal of Biological Chemistry. 2004;279:22624–22634. doi: 10.1074/jbc.M401797200. [DOI] [PubMed] [Google Scholar]
- Nyska A, Maronpot RR. Cache River Press; Vienna, IL, USA: 1999. Pathology of the Mouse. [Google Scholar]
- Pannett AA, Thakker RV. Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis. Journal of Clinical Endocrinology and Metabolism. 2001;86:4371–4374. doi: 10.1210/jcem.86.9.7844. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Crabtree JS, Novotny EA, Garrett-Beal L, Chen A, Edgemon KA, Marx SJ, Spiegel AM, Chandrasekharappa SC, Collins FS. Bidirectional transcriptional activity of PGK-neomycin and unexpected embryonic lethality in heterozygote chimeric knockout mice. Genesis. 2001;30:259–263. doi: 10.1002/gene.1072. [DOI] [PubMed] [Google Scholar]
- Thakker RV 2006 Multiple endocrine neoplasia type 1. In Endocrinology, edn 5, pp 3509–3531. Eds LJ DeGroot & JL Jameson. Philadelphia: Elsevier Saunders.
- Thakker RV, Bouloux P, Wooding C, Chotai K, Broad PM, Spurr NK, Besser GM, O'Riordan JL. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. New England Journal of Medicine. 1989;321:218–224. doi: 10.1056/NEJM198907273210403. [DOI] [PubMed] [Google Scholar]
- The European Consortium on MEN1. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Human Molecular Genetics. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, Edwards CR, Heath DA, Jackson CE, Jansen S, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1) Quarterly Journal of Medicine. 1996;89:653–669. doi: 10.1093/qjmed/89.9.653. [DOI] [PubMed] [Google Scholar]
- Turner HE, Harris AL, Melmed S, Wass JA. Angiogenesis in endocrine tumors. Endocrine Reviews. 2003;24:600–632. doi: 10.1210/er.2002-0008. [DOI] [PubMed] [Google Scholar]
- Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, Cougard P, Chambe B, Montvernay C, Calender A. Pituitary disease in MEN type 1 (MEN1): data from the France–Belgium MEN1 multicenter study. Journal of Clinical Endocrinology and Metabolism. 2002;87:457–465. doi: 10.1210/jcem.87.2.8145. [DOI] [PubMed] [Google Scholar]
- Vierimaa O, Ebeling TM, Kytola S, Bloigu R, Eloranta E, Salmi J, Korpi-Hyovalti E, Niskanen L, Orvola A, Elovaara E, et al. Multiple endocrine neoplasia type 1 in Northern Finland; clinical features and genotype phenotype correlation. European Journal of Endocrinology. 2007;157:285–294. doi: 10.1530/EJE-07-0195. [DOI] [PubMed] [Google Scholar]