

Abstract
Many human malignancies are associated with aberrant regulation of protein or lipid kinases due to mutations, chromosomal rearrangements and/or gene amplification. Protein and lipid kinases represent an important target class for treating human disorders. This review focus on ‘the 10 things you should know about protein kinases and their inhibitors', including a short introduction on the history of protein kinases and their inhibitors and ending with a perspective on kinase drug discovery. Although the ‘10 things’ have been, to a certain extent, chosen arbitrarily, they cover in a comprehensive way the past and present efforts in kinase drug discovery and summarize the status quo of the current kinase inhibitors as well as knowledge about kinase structure and binding modes. Besides describing the potentials of protein kinase inhibitors as drugs, this review also focus on their limitations, particularly on how to circumvent emerging resistance against kinase inhibitors in oncological indications.
Tables of Links.
| TARGETS | ||
|---|---|---|
| Catalytic receptorsa | Enzymesb | |
| ALK | ABL (Abl) | MAPK |
| AXL | Akt (PKB) | MEK1 |
| CSF1R | AMPK | MLKL |
| EGFR | Aurora kinase | mTOR |
| FGFR1 | B-Raf (BRAF) | PDK1 |
| FLT3 | BTK | PHK |
| HER2 (Neu) | CHEK1 (CHK1) | PI3Kδ |
| IGF1R | ELK (EphB1) | PIK3CA |
| Insulin receptor | FAK | PKCζ |
| KIT | Fes | PTEN |
| MET (c-Met) | Glucokinase | PTK |
| PDGFRα | GSK3β | RAF |
| PDGFRβ | Haspin | Ribosomal S6 kinase |
| RET | Hck | ROCK |
| ROS1 | JAK2 | STK11 |
| TIE2 | JNK1 | STRAD1 |
| TrkB | LKB1 | Src |
| LIGANDS | |
|---|---|
| ADP | Lapatinib |
| ATP | Myristate |
| Afatinib | Nilotinib |
| AZD6244 | Nintedanib |
| Crizotinib | Pertuzumab |
| Cyclosporine | Ponatinib |
| Dabrafenib | Sirolimus (rapamycin) |
| Dasatinib | Sorafenib |
| Erlotinib | Staurosporine |
| Fasudil (HA1077) | Sunitinib |
| Gefitinib | Tofacitinib |
| GNF-2 | Trametinib |
| Ibrutinib | Trastuzumab |
| Imatinib | Vemurafenib |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Short history on protein kinases and inhibitors
Post-translational modifications such as phosphorylation, glycosylation, ubiquitination, nitrosylation, acylation, methylation, lipidation and proteolysis, which are known to increase the diversity of the proteome, influence various aspects of normal and pathological physiology (Walsh et al., 2005; Liu et al., 2013). Kinases carry out the phosphorylation reactions by transferring the gamma phosphate of ATP onto hydroxyl groups of various substrates including lipids, sugars or amino acids and is reversed by the corresponding phosphatases. Phosphorylation plays a pivotal role in many cellular and extracellular processes (Blume-Jensen and Hunter, 2001; Cohen, 2001; Walsh et al., 2005; Kannan et al., 2007; Ubersax and Ferrell, 2007). While bacterial signalling occurs through His-Asp kinases and some eukaryotic-like proteins as well as small molecule kinases [eukaryotic protein kinase (ePK)-like kinases or eukaryotic-like kinase (ELK)], the protein kinases of eukaryotes which include the ePKs phosphorylate either tyrosine (TPKs; tyrosine-specific protein kinases), serine/threonine (STPKs; Ser-/Thr-specific protein kinases) or both tyrosine and threonine (dual-specificity protein kinases) (Cohen, 2001; 2002b,; Kennelly, 2002; 2003,; Kannan et al., 2007; Ubersax and Ferrell, 2007). In addition, eukaryotes have kinases that specifically phosphorylate small molecules, including lipids and sugars (Figure 1A) (Kannan et al., 2007; Yuan and Cantley, 2008; Bornancin, 2011; Kunkel et al., 2013). Aberrant phosphorylation in eukaryotes is associated with a variety of disorders ranging from cancer to inflammatory diseases, diabetes, infectious diseases, cardiovascular disorders, cell growth and survival (Blume-Jensen and Hunter, 2001; Cohen, 2001; Walsh et al., 2005; Ubersax and Ferrell, 2007; Lahiry et al., 2010).
Figure 1.
(A) Reversible phosphorylation and (B) kinase drug discovery. For explanation, see text. DualPase, dual-specificity phosphatases; DualPK, dual-specificity kinases; PI, phosphatidylinositol; PIP, phosphatidylinositolphosphate; PI3K, phosphatidylinositol kinases; STPase, Ser/Thr-specific phosphatases; STPK, Ser/Thr-specific kinases; TPK, Tyr-specific kinases.
The first phosphorylation of proteins was described for casein [by phosphorylase kinase (PHK)] in 1954 (reviewed in Cohen, 2002a). In the late 1970s, only a handful of biochemically characterized STPKs were known against which some inhibitors were identified that were neither potent nor selective (Figure 1A) (Glossmann et al., 1981; Hidaka et al., 1984; Inagaki et al., 1986; Davies et al., 2000). The identification of PKCs as receptors for tumour-promoting phorbol esters, together with the discovery of TPKs as oncogenes in the 1980s, with the advent of molecular cloning led to the initiation of more rational kinase drug discovery approaches. The foundation for the medicinal chemistry on kinase inhibitors at that time was derived from just a few lead compounds, including the natural compound staurosporine and the synthetic tyrphostins (Tamaoki et al., 1986; Levitzki, 1990) (Figure 1B). The first protein kinase inhibitor was fasudil (HA-1077), which was approved in Japan in 1995 for cerebral vasospasm (Shibuya and Suzuki, 1993). Fasudil was followed by sirolimus (Rapamune), the first allosteric kinase inhibitor, which was approved in 1999 for use in combination with cyclosporine for the prevention of organ rejection in patients receiving renal transplants (Kelly et al., 1997; Vasquez, 2000). The target of this natural compound, the kinase mTOR (mammalian target of rapamycin), was discovered by a genetic screen just a few years before (Kunz et al., 1993).
The first economically successful protein kinase inhibitor imatinib (CGP57148, STI571, Glivec, Gleevec) designed to inhibit the Abelson (ABL) kinase in the context of the BCR-ABL translocation was approved in 2001 for chronic myeloid leukaemia (CML) (Figure 1B) (Buchdunger et al., 2001). The success of imatinib is due to its efficacy during the chronic phase of CML, which is an almost monogenic BCR-ABL-driven myeloproliferative disorder. Imatinib is much less effective against the more aggressive disease state of CML, the blast crisis, an acute leukaemia, which marks the fatal end stage of the disease (Goldman and Druker, 2001; Druker et al., 2006). The success of imatinib is also due to its ‘selectivity’ or rather the lack thereof. The poly-pharmacology of imatinib allowed proof of clinical concept in indications other than CML, including GIST (gastrointestinal stromal tumour), HES (hyper-eosinophilic syndrome) and others (Fabbro et al., 2005). These successes convinced the pharmaceutical industry to invest in protein and lipid kinase inhibitors as targeted therapies for various cancers (Fabbro et al., 2002b; 2011; Engelman, 2009; Sellers, 2011; Bartholomeusz and Gonzalez-Angulo, 2012; Workman and Al-Lazikani, 2013a; Workman et al., 2013b). The sequencing of the human kinome in 2002, the steady increase in structural analysis of protein kinases, the advent of cancer genetics in conjunction with the development of high-throughput biochemical and cell-based profiling for protein kinases led to a continuous flow of kinase inhibitor approval into the clinical space (Table 1 and Figure 1B) (Manning et al., 2002; Fedorov et al., 2010; Fabbro et al., 2011; Workman and Al-Lazikani, 2013a; Workman et al., 2013b). Thus, protein kinases have been successfully pursued since the late 1980s in the pharmaceutical industry as potential drug targets mainly for the treatment of cancer indications.
Table 1.
Approved kinase inhibitors as of February 2015
| Generic name (compound code, trade names) | Kinase target | Disease | Company (year, type) |
|---|---|---|---|
| Fasudil (HA-1077) | ROCK1/2 | Cerebral vasospam, PAH | Asahi Kasei (1995, type-1) |
| Sirolimus (Rapamune) | mTOR | Kidney transplants | Pfizer, Wyeth (1999, type-3) |
| Imatinib (STI571, Glivec, Gleevec) | ABL, PDGFR, KIT | CML, Ph+ B-ALL, CMML, HES, GIST | Novartis (2001, type-2) |
| Gefitinib (ZD1839, Iressa) | EGFR | NSCLC | AZ (2003, type-1) |
| Erlotinib (OSI-774,Tarceva) | EGFR | NSCLC, pancreatic cancer | Roche, OSI (2004, type-1) |
| Sorafenib (BAY 43-9006, Nexavar) | VEGFR2, PDGFR, KIT, FLT3, BRAF | RCC, HCC | Bayer, Onyx (2005, type-2) |
| Sunitinib (SU11248, Sutent) | VEGFR, KIT, PDGFR, RET, CSF1R, FLT3 | RCC, imatinib resistant GIST | Pfizer (2006, type-1) |
| Lapatinib (GW2016, Tykerb) | EGFR, ERBB2 | BC | GSK (2007, type-1.5) |
| Dasatinib (BM-354825,Sprycel) | ABL], PDGFR, KIT, SRC | CML | BMS (2007, type-1) |
| Nilotinib (AMN107,Tasigna) | ABL, PDGFR, KIT | CML | Novartis (2007, type-2) |
| Everolimus (Rad001, Certican, Zortress, Afinitor, Votubia) | mTOR | RCC, SEGA, Transplantation | Novartis (2009, type-3) |
| Temsirolimus (CCI-779, Torisel) | mTOR | RCC | Pfizer, Wyeth (2009, type-3) |
| Crizotinib (PF-02341066, Xalcori) | MET and ALK | NSCLC with ALK translocations | Pfizer (2011, type-1) |
| Vandetanib (ZD6474, Caprelsa) | RET, VEGFR1-2, FGFR, EGFR | MTC | AZ (2011, type-1) |
| Ruxolitinib (INC424, Jakafi) | JAK2 | IMF with JAK2V617F mutations | Novartis, Incyte (2011, type-1) |
| Vemurafenib (PLX4032, RG7204, Zelboraf) | BRAF | Metastatic melanoma with BRAFV600E mutations | Roche, Plexxikon (2011, type-2) |
| Axitinib (AG013736, Inlyta) | VEGFR, KIT, PDGFR, RET, CSF1R, FLT3 | RCC | Pfizer (2012, type-1) |
| Regorafenib (BAY 73-4506, Stivarga) | VEGFR2, Tie2 | CRC, GIST | Bayer (2012, type-2) |
| Pazopanib (GW-786034, Votrient) | VEGFR, PDGFR, KIT | RCC | GSK (2012, type-1) |
| Tofacitinib (CP-690550, Xeljanz Tasocitinib) | JAK3 | RA | Pfizer (2012, type-1) |
| Cabozantinib (XL184, BMS907351, Cometriq) | VEGFR2, PDGFR, KIT, FLT3 | MTC | Exelexis (2012, type-1) |
| Ponatinib (AP24534, Iclusig) | ABL | Imatinib resistant CML with T315I mutations | Ariad (2012, type-1) |
| Bosutinib (SKI-606, Bosulif) | ABL | CML resistant/ intolerant to therapy | Pfizer (2012, type-1) |
| Dabrafenib (Tafinlar) [6494] | BRAF | Metastatic melanoma with BRAFV600E mutations | GSK (2013, type-2) |
| Trametinib (Mekinist) [6495] | MEK | Metastatic melanoma with BRAFV600E mutations | GSK (2013, type-3) |
| Afatnib (Gilotrif, Tomtovok, Tovok) | EGFR | NSCLC with EGFR activating mutations | BI (2013, covalent) |
| Ibrutinib (PCI-32765, Imbruvica) | BTK | MCL, CLL | Janssen, Pharmacyclic (2013, covalent) |
| Ceritinib (LDK378, Zykadia) | ALK | NSCLC with ALK translocations | Novartis (2014, type-1) |
| Idelalisib (CAL101, GS1101, Zydelig) | PI3Kdelta | CLL, FL and SLL | Gilead, Calistoga, ICOS (2014, type-1) |
| Nintedanib (BIBF 1120, Vargatef, Intedanib) | VEGFR, PDGFR, FGFR | Idiopathic Pulmonary Fibrosis | BI (2014, type-1) |
| Alectinib (AF802, RO5424802) | ALK | ALK-rearranged NSCLC | Roche (2014) |
| Palbociclib (PD-0332991, Ibrance) | CDK4/6 | Advanced (metastatic) BC | Pfizer (2015) |
| Lenvatinib (E7080) | VEGFRs | Thyroid cancer | Eisai Co (2015) |
The biochemical profiles of the 33 approved kinase inhibitors are stored in the IUPHAR database (http://www.guidetopharmacology.org/GRAC/LigandListForward?type=Approved&database=all). The 33 kinase inhibitors approved to date are shown with generic compound name, compound code, trade name, primary indications, company and mode of binding. The approved kinase inhibitors include fasudil (HA-1077) (Shibuya and Suzuki, 1993; Shibuya et al., 2001), sirolimus (Rapamycin, Rapamune®) (Kelly et al., 1997; Vasquez, 2000), imatinib (Glivec®) (Druker et al., 1996), gefitinib (Iressa™ ) (Barker et al., 2001), erlotinib (Tarceva™) (Perez-Soler, 2004), lapatinib (Tykerb®) (Gaul et al., 2003), sorafenib (Nexavar®) (Lowinger et al., 2002), sunitinib (Sutent®) (Sun et al., 2003), dasatinib (Sprycel®) (Lombardo et al., 2004), nilotinib (Tasigna®) (Weisberg et al., 2005), torisel (Temsirolimus®) (Galanis et al., 2005), everolimus (Rad001) as Afinitor® (Chan et al., 2010; Baselga et al., 2012; Beck et al., 2014) as Zortress® and Certican™ (Cibrik et al., 2013) as Votubia® for SEGA (Krueger et al., 2010), crizotinib (Xalcori®) (Shaw et al., 2011), vandetanib (Caprelsa®) (Carlomagno and Santoro, 2004; Chau and Haddad, 2013), ruxolitinib (Jakafi®) (Harrison et al., 2012), vemurafenib (Zelboraf®) (Flaherty et al., 2010), axitinib (Inlyta®) (Ansari et al., 2013; Rini et al., 2013), regorafenib (Stivarga®) (Shahda and Saif, 2013), pazopanib (Votrient™) (Sternberg, 2009), tofacitinib (Xeljanz) (Simmons, 2013), cabozantinib (Cometriq) (Viola et al., 2013), ponatinib (Iclusig®) (Nicolini et al., 2013), bosutinib (Bosulif®) (Amsberg and Koschmieder, 2013), dabrafenib (Tafinlar®) (Ballantyne and Garnock-Jones, 2013; King et al., 2013), trametinib (Mekinist®) (Salama and Kim, 2013; Wright and McCormack, 2013), afatinib (Gilotrif®) (Nelson et al., 2013; Ninomiya et al., 2013), ibrutinib (Imbruvica®) (McDermott and Jimeno, 2014), ceritinib (Zykadia®) (Friboulet et al., 2014), idelalisib (Zydelig®) (Gopal et al., 2014) and nintedanib (Vargatef®, Ofev™) (Reck et al., 2014; Richeldi et al., 2014), alectinib (Yang, 2013), palbociclib (Ibrance®) (http://www.onclive.com/web-exclusives/FDA-Approves-Palbociclib-for-Metastatic-Breast-Cancer) and levantinib (http://www.eisai.com/news/enews201407pdf.pdf). All compounds are commercially available. AZ, Astra-Zeneca; BI, Boehringer-Ingelheim; GSK, Glaxo-Wellcome.
CLL, chronic lymphocytic leukaemia; CML, chronic myeloid leukaemia; CMML, chronic myeloid monocytic leukaemia; CSF1R, colony stimulating factor 1 receptor; FL, folliclular lymphoma; HCC, hepatocellular cancer; IMF, idiopathic myelofibrosis; MCL, mantle cell lymphoma; MTC, medullary thyroid cancer; NSCLC, non-small-cell lung cancer; PAH, pulmonary arterial hypertension; RCC, renal cell carcinoma.
The ePK and PI3K
According to the latest counts, the human kinome contains 538 ePK genes, which are subdivided into seven families of typical and seven families of atypical protein kinases (http://kinase.com/kinbase/; http://kinase.com/human/kinome/) (Hanks and Hunter, 1995; Hunter, 2000; Manning et al., 2002). The majority of ePKs are STPKs, a fact that is reflected in the ratio of cellular phosphorylation (pSer : pThr : pTyr = 1000:100:1) (Hanks and Hunter, 1995; Hunter, 2000; Cohen, 2001; 2002b,; Manning et al., 2002; Ubersax and Ferrell, 2007). Although only a minor number of substrates are phosphorylated by TPKs, the importance of tyrosine phosphorylation is demonstrated by the many gain of function (GOF) and/or loss of function (LOF) mutations that are found in TPKs (Hunter, 2000; Blume-Jensen and Hunter, 2001; Cohen, 2001; 2002b,; Greenman et al., 2007; Thomas et al., 2007; Fedorov et al., 2010; Lahiry et al., 2010; Fabbro et al., 2011; Workman et al., 2013b). TPKs can be subdivided into two main classes (receptor TPKs and non-receptor TPKs). In contrast, the STPKs are a more heterogeneous class of enzymes and are divided into six main families of typical ePKs, including the TKL (the Tyrosine Kinase Like group closely related to the TPKs), the CMGC (the cyclin-dependent kinases, MAP kinases, Glycogen synthase kinases, Casein kinases 2), the AGC (PKA, PKG and PKC), the CAMK (CAlcium/calModulin-dependent Kinases), the STE20 [homologues of yeast Sterile 7, Sterile 11, STErile 20 kinases which include the MAP2Ks (mitogen activated kinase kinase), MAP3Ks (mitogen activated kinase kinase kinase) and MAP4Ks (mitogen activated kinase kinase kinase kinase)] and finally the CK1 (Casein Kinases 1). Most of the atypical ePKs are STPKs, which indicates that the ePK domain phylogeny may reflect substrate specificity and/or mode of regulation (http://kinase.com/kinbase/) (Manning et al., 2002). About 10% of the human protein kinases are so-called pseudo-kinases because they are either only weakly active or presumed to be inactive. These pseudo-kinases are evenly distributed over the human kinome (Boudeau et al., 2006; Kannan and Taylor, 2008). They lack at least one of three motifs in the catalytic domain that are essential for catalysis (Figure 2B). Although the non-catalytic functions are poorly understood, the pseudo-kinases can bind ATP and appear to have important regulatory functions as exemplified by the regulation of the LKB1 (serine/threonine-protein kinase STK11) by the pseudo-kinase STRAD1 (STE20-related adapter alpha) or the ‘activation’ of Janus kinase 2 (JAK2) catalytic domain (JH1) via a single amino acid substitution (V617F) in its JH2 (JAK homology domain-2)-pseudo-kinase domain (Kralovics et al., 2005; Boudeau et al., 2006; Kannan and Taylor, 2008; Zeqiraj et al., 2009; Rajakulendran and Sicheri, 2010).
Figure 2.

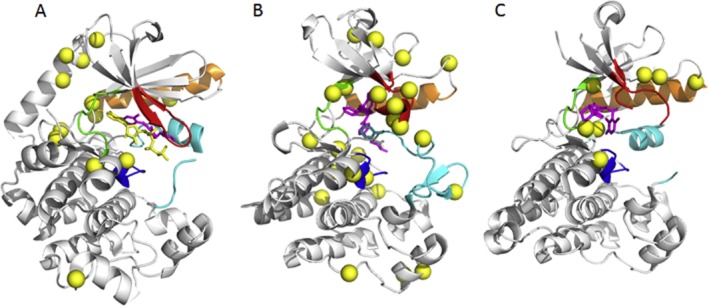
Kinase (inactive), pseudokinase and atypical kinase conformational states. Various examples of the positions and forms of structural elements in different kinases. The same colour scheme as in Figure 2 is used. Panels (A)–(C) show the inactive states of three kinases, with only those structural elements where there are large shifts compared with the active state coloured. (A) Inactive conformation of Hck: C-helix out and closed A-loop (PDB entry 1HCK). (B) Inactive conformation of Abl1 kinase: Collapsed P-loop and closed A-loop with DFG-motif out (PDB entry 1IEP). (C) Inactive conformation of c-Met: C-helix out and yet another conformation of the A-loop (PDB entry 3CCN). Panels (D) and (E) show the structures of the pseudokinases JAK2 (PDB entry 4FVR), where the HRD sequence motif is not conserved, and MLKL (PDB entry 4MWI), where both the DFG and HRD sequence motifs are not conserved respectively. Panel (F) shows the structure of the atypical kinase, haspin (PDB entry 2VUW).
Another important set of kinases that play essential roles in the eukaryotic signalling and which share the protein kinase-like (PKL) fold with the ePKs include the phosphatidyl-inositol (PI) kinases and related protein kinases (Kannan et al., 2007). In particular, the PI3Ks, which phosphorylate PI together with the atypical STPK mTOR, have been implicated in cancer and immunological disorders (Engelman, 2009; Courtney et al., 2010; Rommel, 2010; Fruman and Rommel, 2014).
The physiological activation of kinase occurs in many different ways and their mechanisms of activation have been summarized in excellent reviews (Taylor et al., 2005; Murray, 2007; Rommel et al., 2007; Schmierer and Hill, 2007; Engelman, 2009; Malumbres and Barbacid, 2009; Kawai and Akira, 2010; Lemmon and Schlessinger, 2010; Mendoza et al., 2011; Hoesel and Schmid, 2013; Brooks et al., 2014; Hardie, 2014). Kinases are organized in cascades, which are typically initiated by various receptors including receptor and non-receptor TPKs or STPKs, which further pass their signals through various downstream effectors such as the PI3K/mTOR, the RAS-RAF-MAPK, the SMAD [composite of MAD from drosophila Mothers Against Decapentaplegic and SMA of Caenorhabditis elegans (from gene sma for small body size)], the STAT to the cell cycle kinases and kinases regulating transcription (Lahiry et al., 2010; Lemmon and Schlessinger, 2010). Besides transferring the gamma phosphate of ATP onto hydroxyl groups of substrates protein and lipid kinase, protein kinases also utilize non-catalytic functions for scaffolding, relocation, allosteric effects, subcellular targeting, DNA binding as well as protein–protein interactions (Rauch et al., 2011). Abnormal hyperactivity, due to mutations, chromosomal rearrangements and/or gene amplification or LOFs of protein and PI3K kinases, plays a role in a wide variety of diseases, including cancer, inflammatory diseases, diabetes, atherosclerosis and immunological disorders (Blume-Jensen and Hunter, 2001; Cohen, 2001; Chico et al., 2009; Lahiry et al., 2010; Muller and Knapp, 2010; Rommel, 2010; Fabbro et al., 2011; Angulo et al., 2013). One-third of all protein targets under investigation in the pharmaceutical industry are protein or PI3K kinases, although their potential has so far not been fully exploited (Fedorov et al., 2010). In summary, at present, a set of divergent protein and PI3Ks represent an important class of enzymes for treating human disorders.
Approved protein kinase inhibitors to date
Since the approval of fasudil in 1995, the number of approved kinase inhibitors has increased to 33 with many others still in preclinical development (Figure 1B and Table 1). More than 130 kinase inhibitors are reported to be in Phase-2/3 clinical trials (Vieth et al., 2005) (http://www.clinicaltrials.gov/) (http://chembl.blogspot.ch/2013/09/the-clinical-kinome-in-2013.html). It is beyond the scope of this review to discuss all the protein kinase inhibitors that are in preclinical or in early clinical development. It should be emphasized that all of the mentioned approved and clinically advanced kinase inhibitors (Phase-3) with a few exceptions, like the rapalogs and trametinib, are directed towards the ATP binding site and do not cover more than 20% of the whole kinome (Fedorov et al., 2010).
Most of the approved kinase drugs are active against more than one type of cancer. Only a few of them have been used for the treatment of non-oncological indications, namely tofacitinib for rheumatoid arthritis, sirolimus for organ rejection, fasudil for cerebral vasospasm and more recently nintedanib for idiopathic pulmonary fibrosis (Table 1, http://www.discoverx.com/tools-resources/interaction-maps). In contrast, there are numerous kinase drugs for one single indication. For example, imatinib, nilotinib, dasatinib, bosutinib and ponatinib have all been approved for CML, whereas sorafenib, sunitinib, everolimus, temsirolimus, axitinib or pazopanib are indicated for various stages of renal cell cancer. Ceritinib, crizotinib and alectinib are used for the treatment of non-small-cell lung cancer (NSCLC) with anaplastic lymphoma kinase (ALK) translocations, while gefitinib, erlotinib and afatinib are indicated for NSCLC with activated EGFR. Vandetanib, cabozantinib and levantinib are used for the treatment for medullary thyroid carcinoma, while imatinib, sunitinib and regorafenib are indicated also for GIST. Finally, vemurafenib or dabrafenib in combination with trametinib is indicated for metastatic melanoma with BRAFV600 mutations (Table 1).
The conservation of the ATP binding site in the human kinome often causes these ‘ATP-mimetics’ to cross-react with many other different kinases, resulting in compounds with promiscuous profiles. Promiscuous compounds like, for example, dasatinib (Lombardo et al., 2004) or sunitinib (Motzer et al., 2006; Faivre et al., 2007) have been termed multi-kinase inhibitors but have some toxicological liabilities (Cheng and Force, 2010). In contrast, kinase inhibitors targeting the ATP site, such as lapatinib, tofacitinib or imatinib, are reasonably selective (http://www.discoverx.com/tools-resources/interaction-maps).
These ATP-site-directed inhibitors may be viewed as first generation, as they have demonstrated appropriate selectivity, potency and pharmacokinetic (PK) properties. However, the usually poor physicochemical properties, the limited selectivity and the relatively restricted ATP pharmacophore with an extensive coverage of chemo-types remain as the main challenges for kinase drug discovery (Traxler et al., 2001; Cowan-Jacob, 2006; Engelman, 2009; Zhang et al., 2009; Fabbro et al., 2011; Liu et al., 2013). We are just beginning to have a molecular and structural understanding of the regulation of the kinase activity, both at the level of the kinase domain as well as at the level of the full-length protein kinases. There is now increasing interest in identifying inhibitors that do not compete with ATP. Kinase inhibitors with outstanding selectivity are likely to become important not only for minimizing side effects and allowing chronic treatment of non-life-threatening diseases, but also to better understand the on- and off-target pharmacology of kinase inhibitors (Robert et al., 2005; Force et al., 2007; Fabbro et al., 2011; Moebitz and Fabbro, 2012; Cowan-Jacob et al., 2014).
While the mutational status of kinases may be associated with various cancer conditions, the identification and validation of the driver kinase(s) in these diseases by genome-wide screening for kinase amplifications, translocations and/or mutations as well as studying the multiple mechanisms of resistance is an area of intense research to improve the efficacy of these targeted therapies (Hunter, 2000; Blume-Jensen and Hunter, 2001; Cohen, 2002b; Weinstein, 2002; Bardelli et al., 2003; Sawyers, 2004; Vieth et al., 2004; Takano et al., 2005; Ventura and Nebreda, 2006; Wolf-Yadlin et al., 2006; Ali and Ali, 2007; Engelman et al., 2007; Greenman et al., 2007; Thomas et al., 2007; Luo et al., 2009; Stransky et al., 2014).
In conclusion, the actual landscape of kinase inhibitor drugs developed over the last two decades shows that
only a small number of protein and lipid kinase targets (about 80) out of the 500+ protein kinases in the human kinome have been successfully targeted
most of the kinase inhibitor drugs are used for oncological indications
many kinase inhibitor drugs are used to target the same indication (mainly due to the generation of resistance)
The structure and catalytic mechanisms of ePKs
The ePK protein kinase domain has evolved to have many different regulatory mechanisms and is often associated with a large variety of other protein domains that directly or indirectly contribute to the regulation of the kinase activity (Nolen et al., 2004; Cowan-Jacob et al., 2009; 2014,; Scott and Pawson, 2009; Zhang et al., 2009; Taylor and Kornev, 2011; Jin and Pawson, 2012). The ePKs and ELKs share the PKL-fold and similar catalytic mechanisms, although ELKs generally display very low sequence identity with ePKs and with each other (Kannan et al., 2007). The overall structural organization of the ca. 300 residue protein kinase domain is conserved with 10 key residues mediating the core functions of the catalytic domain (Hanks et al., 1988; Manning et al., 2002; Kannan et al., 2007). All the other structural elements outside the kinase domains typically serve either as regulatory or as targeting modules (Scott and Pawson, 2009; Jin and Pawson, 2012).
All protein kinase domains consist of a small, mostly β-stranded N-lobe, connected by a short hinge region to a larger α-helical C-lobe (Figure 3). ePKs bind the ATP in the cleft between the N- and C-terminal lobes of the kinase domain where the adenine group of ATP is sandwiched between hydrophobic residues and makes contact via hydrogen bonds to the hinge region (Figure 3A and B) (Nolen et al., 2004; Taylor and Kornev, 2011; Cowan-Jacob et al., 2014).
Figure 3.

The active conformation of protein kinases. Front (A) and side views (B) of a typical active kinase conformation displaying the ternary complex of the insulin receptor (InsR), ATP and peptide substrate (pdb 1ir3). The helices and β-sheets forming the canonical kinase fold are labelled, as well as important secondary structure elements which are shown colour coded. (C) The ATP-site pharmacophore: The hydrophobic channel, the sugar pocket, the hinge and the hydrophobic back-pocket are the major pharmacophores. (D) Close-up of the active ATP site of the InsR. For explanations, see text. The two Mg2+ ions are in magenta.
The N-lobe contains a five-stranded β-sheet (β1–β5) with a single α-helix (the C-helix, αC). The Gly-rich loop (also known as P-loop ort G-loop) lies between the β1 and β2 strands and contains an important hydrophobic residue at its tip, which contributes to coordination of the phosphates of ATP (Figure 3) (Nolen et al., 2004; Cowan-Jacob, 2006; Taylor and Kornev, 2011). This is the most flexible part of the N-lobe, which folds over the nucleotide positioning the γ-phosphate of ATP for catalysis. The C-terminus of the C-helix is anchored to the core of the C-lobe by the β4-loop via the β5 strand, which continues into the hinge region, whereas its N-terminus interfaces with the activation loop (also called activation segment or A-loop). The A-loop occurs either in an open (the hallmark for the active ATP-bound state of the kinase) or various closed conformations, indicating the inactive state of the kinase by occluding the access of the protein substrate sites (Figure 3) (Nolen et al., 2004; Cowan-Jacob, 2006). The N-terminus of the C-helix has to be positioned correctly for efficient catalysis facilitating the interaction between the active site Lys (of the AXK-motif in the β3-strand) and the Glu from the C-helix (‘C-helix-in’). Rotating the N-terminus of the C-helix in a suboptimal position for catalysis (‘C-helix-out’) results in an inactive state of the kinase (Cowan-Jacob, 2006; Kannan et al., 2007; Taylor and Kornev, 2011; Moebitz and Fabbro, 2012). N-terminal to the hinge, deep in the ATP pocket, is an important residue called the ‘gatekeeper’, which controls the access to the ‘back-pocket’ of the kinase and which is often mutated in kinases resistant to inhibitors (Figure 3C) (Nolen et al., 2004; Kornev et al., 2006; Cowan-Jacob et al., 2009; Taylor and Kornev, 2011; Moebitz and Fabbro, 2012).
The larger lobe or C-terminal lobe of the kinase domain is mostly helical. There are four β-strands in the active state: β6 and β7 contain the catalytic loop with most of the catalytic machinery (Y/HRD or Tyr/His-Arg-Asp), whereas β8 and β9 flank the DFG-motif where the Asp recognizes one of the ATP-bound Mg2+. The Phe of the DFG-motif (Aspartate-Glycine-Phenylalanine or Asp-Gly-Phe) makes hydrophobic contacts with the C-helix and the nearby Y/HRD-motif from the catalytic loop (Figure 3). The Asp of the Y/HRD, one of the most conserved residues (present in all ePKs), is responsible for correct orientation of the P-site hydroxyl acceptor group in the peptide substrate. Similarly, the Tyr/His in Y/HRD is conserved throughout all ePKs and ELKs and serves as a central scaffold for binding both to the carbonyl group of Asp and making a hydrophobic contact to the Phe of the DFG-motif. The Mg2+-binding loop, which is followed by the β9 strand, forms an antiparallel β-sheet with the β6 strand that precedes the catalytic loop (Y/HRD-motif). This portion of the sheet is disordered in the inactive kinases and is believed to be important for the correct Mg2+-binding loop configuration. The A-loop, which includes the β9 strand, extends from the DFG-motif (at the very N-terminus of the A-loop) to a conserved Asp at the beginning of the F-helix. The Phe of the DFG-motif is responsible for proper positioning of the Asp and accommodation of the C-helix facilitating the Lys–Glu salt bridge. The flexible A-loop regulates the on and off state of the kinase by providing the platform together with the helical subdomains of the C-lobe for binding and positioning of the hydroxyl group residue of the peptide substrate (Nolen et al., 2004; Ubersax and Ferrell, 2007). The extended helical element that follows the F-helix is unique to the ePKs and includes the G-helix through the I-helix (GHI domain). Many substrate proteins and regulatory proteins are tethered to the GHI domain (Figure 3A) (Taylor and Kornev, 2011).
In summary, three sequence motifs are essential for catalysis (Hanks and Hunter, 1995; Cowan-Jacob, 2006; Taylor and Kornev, 2011):
The AXK-motif (β3 strand) with the active site Lys forming a salt bridge with the conserved Glu from the C-helix, which interact with the α and β phosphates of ATP to anchor and orient the ATP
The Y/HRD-motif or catalytic loop (β6/β7), in which the Asp is the catalytic residue functioning as a base acceptor for the proton transfer
The DFG-motif of the A-loop where the Asp binds the Mg2+ ions that coordinate the β and γ phosphates of ATP in the ATP binding cleft positioning the latter for the phosphate transfer
Activation of protein kinases results in the re-orientation of the C-helix (‘C-helix-in’) to bring the conserved Glu into the proximity of the active site Lys of the AXK-motif as well as the A-loop. In many instances, the Phe of DFG moves from the DFG-out (an inactive conformation) into the DFG-in position (usually an active conformation) (Figure 2). The extended β-sheet conformation of the G-loop helps coordinating the phosphates of ATP, whereas the β6 strand forms part of the catalytic loop that facilitates the phosphor transfer. The catalytic loop (Y/HRD-motif) is the only conserved element that does not differ between the active and inactive states of the protein kinase. The short EF-helix at the end of the A-loop with the conserved Glu of the APE-motif forms the peptide substrate binding site. The A-loop in the active conformation can be stabilized by phosphorylation or interactions with accessory regulatory proteins (Figure 2) (Nolen et al., 2004; Cowan-Jacob et al., 2009).
The regulation of the catalytic mechanism of protein kinases may further involve a regulatory (R) and a catalytic (C) spines, which are each built up by two conserved residues from and the N- and C-lobes respectively. The R-spine is formed by four hydrophobic residues, one from the β4 strand, one from the C-helix, the Phe from the DFG and the Tyr/His from the catalytic loop. Proper alignment of these hydrophobic residues results in the formation of the R-spine linking the N- and C-lobes for optimal protein kinase activity. The C-spine comprises two residues from both lobes and is completed by the adenine ring of ATP. The Val in the β2 strand and the Ala from the AXK-motif of the β3 strand are docked directly onto the adenine ring of ATP. The hydrophobic residue lies in the middle of β7 strand of the C-lobe and which, on the one hand, docks directly onto the adenine ring and, on the other hand, rests on a hydrophobic residue from the D-helix, which, in turn, is bound to the F-helix (Kornev et al., 2006; Taylor and Kornev, 2011).
The different modes to inhibit kinases
Low-molecular-weight kinase inhibitors can bind either covalently or reversibly to kinases (Zhang et al., 2009; Liu et al., 2013; Cowan-Jacob et al., 2014).
Covalent inhibitors
Covalent inhibitors usually have a binding, a linker and a warhead module that can bind in or close to the ATP binding sites. Depending upon the reactivity of the warhead, the covalent binding can be reversible (Wymann et al., 1996; Liu et al., 2013). It should be mentioned that drugs that bind covalently to their targets have always been perceived as being potentially toxic. However, it should be emphasized that many marketed drugs bind covalently to their targets (Singh et al. 2011). Covalent kinase inhibitors usually target the active site Lys or a Cys in or around the ATP binding site (Figure 4D) (Wymann et al., 1996; Rabindran et al., 2004; Kwak et al., 2005; Zhou et al., 2009). Various covalent kinase inhibitors have been identified for various protein kinases, including Fes (Feline sarcoma oncogene kinase) (Filippakopoulos et al., 2008), VEGFR-2 (Wissner et al., 2007), ribosomal S6 kinase (Cohen et al., 2007) and Bruton tyrosine kinase (BTK) (Pan et al., 2007). Some of them have progressed into the clinic like AVL-292, an orally available, selective covalent inhibitor of BTK that is currently undergoing Phase-2 clinical trials for chronic lymphocytic leukaemia and non-Hodgkin lymphoma (Robak and Robak, 2012). Other covalent inhibitors like ibrutinib, targeting BTK, and afatinib, targeting the gefitinib-resistant EGFR, have been recently approved by the Food and Drug Administration (Figure 4D and Table 1) (Minkovsky and Berezov, 2008; Ninomiya et al., 2013; Akinleye et al., 2014). Although these covalent kinase inhibitors have shown impressive clinical results, it should be mentioned that mutation of the Cys, which is not essential for the kinase structure and activity, can prevent the covalent binding and their clinical efficacy (Furman et al., 2014).
Figure 4.
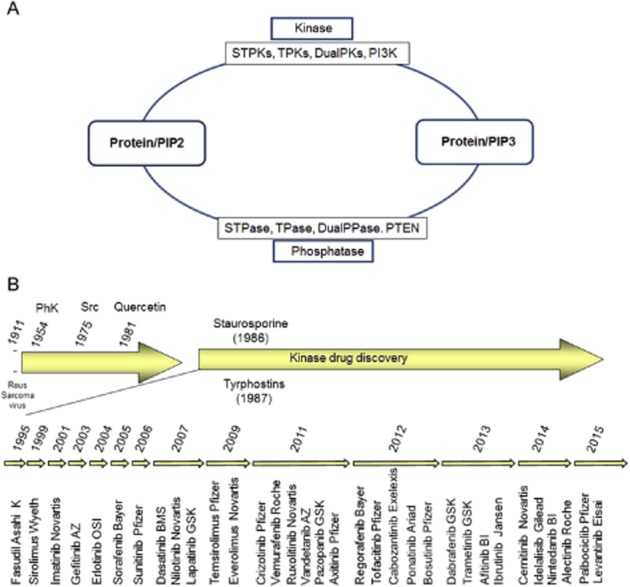
Representative binding modes of the four classes of kinase inhibitors. Representative binding modes of the four classes of kinase inhibitors with ligand in blue sticks and key polar interactions shown as red dotted lines: (A) gefitinib bound to EGFR (type 1, pdb 2ity); (B) vemurafenib bound to B-Raf (type 1.5, pdb 3og7); (C) imatinib bound to ABL (type 2, pdb 1iep); and (D) afatinib bound covalently to EGFR (pdb 4g5j). Activation states of helix-C and the DFG-motif are annotated.
Non-covalent inhibitors
The non-covalent kinase inhibitors can be further classified into those that either bind or do not bind to the hinge region of the kinase (Figure 3C), leading to the classification of type-1, type-2 and type-3 reversible kinase inhibitors (Traxler et al., 2001; Li et al., 2004; Liu and Gray, 2006; Cowan-Jacob et al., 2009; Zhang et al., 2009; Moebitz and Fabbro, 2012).
Type-1 and type-1.5 inhibitors
The vast majority of the ATP-competitive inhibitors bind to active conformations with the conserved Phe residue of the DFG-motif buried in a hydrophobic pocket in the groove between the two lobes of the kinase (Figures 3 and 4A and B) (Pargellis et al., 2002; Li et al., 2004; Liu and Gray, 2006; Cowan-Jacob et al., 2009; Zhang et al., 2009; Moebitz and Fabbro, 2012). The ATP binding site of active protein kinases (and PI3Ks) is very similar, despite the fact that they have different substrate specificities and different modes of regulation. In the active conformation, the A-loop adopts an open conformation typical for the ATP-bound state of the kinase where the Asp in the DFG-motif coordinates the phosphates of ATP, whereas the Phe stabilizes the C-helix and the A-loop for catalysis (Figure 3D) (Nolen et al., 2004; Cowan-Jacob, 2006; Liu and Gray, 2006; Zhang et al., 2009; Cowan-Jacob et al., 2014). Type-1 inhibitors utilize variation in the size, shape and polarity of the gatekeeper residue to gain selectivity (Figure 3C). Finding compounds that target the active conformation of the kinase by ATP mimetics is best achieved using enzymatic kinase assays displaying the highest level of activity. Classical examples for this type of approved kinase inhibitor class are gefitinib, erlotinib, dasatinib and sunitinib (Table 1). The type-1.5 inhibitor, exemplified by vemurafenib, is a subtype of the type-1 inhibitor that binds to an inactive kinase conformation (Figure 4B) (Tsai et al., 2008; Zuccotto et al., 2010). In this case, the BRAF adopts a DFG-in conformation, typical of an active kinase, but with the C-helix being pushed out (‘C-helix-out’) by vemurafenib effectively disrupting the ion pairing between the active site Lys and the Glu from the C-helix (Tsai et al., 2008). This type-1.5 inhibitor with a ‘DFG-in’ inactive conformation has also been observed in other kinases (Figure 2A and C) (Cowan-Jacob et al., 2014).
Selective type-1 or type-1.5 inhibitors use additional sites close to the ATP binding site, like the adjacent hydrophobic pockets (Figure 3C) whose entry is regulated by the gatekeeper (Zuccotto et al., 2010), or additional sites close to the peptide binding site, like the bivalent/bitopic inhibitors (Hill et al., 2012), the macrocycles (Tao et al., 2007) or some of the covalent inhibitors (Liu et al., 2013). The success of type-1 inhibitors in the clinic demonstrates that, despite the highly conserved ATP binding site, it is feasible to optimize selectivity for kinases by following appropriate strategies, which is reflected in the fact they represent the vast majority of the kinase inhibitors. The most rational way to obtain selectivity is by targeting poorly conserved residues, particularly residues flanking the hinge. Although typically discovered by serendipity rather than rational design, the interplay of sequence and conformational penalty can lead to exceptional selectivity. The balance between sequence and conformational contributions comes in different flavours. In one extreme, optimization of the compound leads from an active to an inactive, high-energy conformation of the kinase such that the additional interactions make up for the conformational penalty, but only on the target kinase. Examples are vemurafenib (‘C-helix-out’ and ‘DFG-in’, type-1.5 inhibitor) versus the relatively unselective type-1 inhibitors gefitinib (Figure 4A and B) (Wood et al., 2004). Another example is the MET (mesenchymal epithelial transition factor or hepatocyte growth or scatter factor receptor) kinase whose native, inactive conformation offers the potential for a unique, crucial stacking interaction with a Tyr in the A-loop where several unique sequence features play together to stabilize an otherwise high-energy conformation (Figure 2C) (Albrecht et al., 2008).
The ability of the G-loop of kinases to partially collapse onto the ligand, thus creating a more buried, less solvent-exposed cavity with high intrinsic ligand efficiency, is another element for selectivity like in ABL (Figure 5B), which include other kinases that show high intrinsic ligand efficiency like the Aurora kinase and GSK3β (glycogen synthase kinase-3β). Lastly, there are cases of excellent selectivity, which arise from a multitude of subtle interactions as exemplified by the JAK inhibitor CP-690,550, which, again, builds upon a standard, pan-kinase-inhibitor-pyrrolo-pyrimidine scaffold (West, 2009; Williams et al., 2009) (http://www.discoverx.com/tools-resources/interaction-maps).
Figure 5.
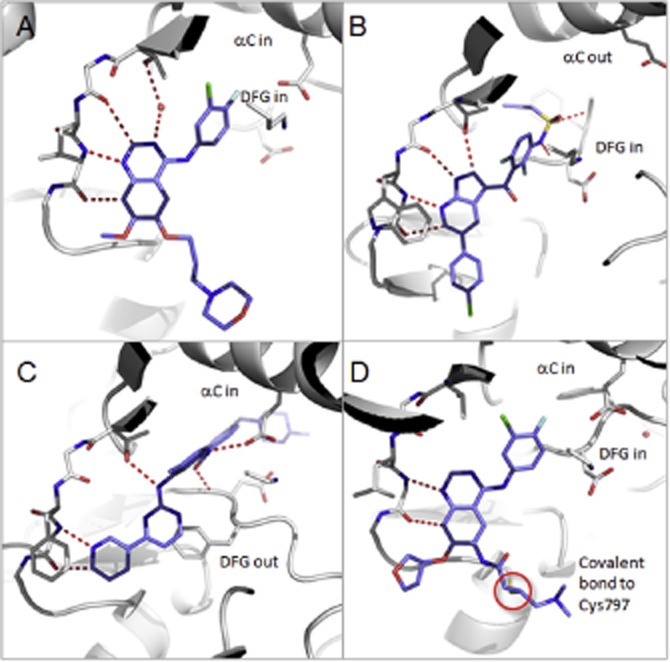
Clinically relevant resistance mutations of MEK1, ABL1 and ALK. (A) MEK1 and MEK2 mapped onto pdb 3eqc. The majority of the mutations cluster at the interface with the autoinhibitory N-terminal helix (top left in this view). (B) ABL mapped onto pdb 1iep. The imatinib-resistant mutants are spread all over the kinase domain; however, the most resistant against imatinib is the gatekeeper mutant T315I in the hinge (green) and the mutations located in the P-loop (red). (C) ALK mapped onto pdb 2xp2. Crizotinib-resistant mutations. The most resistant mutation is L1196M in the hinge (green) region.
The type-2 inhibitors
The type-2 kinase inhibitors preferentially bind to the inactive conformation of the protein kinase and still have contact with the hinge (Liu and Gray, 2006; Cowan-Jacob et al., 2009; Zhang et al., 2009). They usually score as ATP-competitive and bind to the inactive, the so-called ‘DFG-out’, conformation (Figure 4C) (Nolen et al., 2004; Cowan-Jacob, 2006; Liu and Gray, 2006; Zhang et al., 2009). The transition from the ‘DFG-in’ to the ‘DFG-out’ conformation exposes an additional hydrophobic pocket adjacent to the ATP site that is utilized by type-2 inhibitors locking the kinase in the inactive conformation (Nolen et al., 2004; Cowan-Jacob, 2006; Liu and Gray, 2006; Zhang et al., 2009; Cowan-Jacob et al., 2014).
Type-2 inhibitors are in general less promiscuous than type-1 inhibitors as revealed by several selectivity profiles. Although some type-1 inhibitors can be very specific, there are also examples of type-2 inhibitors that are rather promiscuous (Goldstein et al., 2008; Karaman et al., 2008; Anastassiadis et al., 2011). Approved kinase inhibitors binding to or stabilizing the ‘DFG-out’ conformations are imatinib, nilotinib or sorafenib (Table 1).
In addition to the DFG-out, combinations of different conformational states of C-helix, the A- and/or the P-loop can generate various inactive conformations of the kinase domain (Cowan-Jacob, 2006; Cowan-Jacob et al., 2014). Each individual kinase has a preferred inactive conformation, depending upon its phosphorylation state and regulatory mechanisms involving structures outside the kinase domain (Cowan-Jacob, 2006; Cowan-Jacob et al., 2014).
Another interesting mechanism of inhibition that requires an ATP-site-directed kinase inhibitor, irrespective of its type, concerns the interactions of the molecular chaperone HSP90–CDC37 system and mutated versions of kinases. CDC37 appears to inhibit the binding of ATP to the kinases they regulate. This interaction can be inhibited with ATP-site-directed kinase inhibitors, thereby destabilizing the influence of the HSP90–CDC37 chaperone system on the kinase, resulting in the destruction of the kinase through an HSP90-independent degradation pathway (Polier et al., 2013).
Type-3 (allosteric) inhibitors
The type-3 inhibitors are a heterogeneous group of kinase inhibitors that bind to allosteric or remote sites on the kinase and include, for example, inhibitors targeting MEK1 (mitogen activated kinase kinase-1), CHEK1 (checkpoint kinase-1), ABL, FAK (focal adhesion kinase) or Akt (protein kinase B or kinase from the transforming oncogene AKT8) (Figure 6) (Ohren et al., 2004; Barnett et al., 2005; Lindsley et al., 2005; Adrian et al., 2006; Converso et al., 2009; Vanderpool et al., 2009; Wang and Sun, 2009). The type-3 inhibitors are non-ATP site (allosteric) kinase inhibitors that have no physical contact with the hinge and show the highest degree of selectivity by exploiting binding sites and regulatory mechanisms that are unique to a particular kinase (McIntyre et al., 2003; Ohren et al., 2004; Barnett et al., 2005; Lindsley et al., 2005; Adrian et al., 2006; Converso et al., 2009; Cowan-Jacob et al., 2009; Vanderpool et al., 2009; Wang and Sun, 2009; Zhang et al., 2009; Fabbro et al., 2012). While most type-3 inhibitors are non-ATP-competitive or ATP-uncompetitive, some compete with ATP indirectly by binding to mutually exclusive conformations. The non-catalytic roles of kinases involve unique non-conserved interactions and increase the target space on the kinome (Rauch et al., 2011; Cowan-Jacob et al., 2014). In addition to the ‘DFG-in’ and ‘DFG-out’ combinations of different states of the C-helix, the A-loop and/or the G-loop can generate various inactive conformations of the kinase domain. Moreover, elements outside the kinase domain like the juxta-membrane region of the receptor PTKs or other N- or C-terminal elements, linkers and/or other regulatory domains required for protein–protein interactions are all important elements in the regulation of the catalytic domain (Cowan-Jacob et al., 2009; Zhang et al., 2009; Fabbro et al., 2012; Moebitz and Fabbro, 2012). The unique combinations of all these structural elements create a structural diversity that can be used to design selective inhibitors with clear advantages over the regular type-1 and type-2 ATP site inhibitors. This includes improved selectivity and slower off-rates, which increase the residence time of the inhibitor bound to the kinase (Wood et al., 2004; Tummino and Copeland, 2008). However, the paucity of available structures for the inactive protein kinase (apo-form), along with the lack of a general method to assay for such inhibitors, represents a major difficulty in identifying inhibitors targeting the inactive conformations. Type-3 inhibitors can bind either to the kinase domain (close to or removed from the ATP site) or to sites that are located outside the kinase domain.
Figure 6.
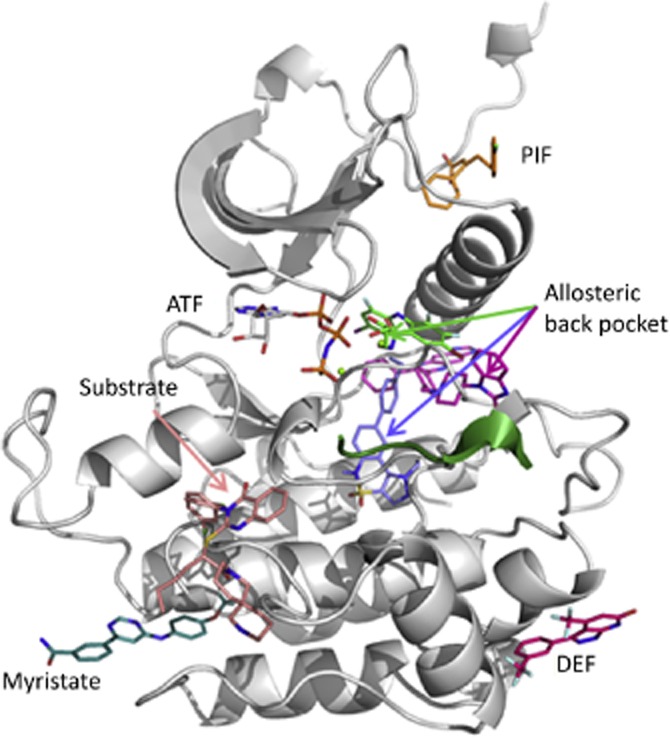
Allosteric pockets. Examples of allosteric ligands mapped onto an active kinase conformation, comprising the myristate site of ABL (pdb 3k5v), the PIF pocket in PDK1 (3hrf), the substrate site in CHK1 (pdb 3f9n), the DEF (docking site for ERK) site in p38 (pdb 3new) and the allosteric back-pocket in MEK1 (DFG-in, pdb 1s9j, green), Akt1 (DFG-out, pdb 3o96, magenta) and FAK (DFG-out, pdb 4ebw, blue).
The type-3 inhibitors include very diverse compounds ranging from the MEK1 inhibitors to rapamycin derivatives. For example, the allosteric type-3 inhibitors of MEK1 bind to a pocket adjacent to the ATP binding site, referred to as ‘allosteric back-pocket’ (Ohren et al., 2004), in the presence of ATP and are referred to as ‘allosteric back-pocket-DFG-in’ inhibitors (Figure 6). Other type-3 inhibitors bind to the ‘allosteric back-pocket’ in the absence of ATP in the ‘DFG-out’ conformation like the IGF1R (Heinrich et al., 2010), FAK (Tomita et al., 2013) or p38 (Over et al., 2013) and are referred to as ‘allosteric back-pocket-DFG-out’ inhibitors (Figure 6). In the case of IGF1R, the inhibitor binds the ‘allosteric back-pocket’ and extends over towards the substrate binding site and the catalytic loop. In contrast, the type-3 FAK inhibitors extend from the ‘allosteric back-pocket-DFG-out’ into a pocket formed in the C-lobe by displacing the normally rigid catalytic loop (Tomita et al., 2013). The allosteric Akt inhibitors are a special case of the ‘allosteric back-pocket-DFG-out’ as they only bind to this site when the pleckstrin homology domain of Akt is present (Figure 6). Therefore, their identification required the full-length protein for the kinase assay (Barnett et al., 2005; Lindsley et al., 2005). While lack of competition with ATP has, in some cases, proven to be a useful way to identify type-3 inhibitors, it should be pointed out that the allosteric back-pocket DFG-out inhibitors will score as ATP-competitive.
Type-3 inhibitors that are further away from the ATP site are, for example, the ABL myristate-pocket (Myr-pocket) binders (Adrian et al., 2006; Zhang et al., 2009; Fabbro et al., 2010), the CHEK1 inhibitors occupying part of the substrate binding site (Converso et al., 2009) and the Jun kinase-1 (JNK1) inhibitors, which bind in part to the MAPK insert region and A-loop (Comess et al., 2011) or to the DEF (docking site for ERK) domain (Tzarum et al., 2013) to only cite a few (Figure 6). A more comprehensive review on the type-3 inhibitors has been recently assembled by Cowan-Jacob et al. (2014).
Rapamycin and its derivatives (rapalogs), which target specifically mTOR kinase in the context of the mTORC1 complex, appear to be further removed from the kinase domain as they seem to act in the context of the mTORC1 complex (Wang and Sun, 2009; Yang et al., 2013). Targeting the extracellular domains of the receptor TPKs and others by peptide-mimetics, ‘peptoids’ or antibodies is another special case of type-3 inhibitors (Fleishman et al., 2002; Udugamasooriya et al., 2008; Cazorla et al., 2010; Jura et al., 2011; Christopoulos et al., 2014). The extracellular domains of RTPKs can be targeted by monoclonal antibodies trastuzumab (Herceptin, Roche, Basel, Switzerland) and pertuzumab (Perjeta, Roche, Basel, Switzerland), which act at different domains with trastuzumab binding to domain IV and pertuzumab to subdomain II of the extracellular segments of the HER2 (neu) receptor respectively (Cho et al., 2003; Hynes and Lane, 2005; Hsieh and Moasser, 2007). In contrast, small molecules such as SSR128129E, which target the extracellular D2D3 domains of the fibroblast growth factor receptor (FGFR), modulate signalling of the FGFR-RTKs (Bono et al., 2013; Herbert et al., 2013). Examples of approved type-3 inhibitors are trametinib and the rapamycins (Table 1). There are many potential topographically distinct binding sites on kinases between which allosteric interactions can occur; the point from which the interaction is viewed drives classification of interacting ligands.
Summary of binding modes
While it is undisputed that type-3 inhibitors display the highest degree of selectivity, the implication that type-2 inhibitors are generally more selective than type-1 lacks quantitative arguments. In fact, there are examples of exquisitely selective inhibitors known for both types. Selectivity rests on particular features of a particular protein kinase, regardless of the binding mode of the inhibitor. In contrast to type-1 and type-2 inhibitors, the high level of selectivity of type-3 inhibitors reflects the unique binding sites and is off-set by the difficulty in obtaining and optimizing chemical matter (Converso et al., 2009; Vanderpool et al., 2009). So far, there are only sparse hints that allosteric sites can be generalized and chemical matter transferred to other kinases (Tecle et al., 2009). There is currently no general strategy for the identification of allosteric kinase inhibitors or activators as most of them have been discovered serendipitously by diverse approaches ranging from phenotypic screening to sophisticated structure-based drug design.
Activators, paradoxical activation and priming
In addition, targeting allosteric sites on protein kinases may provide a means also to identify activators rather than inhibitors, which could be useful for therapeutic intervention as is the case for the glucokinase and the AMP-dependent protein kinase (AMPK) (Guertin and Grimsby, 2006; Sanders et al., 2007). This can be useful for therapeutic intervention or as a pharmacological tool to better understand the biology of the protein or lipid kinase.
In particular, compounds targeting the PIF (PDK1 interacting fragment) pocket (the hydrophobic motif present in the N-terminal lobe of the AGC kinases) of either PDK1 (3-phosphoinositide-dependent protein kinase-1) or PKCζ can either act as activators (Hindie et al., 2009) or as substrate selective inhibitors (Figure 6) (Lopez-Garcia et al., 2011; Sadowsky et al., 2011; Busschots et al., 2012). Similarly, the Myr-pocket binders of ABL can be converted into activators if they are designed not to allow bending of the I-helix of the ABL kinase domain (Jahnke et al., 2010; Yang et al., 2011).
However, there are a few protein kinases that require activation rather than inhibition to fulfil their therapeutic need, like the AMPK or the insulin receptor for which activators have been identified (Li et al., 2001; Pender et al., 2002; Sanders et al., 2007; Lee et al., 2011; Salt and Palmer, 2012; Hardie, 2014). PKC activation by exogenous compounds can have tumour-promoting or tumour-suppressing effects by acting via the DAG binding site (Martiny-Baron and Fabbro, 2007). These include phorbol esters, bryostatin and other compounds acting as DAG mimetic (Martiny-Baron and Fabbro, 2007). Other examples of kinase activators include a mimetic of the brain-derived neurotrophic factor that activates TrkB [tropomyosin receptor kinase B or neurotrophin receptor kinase-2 (NTRK-2)] (Massa et al., 2010).
In some cases, kinase inhibitors can lead to unintended paradoxical activation either directly or via modulation of feedback loops. Evolution has endowed the signalling cascades of kinases with a high degree of robustness, which is achieved through redundancy at various levels, like compensatory pathways or protein expression, counteracting phosphatases and feedback loops. Therapeutic inhibition of kinases struggles with this innate inertia of kinase signalling. The most striking example is the paradoxical activation of selective BRAF inhibitors, which can activate the MAPK pathway in certain genetic backgrounds (Hall-Jackson et al., 1999). This phenomenon is linked to a complex regulation of BRAF and cRAF due to cross-activation of the wild-type (wt) rapidly accelerated fibrosarcoma (RAF) isoforms, which is just beginning to be understood, almost a decade after the first so-called RAF inhibitor sorafenib was approved (Hall-Jackson et al., 1999; Hatzivassiliou et al., 2010; Poulikakos et al., 2010; Holderfield et al., 2013).
Another phenomenon is priming, which can lead to activation via kinase inhibitors and which has been observed for several kinases such as Akt, MEK and JAK (Okuzumi et al., 2009; Andraos et al., 2012; Hatzivassiliou et al., 2013; Holderfield et al., 2013). Priming describes the up-regulation of the phosphorylated form of the targeted kinase upon inhibition, which can lead to the activation of the pathway once the inhibitor is removed. Priming depends upon the mode of action of the kinase inhibitor. Inhibitors binding to the active conformation of Akt cause priming, whereas allosteric inhibitors targeting the inactive conformation of Akt do not (Lin et al., 2012). This distinction was shown to depend upon the accessibility of the complex to its phosphatase PP2A (protein Ser/Thr-specific phosphatase-2). A broader understanding of priming and its impact on the efficacy of kinase inhibition is in its infancy, but there are reports that it could contribute to the lack of efficacy for certain inhibitors.
Another way by which kinase inhibitors influence the target kinase is by stabilizing and increasing its expression. In the case of lapatinib, this leads to the expected inhibition of HER2, which at the same time resulted in an accumulation of HER2 due to decreased degradation, which leads to enhanced trastuzumab-dependent cytotoxicity (Scaltriti et al., 2009). This is mainly due to the inhibition of dimerization, which is not common to all EGFR inhibitors (Sanchez-Martin and Pandiella, 2012).
Methods for discovering and profiling kinase inhibitors
In vitro biochemical and cellular assays followed by in vivo efficacy are the traditional pillars for drug discovery approaches (Knight et al., 2013). The currently available technologies for the discovery and profiling of kinase-based drugs are numerous and it should be emphasized that the assessment of biochemical kinome-wide selectivity has only become available recently. There are a variety of biochemical protein kinase assays, including detection of radiolabelled transfer of phosphate to the substrate, ATP consumption or ADP production measurement, time-resolved FRET, peptide array-based, microfluidic technologies and label-free analysis (biophysical methods such as isothermal titration calorimetry and differential scanning fluorometry) (Jia et al., 2008; Ma et al., 2008). Among the various providers that offer a kinase selectivity panel are KinomeScan™ of DiscoverX (http://www.discoverx.com/targets/kinase-target-biology), Millipore's Kinase Profiler (http://157.93.252.5/life_sciences/flx4/ld_kinaseprofiler_service) and Reaction Biology (http://www.reactionbiology.com/webapps/site/), which, to date, have the broadest kinome coverage (Karaman et al., 2008; Anastassiadis et al., 2011; Davis et al., 2011; Gao et al., 2013). For the assessment of biochemical selectivity, of course, the various formats of the various assays may change the overall selectivity. Although the biochemical assays of the above three providers are quite different, the IC50 of a series of kinase inhibitors have been shown to be in reasonable agreement. Besides the caveats regarding assay formats, correlation to cellular selectivity, activation state of the recombinant kinase and the usually poor physicochemical properties of most inhibitors requires cautious interpretation of results. Ideally, the biochemical selectivity should match the cellular selectivity, which, to date, cannot be achieved with the same coverage as with the biochemical profile (Knight and Shokat, 2005; Knight et al., 2013). Similar to the biochemical assay, the cellular assays for screening and profiling of kinase inhibitors come in different formats. Target profiler assays detect kinase proximal substrates by methods such as Western blots, phospho-elisas, reverse phase arrays, ALPHA (amplified luminescent proximity homogeneous assay)-screen assay and high content cellular analysis, and are being offered by various providers (Chen et al., 2005; Warmuth et al., 2007; Eglen et al., 2008; Jia et al., 2008). In addition, engineered cellular assays, such as BaF3, reporter gene assays, cell encyclopaedias and others, are particularly suited to obtain an integrated readout of the signalling cascade (Melnick et al., 2006; Warmuth et al., 2007; Barretina et al., 2012). Cellular assay with a high kinome coverage may use biotinylated acyl phosphates of ATP and ADP that irreversibly react with protein kinases on the conserved active site lysine residues in the ATP binding pocket followed by quantitative mass spectrometry (http://www.kinativ.com/technology.html) (Patricelli et al., 2007; 2011,). Alternatively, chemical proteomics can also probe the effectiveness of kinase inhibitors in cells and tissues (Bantscheff et al., 2007). Finally, the function of the inhibition of the kinase target is being studied in specific tailor-made cell-based assays.
Taken together, the biochemical, cell-based phosphorylation and functional assays, most likely multiplexed, deliver a direct readout on the kinase activity in a cellular context and allow an in vitro activity of kinase inhibitors revealing on-target and off-target effects. Thus, the systematic profiling of inhibitors in broad arrays of biochemical and cellular assays has provided novel ways to better define the selectivity profile of drug candidates, including the potential for the discovery of novel mechanisms of actions. The most important contribution of profiling of compounds in large kinase panels is probably the cross-fertilization between protein kinase projects.
Major issues in kinase drug discovery
Kinase inhibitors are the prototypes of the targeted therapy and are therefore plagued by the fact that they are, like all targeted therapies, tailor-made to a particular patient population with the particular abnormal molecular or cellular defect. Targeted therapies are the cornerstone of precision medicine, which has improved the diagnostic, stratification and targeted treatment of patients as well as to better predict the outcome of the disease treatment (http://cancergenome.nih.gov/, https://www.broadinstitute.org/, https://www.sanger.ac.uk/) (Sellers, 2011; Garay and Gray, 2012; Plenge et al., 2013). Thus, successful targeting is ultimately assessed by producing selective pharmacological responses, which reduce or eliminate side effects that are not mechanism-related. The idea that molecular information improves the precision with which patients are categorized and treated has led to a fragmentation of the patient population most likely to respond to the target agents (Figure 6B). In contrast, the duration of responses to certain targeted therapies has been shown to be limited, resulting in a poor benefit for the treated patients (Engelman and Settleman, 2008a; Engelman, 2009; Corcoran et al., 2011; Chong and Janne, 2013). Therefore, correcting one molecular or cellular target by targeted therapies may be effective in diseases that strongly depend upon this one target. The reality of advanced cancers or other diseases are that they have multiple molecular abnormalities resulting in the potential for short-term efficacy (limited clinical benefit), which are usually associated with high costs (Pao and Hutchinson, 2012; Kantarjian et al., 2013) (Figure 7B). In addition, in many cases, targeted therapies require chronic treatment and therefore the dependency on drugs to maintain molecular and cellular changes for the balance of life (Druker et al., 2006).
Figure 7.

(A) Major issues in kinase drug discovery. (B) Pie chart showing the percentage distribution of clinically relevant driver mutations in lung adenocarcinoma [adapted from with high costs ‘Chipping away at the lung cancer genome’ by Pao and Hutchinson (2012)]. (C) Selectivity of selected approved protein kinase inhibitors as determined by the DiscoverX KinomeScan. The human kinome is represented as circular phylogenetic tree without the atypical protein kinases and results are reported as a map (Treespot), which allows visualizing compound interactions across the human kinome panel. AZD6244 (selumetinib) is an allosteric MEK inhibitor which displays the same selectivity as trametinib. Data are taken from http://www.discoverx.com/tools-resources/interaction-maps.
The degree of selectivity a protein kinase inhibitor should ideally have has been and remains a controversial issue. The ideal kinase inhibitor should inhibit only the target kinase, which is usually almost impossible to achieve in the face of the over 500 protein kinases of the human kinome. Nevertheless, protein kinase inhibitors with a lower degree of selectivity have been hailed as ideal for oncological indications due to their potential for poly-pharmacology (Figure 7C) (Knight and Shokat, 2005; Force et al., 2007; Morphy and Rankovic, 2007; Goldstein et al., 2008; Karaman et al., 2008; Anastassiadis et al., 2011; Davis et al., 2011; Gao et al., 2013). A look at clinically advanced kinase inhibitors reminds us that a promiscuous selectivity profile, such as that of sunitinib, may be tolerated in oncological settings, albeit with some side effects (Figure 7C) (http://www.discoverx.com/tools-resources/interaction-maps). However, even some of the most selective inhibitors, particularly the allosteric inhibitors for mTOR or MEK1, can have serious dose-limiting on-target toxicity (Chhajed et al., 2006; Akinleye et al., 2013). With the exception of the highly selective lapatinib, tofacitinib, the rapalogs and trametinib, most of the other ATP-site-directed protein kinase inhibitors marketed derive their efficacy, at least in part, from their poly-pharmacology (http://www.discoverx.com/tools-resources/interaction-maps) (Figure 7C). In any case, for pharmacological target validation as well as chronic administration of kinase inhibitors in non-oncological indications, a reasonable selectivity is a prerequisite (Knight and Shokat, 2005; Goldstein et al., 2008; Karaman et al., 2008; Anastassiadis et al., 2011; Davis et al., 2011; Gao et al., 2013).
Despite several successes over the past few years with kinase inhibitors, in most cases, the dependence of a disease state on the target kinase is either not known, poorly understood or displays a high degree of complexity, particularly in cancer. This often makes the selection of patients most likely to respond to a given kinase inhibitor treatment an almost impossible task (Fabbro et al., 2012). Ongoing efforts using genome-wide screening, analysis of driver mutations in conjunction with the use of sophisticated disease models will unravel new disease associations and will pave the way for the discovery of many more new protein kinase targets in the coming years (Sellers, 2011).
In addition, understanding and predicting the cross-reactivity of kinase inhibitors in conjunction with the knowledge about the disease dependency of the target kinase would allow a more rapid proof of concept in the clinic. As discussed before, the selectivity of kinase inhibitors remains controversial. Unfortunately, we still poorly understand the selectivity profile with respect to their liabilities regarding preclinical toxicity findings and their relevance in patients (Yang et al., 2010). The recent progress made in molecular profiling in conjunction with precision medicine will further our understanding towards a better assessment and prediction of efficacy/toxicity of these inhibitors in disease models [pharmacokinetic/pharmacodynamic (PK/PD)] and patients (Gray-Schopfer et al., 2007; Zhang et al., 2009; Courtney et al., 2010; Fabbro et al., 2012; London, 2013).
While we can expect more approvals for kinase inhibitors to come, the challenges of finding selective compounds with good physicochemical and PK properties remain and the intellectual property space is crowded.
Despite their central role in biology and their sizable potential as therapeutic targets, only a small fraction of the human protein kinases have been functionally annotated. In addition, we are short of selective small molecule kinase inhibitors to address unmet medical need in cancer, metabolism, inflammation and other diseases (Fedorov et al., 2010; Knapp et al., 2013). On the contrary, there are many ‘specific’ protein kinase inhibitors that cannot be used as drugs for reasons of toxicity or solubility but which are extremely useful as research reagents to better understand the cellular networking in normal and diseased tissues (Robert et al., 2005; Force et al., 2007). Using low-molecular-weight compounds offers considerable advantages in experimental demands and interpretation of results over RNA interference techniques and genetic knockout or knock-in models, which are limited by the kinetics of their effects and the inability to discriminate between scaffolding and catalytic roles of the target protein. Thus, selective chemical probes to functionally annotate, in particular, the untapped kinome could stimulate new drug discovery efforts to address unmet medical needs. Since the size of the human kinome combined with the high cost associated with probe generation severely limits access to new probes, potentially a large-scale public–private partnership may minimize redundancy and sharing of risk and cost (Knapp et al., 2013).
The major challenge for kinase drug discovery is not only to better understand the disease dependence of the target kinase but also to anticipate the emerging resistance to kinase inhibitors under treatment. Kinase inhibitors are being and have been designed to specifically target kinase alleles with GOFs (Blume-Jensen and Hunter, 2001; Fabbro and Garcia-Echeverria, 2002a). Despite these successes, it should be emphasized that patients most likely to benefit from these kinase inhibitors often relapse after an initial response. Thus, emergence of drug resistance is not limited to conventional chemotherapeutic drugs but extends to drugs with a targeted mode of action (Engelman and Settleman, 2008a).
Resistance to kinase inhibition
The mechanisms of multidrug resistance (MDR) to chemotherapeutic drugs have been studied and are not only limited to reduced drug accumulation but also involve changes in the level of target proteins, mutations which diminish drug binding, trapping of drugs in acidic vesicles, enhanced metabolism of drugs by cytochrome P450 (CYP) mixed function oxidases, increased tolerance of cellular DNA damage and diminished apoptotic signalling (Gottesman, 2002; Szakacs et al., 2006; Hall et al., 2009). Apart from the usual mechanisms of drug inactivation in cancer as well as the findings that quiescent tumour stem cells are refractory to kinase inhibitors (Graham et al., 2002), there are additional target-related mechanisms for resistance that are not based upon mutations of the target kinase. Drug resistance to targeted agents such as kinase inhibitors can occur either by compensatory mechanisms or by reducing the affinity of the kinase to its inhibitors (Szakacs et al., 2006; Fabbro et al., 2011).
In its simplest way, protein kinases escape inhibition by mutating key residues in their catalytic domains (Hunter, 2000; Gorre et al., 2001; Kobayashi et al., 2005; Takano et al., 2005; Ventura and Nebreda, 2006; Ali and Ali, 2007; Engelman et al., 2007; Chandarlapaty et al., 2011). The most commonly found point mutation leading to resistance concomitant with relapses affects the gatekeeper residue whose size and shape regulate the properties of the hydrophobic pocket located at the back of the ATP binding site. These mutations include the Thr-gatekeeper of BCR–ABL1 (T315I) (Gorre et al., 2001; Sawyers, 2004; Fabbro et al., 2005), KIT (T670I) (Heinrich et al., 2003; Fletcher and Rubin, 2007), platelet-derived growth factor receptor-α (PDGFRα) (T674I) (Cools et al., 2003), PDGFRβ (T681I) (Daub et al., 2004) and Src (proto-oncogene tyrosine-protein kinase Src) T341M (Bishop, 2004), as well as other types of gatekeepers such as L1196M in ALK (Katayama et al., 2012), G697R in FLT3 (fetal liver kinase-3) (Cools et al., 2004) and V561M in FGFR1 (fibroblast growth factor recptor-1) (Blencke et al., 2004). Loss of affinity to the kinase inhibitor is either due to a steric clash between inhibitor and the mutated gatekeeper, like in the case of BCR-ABL, or by significantly increasing the affinity for ATP and thereby reducing the affinity for the kinase inhibitors, like in the case of the EGFR (Daub et al., 2004; Kobayashi et al., 2005; Pao et al., 2005). Inhibitors targeting the inactive conformation of protein kinases are generally more prone to resistance mutations. In this case, a single mutation can act both by destroying crucial interactions and/or destabilizing the target conformation. Two frequent types of activating mutation in the gatekeeper and in the A-loop serve to illustrate the link between mutation and conformation. While the gatekeeper mutation is well conserved (Azam et al., 2008), the A-loop mutations are diverse (Dibb et al., 2004). Both types of mutations may activate the kinase (Azam et al., 2008). In addition to blocking access to the hydrophobic back-pocket (which is detrimental to ligand binding), mutation of a small to a large hydrophobic gatekeeper also stabilizes the active conformation, presumably by stabilization of the R-spine (Kornev et al., 2006; Taylor and Kornev, 2011).
In addition, mutations in MEK1 and ABL illustrate the diversity of the resistance mechanisms: (i) although many different mutations have been reported for MEK1 also in the context of the RAF resistance (Van Allen et al., 2014), they seem to work by a common mechanism in which the interface with the autoinhibitory N-terminal helix is disrupted or (ii) in the case of ABL, the most common and most resistant mutations interfere directly or indirectly with the binding of the drug, but other mutations map all over the kinase domain and it is unclear how some of these confer resistance (Apperley, 2007). The most dominant mutation in terms of resistance in both ABL and ALK is due to the gatekeeper mutations T315I and L1196M, which make them insensitive to imatinib and crizotinib respectively (Figure 5).
Mutations of the gatekeeper as well as other kinase domain mutations confer resistance to a wide spectrum of kinase inhibitors without affecting the kinase activity and may explain a fraction of cases of acquired resistance. The resistance mechanisms to kinase inhibitors are multiple and aim, in cancer, in the large part to restore the activity of the original ‘cancer-addicting’ pathway. This can occur either by conformational changes in the kinase domain or by reactivating the pathway downstream and/or parallel to the targeted kinase (Hunter, 2000; Gorre et al., 2001; Sawyers, 2004; Kobayashi et al., 2005; Takano et al., 2005; Rubin and Duensing, 2006; Ventura and Nebreda, 2006; Ali and Ali, 2007; Engelman et al., 2007; Chandarlapaty et al., 2011; Fabbro et al., 2011; Serra et al., 2011; Logue and Morrison, 2012; Trusolino and Bertotti, 2012; Workman et al., 2013b). Compensatory changes in the signalling pathways bypassing the drug-mediated inhibition and restoring the inhibited signalling pathway include the following:
amplification of the target kinase like BCR–ABL in CML (le Coutre et al., 2000) or dimerization of aberrantly spliced BRAF(V-600E) (Poulikakos et al., 2011)
up-regulation of receptor TPKs following either inhibition of PI3K (Serra et al., 2011; Rodon et al., 2013) or up-regulation of MET, IGF1R or AXL (AXL tyrosine kinase) in the acquisition of resistance to EGFR kinase inhibition (Engelman et al., 2007; Turke et al., 2010; Logue and Morrison, 2012)
activation of the RAS-RAF-MAPK and/or PI3K/Akt pathways by several mechanisms can override the effects of receptor TPK inhibitors by activating point mutations in PI3K, LOF/deletions of the PTEN (phosphatase and tensin homologue) phosphatase, activation of RAS isoforms, activation of COT (cancer Osaka thyroid aka MAP3K8 aka Tpl2; the immunological counterpart of RAF) (She et al., 2003; Johannessen et al., 2010; Corcoran et al., 2011; Prahallad et al., 2012)
signalling redundancies, interconnections through pathway crosstalk and feedback loops have also been identified as contributors to drug resistance (Janne et al., 2009; O'Reilly and McSheehy, 2010; Mendoza et al., 2011; Rodrik-Outmezguine et al., 2011; Chandarlapaty, 2012; Logue and Morrison, 2012; Trusolino and Bertotti, 2012). Allosteric inhibition of mTORC1 by rapamycins leads to disruption of a negative feedback loop, which activates Akt counteracting its anti-proliferative effects (Chandarlapaty, 2012). Inhibition of PI3K/mTOR signalling may lead to activation of the JAK/STAT5 pathway (Britschgi et al., 2012), while inhibition of mutant V600E-B-RAF by vemurafenib in cells with oncogenic RAS causes unexpected activation of the MAPK cascade by favouring the formation of wt BRAF and CRAF dimerization which can result in kerato-acanthomas in patients (Chapman et al., 2011; Poulikakos et al., 2011).
Factors regulating the bioavailability and intracellular concentration of inhibitors, such as poor intestinal absorption, tight binding to blood plasma proteins, overexpression of the MDR genes and/or increased metabolism of the drug by liver cytochrome P450 proteins, have also been linked to primary resistance (Mahon et al., 2003; Apperley, 2007).
All of these mechanisms demonstrate the plasticity of cancer cells and the many ways by which a tumour can evade targeted therapies. Strategies have been deployed to override these various types of resistances, including compounds capable of circumventing the target-related drug resistance by developing ‘second-generation’ kinase inhibitors (Lombardo et al., 2004; Weisberg et al., 2005; Adrian et al., 2006; Quintas-Cardama et al., 2007; Engelman et al., 2008b; Fabbro et al., 2010; Zhang et al., 2010).
For example, inhibitors that bind covalently to the ATP binding site of EGFR have been developed for the emerging resistance to gefitinib and erlotinib (Kwak et al., 2005; Heymach et al., 2006; Felip et al., 2007; Zhou et al., 2011). Several of these covalent inhibitors are in late stage clinical trials (Zhang et al., 2009; Liu et al., 2013). Alternatively, these type of covalent inhibitors, as in the case of ibrutinib, have been designed upfront to bind covalently to Cys481 of BTK and recently approved for B-cell malignancies (Byrd et al., 2013; Wiestner, 2013; Akinleye et al., 2014). Although ibrutinib has shown impressive clinical results, patients that have disease progression revealed a C481S mutation in their BTK that abrogates the covalent binding to ibrutinib (Furman et al., 2014).
Non-covalent inhibitors that can tolerate the amino acid exchange at the gatekeeper position have also been developed and, like ponatinib, approved for the T315I ABL gatekeeper mutant (O'Hare et al., 2009; Hoy, 2014). Targeting the gatekeeper mutation usually leads to low selectivity with deleterious side effects, leading to retraction from the market due to safety issues (Force et al., 2007; Cheng and Force, 2010; Dalzell, 2013).
A further approach is to target the kinase outside the ATP binding sites with the goal of combining the ATP-site-directed inhibitors (type-1 and type-2) with the type-3 inhibitors (Cowan-Jacob et al., 2014). A remote binding site on the kinase domain is addressed by the GNF-2 compound, which was found by a phenotypic screen shown to target the Myr-pocket binding site of ABL (Adrian et al., 2006; Fabbro et al., 2010; Zhang et al., 2010). Exploration of the combined efficacy between the Myr-pocket and ATP binding sites significantly increased the survival of mice in bone marrow transplantation CML models compared to treatment with either agent alone (Zhang et al., 2010). In addition, the improved potency of second-generation Myr-pocket binder against wt ABl and T315-ABL also translated into a high level of degree of synergy in BaF3 cells transformed with BCR-ABL-T315I when combined with ATP-site-directed inhibitors such as nilotinib or dasatinib, as has been noted in previous studies (Fabbro et al., 2002b; Zhang et al., 2010). Surprisingly, NMR and small angle X-ray scattering analyses revealed an open state of the ABL when bound to ATP-site-directed inhibitors, such as imatinib, leading to the detachment of the SH3-SH2 domains from the kinase domain and the formation of an ‘open’ inactive state, which is inhibited in the ATP site, which can be reversed by the addition of the Myr-pocket binder (Skora et al., 2013). Whether these data explain the synergy between the Myr-pocket binder and ATP-directed inhibitors which appear to overcome the T315I-ABL-mediated drug resistance remains to be seen. The findings on the actions of the two classes of inhibitors on a single target kinase may help to devise new strategies for drug development.
Another approach is to combine different kinase inhibitors targeting kinases of the same pathway like in the case of vemurafenib where the emerging resistance is not due to mutations in the B-Raf (V600E) but rather in the downstream MEK1 (Wagle et al., 2011; Medina et al., 2013). This has recently resulted in the approval of the dabrafenib (a RAF inhibitor) and trametinib (a MEK1 inhibitor) combination for metastatic melanoma (Table 1) (King et al., 2013).
Unfortunately, only a very limited number of non-ATP-competitive kinase inhibitors have thus far been identified, which could also be used to address the resistance caused by mutations in the ATP binding site (Cowan-Jacob et al., 2014). In addition, predicting clinical resistance to the targeted kinase inhibitor therapy is a gamble. Taking the example of BCR-ABL, saturation mutagenesis could predict most of the imatinib-resistant mutants in the kinase domain found in the clinic (Azam et al., 2003; 2008,). In contrast, adopting a similar approach as the B-Raf (V600E) or MEK1 would have failed as the acquired resistance is multiple and mainly due to reactivation of the signalling pathway (Corcoran et al., 2011). It should be emphasized that detecting clinical resistance is difficult to resolve due to the paucity of matched biopsies and limited coverage even of next-generation sequencing panels.
Resistance to kinase inhibitors in non-oncological indications is less likely to occur, as the selection pressure for the disease causing cell survival and the complexity of the destabilized genome, as is often the case in advanced cancers, is lower.
Thus, resistance to protein kinase inhibitors can emerge in several ways under treatment, raising the issue of an endless chase of resistance alleles, with ever more specific inhibitors.
A comprehensive combination of inhibitors, which take care of the resistance in the target kinase as well as of compensatory signalling, will be required to combat the emerging resistance in targeted cancer therapies. The only way to approach these problems is to use a rational combination of drugs.
Future perspectives in kinase drug discovery
Kinase inhibitor drug discovery has evolved into a mature field, with a wealth of structural and biological insights, as well as pharmacological tools. At the same time, we have only scratched the surface of the target space and are continuously humbled by the complexity of signalling pathways. The field has seen a lot of reasons why a target can fail regardless of how compelling the genetics may be. We often discover unexpected biology upon pharmacological inhibition. We have yet to understand feedback, compensatory mechanisms and resistance mechanisms better. We have yet to understand target toxicity and how it translates from preclinical species to man. State-of-the-art kinase drug discovery needs to take into account all of these subtleties and incorporate the lessons learned to succeed with the kinase targets of the future.
Understanding the conformational changes of protein kinases, which as molecular switches transition from the ‘on-’ and ‘off-states’, will allow for a better design of inhibitors and will provide a common framework for understanding the activation of the kinase, disease causality, therapeutic modalities and resistance. Aberrant activation of protein kinases occurs by pushing the equilibrium towards a constitutive active conformation, which is very similar in all protein kinases and in its essence can be defined by the DFG-motif forming the typical turn-hairpin-turn conformation in tight contact with the C-helix.
The large number of kinase inhibitors in clinical development will ensure a constant flow of novel targeted therapies, with increasing numbers in non-oncological indications, to the clinic over the next 10 years. The vast majority of kinase inhibitors are, at present, for various oncology indications, which not only reflects the more acute nature of the disease but also the greater tolerability with respect to potential side effects. The future of protein kinase-targeted therapeutics in cancer appears promising, despite the fact that several protein kinase inhibitors that have entered human clinical trials are not very specific and did not achieve the anticipated results. This situation may be improved by the upcoming second generation of kinase inhibitors with a better selectivity that will be applied to a genetically better defined patient population. The development of kinase inhibitors for non-life-threatening indications where chronic regimens are being used will require a priori a better target selectivity to minimize side effects. Identification of highly selective kinase inhibitors and activators should lead to an expansion of the chemical and biological kinase space, as well as to an improved understanding of their therapeutic limitations and potentials.
Acknowledgments
NC-IUPHAR receives financial support from the Wellcome Trust.
Glossary
- ABL
Abelson kinase
- Akt
protein kinase B or kinase from the transforming oncogene Akt8
- ALK
anaplastic lymphoma kinase
- A-loop
activation loop also called activation segment
- BTK
Bruton tyrosine kinase
- Catalytic loop
Y/HRD or Tyr/His-Arg-Asp
- CHEK1 (CHK1)
checkpoint kinase-1
- DGF-motif
Aspartate-Glycine-Phenylalanine or Asp-Gly-Phe
- EGFR
epidermal growth factor receptor
- ELK
eukaryotic-like kinase
- ePK
eukaryotic protein kinases
- FAK
focal adhesion kinase
- FDA
Food and Drug Administration
- Fes
Feline sarcoma oncogene kinase
- FGFR1
fibroblast growth factor recptor-1
- FLT3
fetal liver kinase-3
- GIST
gastrointestinal stromal tumours
- G-loop
glycine-rich loop also known as P-loop Gly-loop
- GSK3β
glycogen synthase kinase-3beta
- HES
hyper-eosinophilic syndrome
- JH2
Jak homology domain-2
- LKB1
serine/threonine-protein kinase STK11
- MAP2K
mitogen activated kinase kinase
- MAP3K
mitogen activated kinase kinase kinase
- MAP4K
mitogen activated kinase kinase kinase kinase
- MAPK
mitogen activated kinase
- MEK1
mitogen activated kinase kinase-1
- MET
mesenchymal epithelial transition factor or hepatocyte growth or scatter factor receptor
- mTOR
mammalian target of rapamycin
- Myr-pocket
myristate-pocket
- P450
cytochromes P450 (CYPs)
- PDGFR
platelet-derived growth factor receptor
- PDK1
3-phosphoinositide-dependent protein kinase-1
- PI
phosphatidyl-inositol
- PIF
PDK1 interacting fragment
- PK/PD
pharmacokinetic/pharmacodynamic
- PKL
protein kinase-like
- pSer, pThr, pTyr, phospho-Serine (pS), phospho-Threonine (pT)
phosphor-Tyrosine (pY)
- P-site
peptide-site
- PTEN
phosphatase and tensin homologue
- RAF
rapidly accelerated fibrosarcoma
- SMAD
SMAD is the composite of MAD form drosophila Mothers Against Decapentaplegic and SMA of Caenorhabditis elegans (from gene sma for small body size)
- STPK
serine- and threonine-specific protein kinase
- STRAD1
STE20-related adapter alpha
- TPK
tyrosine protein kinase
- TrkB or NTRK-2
tropomyosin receptor kinase B or neurotrophin receptor kinase-2
References
- Adrian FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:27. doi: 10.1186/1756-8722-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinleye A, Furqan M, Adekunle O. Ibrutinib and indolent B-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2014;14:253–260. doi: 10.1016/j.clml.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Albrecht BK, Harmange JC, Bauer D, Berry L, Bode C, Boezio AA, et al. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J Med Chem. 2008;51:2879–2882. doi: 10.1021/jm800043g. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. ). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S, Ali S. Role of c-kit/SCF in cause and treatment of gastrointestinal stromal tumors (GIST) Gene. 2007;401:38–45. doi: 10.1016/j.gene.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Amsberg GK, Koschmieder S. Profile of bosutinib and its clinical potential in the treatment of chronic myeloid leukemia. Onco Targets Ther. 2013;6:99–106. doi: 10.2147/OTT.S19901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012;2:512–523. doi: 10.1158/2159-8290.CD-11-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342:866–871. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari J, Hussain SA, Ansari A, Glaholm J. Critical appraisal of axitinib in the treatment of advanced renal cell carcinoma. Biologics. 2013;7:39–46. doi: 10.2147/BTT.S25862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–1029. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- Azam M, Raz T, Nardi V, Opitz SL, Daley GQ. A screen to identify drug resistant variants to target-directed anti-cancer agents. Biol Proced Online. 2003;5:204–210. doi: 10.1251/bpo63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne AD, Garnock-Jones KP. Dabrafenib: first global approval. Drugs. 2013;73:1367–1376. doi: 10.1007/s40265-013-0095-2. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- Barker AJ, Gibson KH, Grundy W, Godfrey AA, Barlow JJ, Healy MP, et al. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett. 2001;11:1911–1914. doi: 10.1016/s0960-894x(01)00344-4. [DOI] [PubMed] [Google Scholar]
- Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:121–130. doi: 10.1517/14728222.2011.644788. [DOI] [PubMed] [Google Scholar]
- Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JT, Hortobagyi GN, Campone M, Lebrun F, Deleu I, Rugo HS, et al. Everolimus plus exemestane as first-line therapy in HR(+), HER2(-) advanced breast cancer in BOLERO-2. Breast Cancer Res Treat. 2014;143:459–467. doi: 10.1007/s10549-013-2814-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC. A hot spot for protein kinase inhibitor sensitivity. Chem Biol. 2004;11:587–589. doi: 10.1016/j.chembiol.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horvath Z, et al. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11:691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Bono F, De Smet F, Herbert C, De Bock K, Georgiadou M, Fons P, et al. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell. 2013;23:477–488. doi: 10.1016/j.ccr.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Bornancin F. Ceramide kinase: the first decade. Cell Signal. 2011;23:999–1008. doi: 10.1016/j.cellsig.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, et al. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811. doi: 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Brooks AJ, Dai W, O'Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344:1249783. doi: 10.1126/science.1249783. [DOI] [PubMed] [Google Scholar]
- Buchdunger E, Matter A, Druker BJ. Bcr-Abl inhibition as a modality of CML therapeutics. Biochim Biophys Acta. 2001;1551:M11–M18. doi: 10.1016/s0304-419x(01)00022-1. [DOI] [PubMed] [Google Scholar]
- Busschots K, Lopez-Garcia LA, Lammi C, Stroba A, Zeuzem S, Piiper A, et al. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem Biol. 2012;19:1152–1163. doi: 10.1016/j.chembiol.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno F, Santoro M. Identification of RET kinase inhibitors as potential new treatment for sporadic and inherited thyroid cancer. J Chemother. 2004;16(Suppl. 4):49–51. doi: 10.1179/joc.2004.16.Supplement-1.49. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Jouvenceau A, Rose C, Guilloux JP, Pilon C, Dranovsky A, et al. Cyclotraxin-B, the first highly potent and selective TrkB inhibitor, has anxiolytic properties in mice. PLoS ONE. 2010;5:e9777. doi: 10.1371/journal.pone.0009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HY, Grossman AB, Bukowski RM. Everolimus in the treatment of renal cell carcinoma and neuroendocrine tumors. Adv Ther. 2010;27:495–511. doi: 10.1007/s12325-010-0045-2. [DOI] [PubMed] [Google Scholar]
- Chandarlapaty S. Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov. 2012;2:311–319. doi: 10.1158/2159-8290.CD-12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. Akt inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau NG, Haddad RI. Vandetanib for the treatment of medullary thyroid cancer. Clin Cancer Res. 2013;19:524–529. doi: 10.1158/1078-0432.CCR-12-2353. [DOI] [PubMed] [Google Scholar]
- Chen H, Kovar J, Sissons S, Cox K, Matter W, Chadwell F, et al. A cell-based immunocytochemical assay for monitoring kinase signaling pathways and drug efficacy. Anal Biochem. 2005;338:136–142. doi: 10.1016/j.ab.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Cheng H, Force T. Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Prog Cardiovasc Dis. 2010;53:114–120. doi: 10.1016/j.pcad.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Chhajed PN, Dickenmann M, Bubendorf L, Mayr M, Steiger J, Tamm M. Patterns of pulmonary complications associated with sirolimus. Respiration. 2006;73:367–374. doi: 10.1159/000087945. [DOI] [PubMed] [Google Scholar]
- Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, et al. International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66:918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibrik D, Silva HT, Jr, Vathsala A, Lackova E, Cornu-Artis C, Walker RG, et al. Randomized trial of everolimus-facilitated calcineurin inhibitor minimization over 24 months in renal transplantation. Transplantation. 2013;95:933–942. doi: 10.1097/TP.0b013e3182848e03. [DOI] [PubMed] [Google Scholar]
- Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur J Biochem. 2001;268:5001–5010. doi: 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002a;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases – the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002b;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Comess KM, Sun C, Abad-Zapatero C, Goedken ER, Gum RJ, Borhani DW, et al. Discovery and characterization of non-ATP site inhibitors of the mitogen activated protein (MAP) kinases. ACS Chem Biol. 2011;6:234–244. doi: 10.1021/cb1002619. [DOI] [PubMed] [Google Scholar]
- Converso A, Hartingh T, Garbaccio RM, Tasber E, Rickert K, Fraley ME, et al. Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorg Med Chem Lett. 2009;19:1240–1244. doi: 10.1016/j.bmcl.2008.12.076. [DOI] [PubMed] [Google Scholar]
- Cools J, Stover EH, Boulton CL, Gotlib J, Legare RD, Amaral SM, et al. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRα-induced myeloproliferative disease. Cancer Cell. 2003;3:459–469. doi: 10.1016/s1535-6108(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64:6385–6389. doi: 10.1158/0008-5472.CAN-04-2148. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, Settleman J, Engelman JA. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget. 2011;2:336–346. doi: 10.18632/oncotarget.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L, Cabrita G, et al. Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood. 2000;95:1758–1766. [PubMed] [Google Scholar]
- Cowan-Jacob SW. Structural biology of protein tyrosine kinases. Cell Mol Life Sci. 2006;63:2608–2625. doi: 10.1007/s00018-006-6202-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan-Jacob SW, Moebitz H, Fabbro D. Structural biology contributions to tyrosine kinase drug discovery. Curr Opin Cell Biol. 2009;21:280–287. doi: 10.1016/j.ceb.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob SW, Jahnke W, Knapp S. Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med Chem. 2014;6:541–561. doi: 10.4155/fmc.13.216. [DOI] [PubMed] [Google Scholar]
- Dalzell MD. Ponatinib pulled off market over safety issues. Manag Care. 2013;22:42–43. [PubMed] [Google Scholar]
- Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov. 2004;3:1001–1010. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Dibb NJ, Dilworth SM, Mol CD. Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat Rev Cancer. 2004;4:718–727. doi: 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Reisine T, Roby P, Rouleau N, Illy C, Bosse R, et al. The use of AlphaScreen technology in HTS: current status. Curr Chem Genomics. 2008;1:2–10. doi: 10.2174/1875397300801010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008a;18:73–79. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008b;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro D, Garcia-Echeverria C. Targeting protein kinases in cancer therapy. Curr Opin Drug Discov Devel. 2002a;5:701–712. [PubMed] [Google Scholar]
- Fabbro D, Ruetz S, Buchdunger E, Cowan-Jacob SW, Fendrich G, Liebetanz J, et al. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther. 2002b;93:79–98. doi: 10.1016/s0163-7258(02)00179-1. [DOI] [PubMed] [Google Scholar]
- Fabbro D, Fendrich G, Guez V, Meyer T, Furet P, Mestan P, et al. Targeted therapy with imatinib: an exception or a rule? Handb Exp Pharmacol. 2005;167:361–389. [Google Scholar]
- Fabbro D, Manley PW, Jahnke W, Liebetanz J, Szyttenholm A, Fendrich G, et al. Inhibitors of the Abl kinase directed at either the ATP- or myristate-binding site. Biochim Biophys Acta. 2010;1804:454–462. doi: 10.1016/j.bbapap.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Fabbro D, Cowan-Jacob SW, Möbitz H, Martiny-Baron G. Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol Biol. 2011;795:1–34. doi: 10.1007/978-1-61779-337-0_1. [DOI] [PubMed] [Google Scholar]
- Fabbro D, Cowan SW, Möbitz H, Martiny-Baron G. Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol Biol. 2012;795:1–34. doi: 10.1007/978-1-61779-337-0_1. [DOI] [PubMed] [Google Scholar]
- Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- Fedorov O, Muller S, Knapp S. The (un)targeted cancer kinome. Nat Chem Biol. 2010;6:166–169. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- Felip E, Santarpia M, Rosell R. Emerging drugs for non-small-cell lung cancer. Expert Opin Emerg Drugs. 2007;12:449–460. doi: 10.1517/14728214.12.3.449. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Kofler M, Hantschel O, Gish GD, Grebien F, Salah E, et al. Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell. 2008;134:793–803. doi: 10.1016/j.cell.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99:15937–15940. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JA, Rubin BP. KIT mutations in GIST. Curr Opin Genet Dev. 2007;17:3–7. doi: 10.1016/j.gde.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4:662–673. doi: 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370:2352–2354. doi: 10.1056/NEJMc1402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- Gao Y, Davies SP, Augustin M, Woodward A, Patel UA, Kovelman R, et al. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J. 2013;451:313–328. doi: 10.1042/BJ20121418. [DOI] [PubMed] [Google Scholar]
- Garay JP, Gray JW. Omics and therapy – a basis for precision medicine. Mol Oncol. 2012;6:128–139. doi: 10.1016/j.molonc.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaul MD, Guo Y, Affleck K, Cockerill GS, Gilmer TM, Griffin RJ, et al. Discovery and biological evaluation of potent dual ErbB-2/EGFR tyrosine kinase inhibitors: 6-thiazolylquinazolines. Bioorg Med Chem Lett. 2003;13:637–640. doi: 10.1016/s0960-894x(02)01047-8. [DOI] [PubMed] [Google Scholar]
- Glossmann H, Presek P, Eigenbrodt E. Quercetin inhibits tyrosine phosphorylation by the cyclic nucleotide-independent, transforming protein kinase, pp60src. Naunyn Schmiedebergs Arch Pharmacol. 1981;317:100–102. doi: 10.1007/BF00506266. [DOI] [PubMed] [Google Scholar]
- Goldman JM, Druker BJ. Chronic myeloid leukemia: current treatment options. Blood. 2001;98:2039–2042. doi: 10.1182/blood.v98.7.2039. [DOI] [PubMed] [Google Scholar]
- Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–1018. doi: 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin KR, Grimsby J. Small molecule glucokinase activators as glucose lowering agents: a new paradigm for diabetes therapy. Curr Med Chem. 2006;13:1839–1843. doi: 10.2174/092986706777452551. [DOI] [PubMed] [Google Scholar]
- Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol Sci. 2009;30:546–556. doi: 10.1016/j.tips.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Jackson CA, Goedert M, Hedge P, Cohen P. Effect of SB 203580 on the activity of c-Raf in vitro and in vivo. Oncogene. 1999;18:2047–2054. doi: 10.1038/sj.onc.1202603. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2014;33C:1–7. doi: 10.1016/j.ceb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- Heinrich T, Gradler U, Bottcher H, Blaukat A, Shutes A. Allosteric IGF-1R Inhibitors. ACS Med Chem Lett. 2010;1:199–203. doi: 10.1021/ml100044h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert C, Schieborr U, Saxena K, Juraszek J, De Smet F, Alcouffe C, et al. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell. 2013;23:489–501. doi: 10.1016/j.ccr.2013.02.018. [DOI] [PubMed] [Google Scholar]
- Heymach JV, Nilsson M, Blumenschein G, Papadimitrakopoulou V, Herbst R. Epidermal growth factor receptor inhibitors in development for the treatment of non-small cell lung cancer. Clin Cancer Res. 2006;12:4441s–4445s. doi: 10.1158/1078-0432.CCR-06-0286. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Hill ZB, Perera BG, Andrews SS, Maly DJ. Targeting diverse signaling interaction sites allows the rapid generation of bivalent kinase inhibitors. ACS Chem Biol. 2012;7:487–495. doi: 10.1021/cb200387g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindie V, Stroba A, Zhang H, Lopez-Garcia LA, Idrissova L, Zeuzem S, et al. Structure and allosteric effects of low-molecular-weight activators on the protein kinase PDK1. Nat Chem Biol. 2009;5:758–764. doi: 10.1038/nchembio.208. [DOI] [PubMed] [Google Scholar]
- Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, et al. RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell. 2013;23:594–602. doi: 10.1016/j.ccr.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Hoy SM. Ponatinib: a review of its use in adults with chronic myeloid leukaemia or Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs. 2014;74:793–806. doi: 10.1007/s40265-014-0216-6. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer. 2007;97:453–457. doi: 10.1038/sj.bjc.6603910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. Signaling – 2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Kawamoto S, Itoh H, Saitoh M, Hagiwara M, Takahashi J, et al. Naphthalenesulfonamides as calmodulin antagonists and protein kinase inhibitors. Mol Pharmacol. 1986;29:577–581. [PubMed] [Google Scholar]
- Jahnke W, Grotzfeld RM, Pelle X, Strauss A, Fendrich G, Cowan-Jacob SW, et al. Binding or bending: distinction of allosteric Abl kinase agonists from antagonists by an NMR-based conformational assay. J Am Chem Soc. 2010;132:7043–7048. doi: 10.1021/ja101837n. [DOI] [PubMed] [Google Scholar]
- Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8:709–723. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- Jia Y, Gu XJ, Brinker A, Warmuth M. Measuring the tyrosine kinase activity: a review of biochemical and cellular assay technologies. Expert Opin Drug Discov. 2008;3:959–978. doi: 10.1517/17460441.3.8.959. [DOI] [PubMed] [Google Scholar]
- Jin J, Pawson T. Modular evolution of phosphorylation-based signalling systems. Philos Trans R Soc Lond B Biol Sci. 2012;367:2540–2555. doi: 10.1098/rstb.2012.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Taylor SS. Rethinking pseudokinases. Cell. 2008;133:204–205. doi: 10.1016/j.cell.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Taylor SS, Zhai Y, Venter JC, Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007;5:e17. doi: 10.1371/journal.pbio.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian HM, Fojo T, Mathisen M, Zwelling LA. Cancer drugs in the United States: Justum Pretium – the just price. J Clin Oncol. 2013;31:3600–3604. doi: 10.1200/JCO.2013.49.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kelly PA, Gruber SA, Behbod F, Kahan BD. Sirolimus, a new, potent immunosuppressive agent. Pharmacotherapy. 1997;17:1148–1156. [PubMed] [Google Scholar]
- Kennelly PJ. Protein kinases and protein phosphatases in prokaryotes: a genomic perspective. FEMS Microbiol Lett. 2002;206:1–8. doi: 10.1111/j.1574-6968.2002.tb10978.x. [DOI] [PubMed] [Google Scholar]
- Kennelly PJ. Archaeal protein kinases and protein phosphatases: insights from genomics and biochemistry. Biochem J. 2003;370:373–389. doi: 10.1042/BJ20021547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE, et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS ONE. 2013;8:e67583. doi: 10.1371/journal.pone.0067583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp S, Arruda P, Blagg J, Burley S, Drewry DH, Edwards A, et al. A public-private partnership to unlock the untargeted kinome. Nat Chem Biol. 2013;9:3–6. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- Knight JD, Pawson T, Gingras AC. Profiling the kinome: current capabilities and future challenges. J Proteomics. 2013;81:43–55. doi: 10.1016/j.jprot.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci U S A. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–7670. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat Rev Genet. 2010;11:60–74. doi: 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK, et al. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J Biol Chem. 2011;286:39247–39258. doi: 10.1074/jbc.M111.264598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitzki A. Tyrphostins – potential antiproliferative agents and novel molecular tools. Biochem Pharmacol. 1990;40:913–918. doi: 10.1016/0006-2952(90)90474-y. [DOI] [PubMed] [Google Scholar]
- Li B, Liu Y, Uno T, Gray N. Creating chemical diversity to target protein kinases. Comb Chem High Throughput Screen. 2004;7:453–472. doi: 10.2174/1386207043328580. [DOI] [PubMed] [Google Scholar]
- Li M, Youngren JF, Manchem VP, Kozlowski M, Zhang BB, Maddux BA, et al. Small molecule insulin receptor activators potentiate insulin action in insulin-resistant cells. Diabetes. 2001;50:2323–2328. doi: 10.2337/diabetes.50.10.2323. [DOI] [PubMed] [Google Scholar]
- Lin K, Lin J, Wu W-I, Ballard J, Lee BB, Gloor SL, et al. An ATP-site on-off switch that restricts phosphatase accessibility of akt. Sci Signal. 2012;5:ra37. doi: 10.1126/scisignal.2002618. [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26:641–650. doi: 10.1101/gad.186965.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- London CA. Kinase dysfunction and kinase inhibitors. Vet Dermatol. 2013;24:181–187. doi: 10.1111/j.1365-3164.2012.01081.x. , e139–140. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia LA, Schulze JO, Frohner W, Zhang H, Suss E, Weber N, et al. Allosteric regulation of protein kinase PKCζ by the N-terminal C1 domain and small compounds to the PIF-pocket. Chem Biol. 2011;18:1463–1473. doi: 10.1016/j.chembiol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Lowinger TB, Riedl B, Dumas J, Smith RA. Design and discovery of small molecules targeting raf-1 kinase. Curr Pharm Des. 2002;8:2269–2278. doi: 10.2174/1381612023393125. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Deacon S, Horiuchi K. The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin Drug Discov. 2008;3:607–621. doi: 10.1517/17460441.3.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003;101:2368–2373. doi: 10.1182/blood.V101.6.2368. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Fabbro D. Classical PKC isoforms in cancer. Pharmacol Res. 2007;55:477–486. doi: 10.1016/j.phrs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott J, Jimeno A. Ibrutinib for the treatment of chronic lymphocytic leukemia and mantle cell lymphoma. Drugs Today (Barc) 2014;50:291–300. doi: 10.1358/dot.2014.50.4.2133570. [DOI] [PubMed] [Google Scholar]
- McIntyre KW, Shuster DJ, Gillooly KM, Dambach DM, Pattoli MA, Lu P, et al. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003;48:2652–2659. doi: 10.1002/art.11131. [DOI] [PubMed] [Google Scholar]
- Medina T, Amaria MN, Jimeno A. Dabrafenib in the treatment of advanced melanoma. Drugs Today (Barc) 2013;49:377–385. doi: 10.1358/dot.2013.49.6.1968669. [DOI] [PubMed] [Google Scholar]
- Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci U S A. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkovsky N, Berezov A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs. 2008;9:1336–1346. [PubMed] [Google Scholar]
- Moebitz H, Fabbro D. Conformational bias: a key concept for protein kinase inhibition. Eur Pharm Rev. 2012;17:41–51. [Google Scholar]
- Morphy R, Rankovic Z. Fragments, network biology and designing multiple ligands. Drug Discov Today. 2007;12:156–160. doi: 10.1016/j.drudis.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Hoosen S, Bello CL, Christensen JG. Sunitinib malate for the treatment of solid tumours: a review of current clinical data. Expert Opin Investig Drugs. 2006;15:553–561. doi: 10.1517/13543784.15.5.553. [DOI] [PubMed] [Google Scholar]
- Muller S, Knapp S. Targeting kinases for the treatment of inflammatory diseases. Expert Opin Drug Discov. 2010;5:867–881. doi: 10.1517/17460441.2010.504203. [DOI] [PubMed] [Google Scholar]
- Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Nelson V, Ziehr J, Agulnik M, Johnson M. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther. 2013;6:135–143. doi: 10.2147/OTT.S23165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolini FE, Ibrahim AR, Soverini S, Martinelli G, Muller MC, Hochhaus A, et al. The BCR-ABLT315I mutation compromises survival in chronic phase chronic myelogenous leukemia patients resistant to tyrosine kinase inhibitors, in a matched pair analysis. Haematologica. 2013;98:1510–1516. doi: 10.3324/haematol.2012.080234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya T, Takigawa N, Ichihara E, Ochi N, Murakami T, Honda Y, et al. Afatinib prolongs survival compared with gefitinib in an epidermal growth factor receptor-driven lung cancer model. Mol Cancer Ther. 2013;12:589–597. doi: 10.1158/1535-7163.MCT-12-0885. [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, et al. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOr inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol. 2010;3:65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Over B, Wetzel S, Grutter C, Nakai Y, Renner S, Rauh D, et al. Natural-product-derived fragments for fragment-based ligand discovery. Nat Chem. 2013;5:21–28. doi: 10.1038/nchem.1506. [DOI] [PubMed] [Google Scholar]
- Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18:699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender C, Goldfine ID, Manchem VP, Evans JL, Spevak WR, Shi S, et al. Regulation of insulin receptor function by a small molecule insulin receptor activator. J Biol Chem. 2002;277:43565–43571. doi: 10.1074/jbc.M202426200. [DOI] [PubMed] [Google Scholar]
- Perez-Soler R. The role of erlotinib (Tarceva, OSI 774) in the treatment of non-small cell lung cancer. Clin Cancer Res. 2004;10:4238s–4240s. doi: 10.1158/1078-0432.CCR-040017. [DOI] [PubMed] [Google Scholar]
- Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12:581–594. doi: 10.1038/nrd4051. [DOI] [PubMed] [Google Scholar]
- Polier S, Samant RS, Clarke PA, Workman P, Prodromou C, Pearl LH. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nat Chem Biol. 2013;9:307–312. doi: 10.1038/nchembio.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- Rajakulendran T, Sicheri F. Allosteric protein kinase regulation by pseudokinases: insights from STRAD. Sci Signal. 2010;3:pe8. doi: 10.1126/scisignal.3111pe8. [DOI] [PubMed] [Google Scholar]
- Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal. 2011;9:23. doi: 10.1186/1478-811X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15:143–155. doi: 10.1016/S1470-2045(13)70586-2. [DOI] [PubMed] [Google Scholar]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- Rini BI, Garrett M, Poland B, Dutcher JP, Rixe O, Wilding G, et al. Axitinib in metastatic renal cell carcinoma: results of a pharmacokinetic and pharmacodynamic analysis. J Clin Pharmacol. 2013;53:491–504. doi: 10.1002/jcph.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak T, Robak E. Tyrosine kinase inhibitors as potential drugs for B-cell lymphoid malignancies and autoimmune disorders. Expert Opin Investig Drugs. 2012;21:921–947. doi: 10.1517/13543784.2012.685650. [DOI] [PubMed] [Google Scholar]
- Robert C, Soria JC, Spatz A, Le Cesne A, Malka D, Pautier P, et al. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol. 2005;6:491–500. doi: 10.1016/S1470-2045(05)70243-6. [DOI] [PubMed] [Google Scholar]
- Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248–259. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C. Taking PI3Kδ and PI3Kγ one step ahead: dual active PI3Kδ/γ inhibitors for the treatment of immune-mediated inflammatory diseases. Curr Top Microbiol Immunol. 2010;346:279–299. doi: 10.1007/82_2010_79. [DOI] [PubMed] [Google Scholar]
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- Rubin BP, Duensing A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab Invest. 2006;86:981–986. doi: 10.1038/labinvest.3700466. [DOI] [PubMed] [Google Scholar]
- Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc Natl Acad Sci U S A. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama AK, Kim KB. Trametinib (GSK1120212) in the treatment of melanoma. Expert Opin Pharmacother. 2013;14:619–627. doi: 10.1517/14656566.2013.770475. [DOI] [PubMed] [Google Scholar]
- Salt IP, Palmer TM. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin Investig Drugs. 2012;21:1155–1167. doi: 10.1517/13543784.2012.696609. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin M, Pandiella A. Differential action of small molecule HER kinase inhibitors on receptor heterodimerization: therapeutic implications. Int J Cancer. 2012;131:244–252. doi: 10.1002/ijc.26358. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007;282:32539–32548. doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28:803–814. doi: 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they're apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers WR. A blueprint for advancing genetics-based cancer therapy. Cell. 2011;147:26–31. doi: 10.1016/j.cell.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Serra V, Scaltriti M, Prudkin L, Eichhorn PJA, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–2557. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahda S, Saif MW. Regorafenib: from bench to bedside in colorectal cancer. Expert Rev Clin Pharmacol. 2013;6:243–248. doi: 10.1586/ecp.13.11. [DOI] [PubMed] [Google Scholar]
- Shaw AT, Yasothan U, Kirkpatrick P. Crizotinib. Nat Rev Drug Discov. 2011;10:897–898. doi: 10.1038/nrd3600. [DOI] [PubMed] [Google Scholar]
- She QB, Solit D, Basso A, Moasser MM. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res. 2003;9:4340–4346. [PubMed] [Google Scholar]
- Shibuya M, Suzuki Y. Treatment of cerebral vasospasm by a protein kinase inhibitor AT 877. No to Shinkei. 1993;45:819–824. [PubMed] [Google Scholar]
- Shibuya M, Asano T, Sasaki Y. Effect of Fasudil HCl, a protein kinase inhibitor, on cerebral vasospasm. Acta Neurochir Suppl. 2001;77:201–204. doi: 10.1007/978-3-7091-6232-3_42. [DOI] [PubMed] [Google Scholar]
- Simmons DL. Targeting kinases: a new approach to treating inflammatory rheumatic diseases. Curr Opin Pharmacol. 2013;13:426–434. doi: 10.1016/j.coph.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Skora L, Mestan J, Fabbro D, Jahnke W, Grzesiek S. NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc Natl Acad Sci U S A. 2013;110:E4437–E4445. doi: 10.1073/pnas.1314712110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg C. 2009. A randomized, double-blind phase III study of pazopanib in treatment-naive and cytokine-pretreated patients with advanced renal cell carcinoma (RCC). ASCO, Abstract No. 5021.
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46:1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:6829–6837. doi: 10.1200/JCO.2005.01.0793. [DOI] [PubMed] [Google Scholar]
- Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent protein kinase. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- Tao ZF, Wang L, Stewart KD, Chen Z, Gu W, Bui MH, et al. Structure-based design, synthesis, and biological evaluation of potent and selective macrocyclic checkpoint kinase 1 inhibitors. J Med Chem. 2007;50:1514–1527. doi: 10.1021/jm061247v. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Kim C, Vigil D, Haste NM, Yang J, Wu J, et al. Dynamics of signaling by PKA. Biochim Biophys Acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Tecle H, Shao J, Li Y, Kothe M, Kazmirski S, Penzotti J, et al. Beyond the MEK-pocket: can current MEK kinase inhibitors be utilized to synthesize novel type III NCKIs? Does the MEK-pocket exist in kinases other than MEK? Bioorg Med Chem Lett. 2009;19:226–229. doi: 10.1016/j.bmcl.2008.10.108. [DOI] [PubMed] [Google Scholar]
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- Tomita N, Hayashi Y, Suzuki S, Oomori Y, Aramaki Y, Matsushita Y, et al. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg Med Chem Lett. 2013;23:1779–1785. doi: 10.1016/j.bmcl.2013.01.047. [DOI] [PubMed] [Google Scholar]
- Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P, et al. Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev. 2001;21:499–512. doi: 10.1002/med.1022. [DOI] [PubMed] [Google Scholar]
- Trusolino L, Bertotti A. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: six degrees of separation. Cancer Discov. 2012;2:876–880. doi: 10.1158/2159-8290.CD-12-0400. [DOI] [PubMed] [Google Scholar]
- Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzarum N, Komornik N, Ben Chetrit D, Engelberg D, Livnah O. DEF pocket in p38α facilitates substrate selectivity and mediates autophosphorylation. J Biol Chem. 2013;288:19537–19547. doi: 10.1074/jbc.M113.464511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. A peptoid ‘antibody surrogate’ that antagonizes VEGF receptor 2 activity. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderpool D, Johnson TO, Ping C, Bergqvist S, Alton G, Phonephaly S, et al. Characterization of the CHK1 allosteric inhibitor binding site. Biochemistry. 2009;48:9823–9830. doi: 10.1021/bi900258v. [DOI] [PubMed] [Google Scholar]
- Vasquez EM. Sirolimus: a new agent for prevention of renal allograft rejection. Am J Health Syst Pharm. 2000;57:437–448. doi: 10.1093/ajhp/57.5.437. , quiz 449–451. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Nebreda AR. Protein kinases and phosphatases as therapeutic targets in cancer. Clin Transl Oncol. 2006;8:153–160. doi: 10.1007/s12094-006-0005-0. [DOI] [PubMed] [Google Scholar]
- Vieth M, Higgs RE, Robertson DH, Shapiro M, Gragg EA, Hemmerle H. Kinomics-structural biology and chemogenomics of kinase inhibitors and targets. Biochim Biophys Acta. 2004;1697:243–257. doi: 10.1016/j.bbapap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- Vieth M, Sutherland JJ, Robertson DH, Campbell RM. Kinomics: characterizing the therapeutically validated kinase space. Drug Discov Today. 2005;10:839–846. doi: 10.1016/S1359-6446(05)03477-X. [DOI] [PubMed] [Google Scholar]
- Viola D, Cappagli V, Elisei R. Cabozantinib (XL184) for the treatment of locally advanced or metastatic progressive medullary thyroid cancer. Future Oncol. 2013;9:1083–1092. doi: 10.2217/fon.13.128. [DOI] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- Wang X, Sun SY. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets. 2009;13:1193–1203. doi: 10.1517/14728220903225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19:55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- West K. CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders. Curr Opin Investig Drugs. 2009;10:491–504. [PubMed] [Google Scholar]
- Wiestner A. Targeting B-cell receptor signaling for anticancer therapy: the Bruton's tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J Clin Oncol. 2013;31:128–130. doi: 10.1200/JCO.2012.44.4281. [DOI] [PubMed] [Google Scholar]
- Williams NK, Bamert RS, Patel O, Wang C, Walden PM, Wilks AF, et al. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol. 2009;387:219–232. doi: 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Wissner A, Fraser HL, Ingalls CL, Dushin RG, Floyd MB, Cheung K, et al. Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg Med Chem. 2007;15:3635–3648. doi: 10.1016/j.bmc.2007.03.055. [DOI] [PubMed] [Google Scholar]
- Wolf-Yadlin A, Kumar N, Zhang Y, Hautaniemi S, Zaman M, Kim HD, et al. Effects of HER2 overexpression on cell signaling networks governing proliferation and migration. Mol Syst Biol. 2006;2:54. doi: 10.1038/msb4100094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Workman P, Al-Lazikani B. Drugging cancer genomes. Nat Rev Drug Discov. 2013a;12:889–890. doi: 10.1038/nrd4184. [DOI] [PubMed] [Google Scholar]
- Workman P, Al-Lazikani B, Clarke PA. Genome-based cancer therapeutics: targets, kinase drug resistance and future strategies for precision oncology. Curr Opin Pharmacol. 2013b;13:486–496. doi: 10.1016/j.coph.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Wright CJ, McCormack PL. Trametinib: first global approval. Drugs. 2013;73:1245–1254. doi: 10.1007/s40265-013-0096-1. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, et al. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC. A selective ALK inhibitor in ALK-rearranged patients. Lancet Oncol. 2013;14:564–565. doi: 10.1016/S1470-2045(13)70170-0. [DOI] [PubMed] [Google Scholar]
- Yang J, Campobasso N, Biju MP, Fisher K, Pan XQ, Cottom J, et al. Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem Biol. 2011;18:177–186. doi: 10.1016/j.chembiol.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Yang X, Huang Y, Crowson M, Li J, Maitland ML, Lussier YA. Kinase inhibition-related adverse events predicted from in vitro kinome and clinical trial data. J Biomed Inform. 2010;43:376–384. doi: 10.1016/j.jbi.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–1711. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Janne PA, Gray NS. Discovery of selective irreversible inhibitors for EGFR-T790M. ACS Med Chem Lett. 2011;21:638–643. doi: 10.1016/j.bmcl.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccotto F, Ardini E, Casale E, Angiolini M. Through the ‘gatekeeper door’: exploiting the active kinase conformation. J Med Chem. 2010;53:2681–2694. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]