

Abstract
Gene expression is dynamically controlled by epigenetics through post-translational modifications of histones, chromatin-associated proteins and DNA itself. All these elements are required for the maintenance of chromatin structure and cell identity in the context of a normal cellular phenotype. Disruption of epigenetic regulation is a common event in human cancer. Here, we review the key protein families that control epigenetic signalling through writing, erasing or reading specific post-translational modifications. By exploiting the leading role of epigenetics in tumour development and the reversibility of epigenetic modifications, promising novel epigenetic-based therapies are being developed. In this article, we highlight the emerging low MW inhibitors targeting each class of chromatin-associated protein, their current use in preclinical and clinical trials and the likelihood of their being approved in the near future.
Linked Articles
This article is part of a themed section on Epigenetics and Therapy. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-11
Table of Links
| TARGETS | LIGANDS | |
|---|---|---|
| DNA methyltransferases | Anacardic acid | GSK126 |
| DOT1L, histone methyltransferase | 5-azacytidine (azacitidine) | Panobinostat |
| EZH2, histone methyltransferase | 5-aza-2′-deoxycytidine (decitabine) | Quisinostat (JNJ-26481585) |
| HAT, histone acetyltransferases | Belinostat | Romidepsin |
| HDAC, histone deacetylase | BIX-01294 | SAM, S-adenosyl methionine |
| HDAC1 | Butyrate | Trichostatin A |
| HDAC2 | C646 | Vorinostat (SAHA) |
| HDAC3 | Curcumin | UNC0638 |
| HDAC6 | E11 | |
| Histone demethylases | EPZ-5676 | |
| HMT, histone methyltransferase | EPZ004777 | |
| KMT, protein lysine methyltransferase | Garcinol | |
| LSD1, lysine-specific demethylase 1 | Givinostat |
This Table lists key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Cells in an organism contain identical genetic material but all of them have the ability to maintain the specific phenotypes and biological functions of the tissues and organs in which they are embedded. This capability is ensured by the chromatin-associated proteins and the heritable chemical modifications of histones and DNA sequence: the epigenome (Waddington, 1952; Berger et al., 2009). Although these modifications do not involve changes in the linear DNA sequence, they are fundamental to maintaining cell identity and regulating processes such as differentiation, development, proliferation and genome integrity (Kouzarides, 2007). Several factors influence epigenetic gene regulation through changes in chromatin structure, but the covalent modifications of histones and DNA are possibly the most decisive elements coordinating this process (Segal and Widom, 2009).
Chromatin modifications are responsible for changes in the conformation of chromatin through their effect over interactions between DNA sequence and histones, which determine accessibility to specific loci, or through the creation of docking sites for the recruitment of epigenetic regulators. This map of the combinations of covalent modifications is known as the histone code and is defined by four different DNA modifications (Baylin and Jones, 2011; Wu and Zhang, 2011; Pfaffeneder et al., 2014) and at least 16 types of histone modification, such as phosphorylation, acetylation, methylation, ubiquitination and sumoylation (Kouzarides, 2007). Research in the past decade has significantly increased our knowledge of the proteins related to these post-translational modifications (PTMs). These are chromatin-interacting proteins that catalyse, recognize and remove the specific chemical modifications. They are popularly known as writers, readers and erasers respectively (Figure 1). Most of these proteins possess specialized domains that are able to recognize particular regions within the genome, being guided by a specific histone code.
Figure 1.
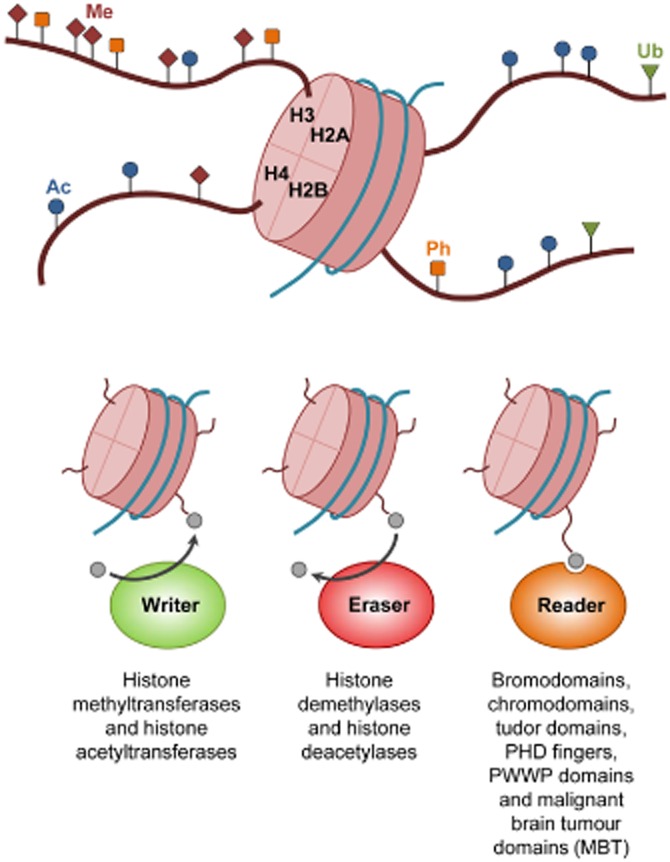
Writers, erasers and readers. The basic functional unit of chromatin is the nucleosome, which is composed of DNA wrapped around histones (H2A, H2B, H3 and H4). Core histone tails are projected from nucleosomes and are subject to PTMs. These include methylation (Me), acetylation (Ac), phosphorylation (Ph) and ubiquitination (Ub). The main epigenetic regulators can be categorized as writers, erasers and readers of PTMs. Epigenetic writers are responsible for the addition of chemical modifications. Epigenetic erasers catalyse the removal of the covalent modifications. Epigenetic readers are proteins with specific domains that recognize and bind to particular modifications.
Deregulation of epigenetic control has been frequently associated with several human diseases, such as cancer (Baylin and Jones, 2011). Tumour cells suffer global epigenetic reorganization resulting in the CpG-specific hypermethylation of tumour suppressor gene promoters and a generalized loss of DNA methylation at microsatellite regions, repetitive sequences and oncogene promoters (Esteller, 2008). Moreover, genes encoding epigenetic regulators undergo several aberrations in cancer, such as point mutations, translocations, amplifications and deletions (Simó-Riudalbas and Esteller, 2013). Depending on the protein involved and the pathway affected, these alterations could lead to changes in gene transcription or to more global changes in chromatin structure.
In contrast with the irreversible genomic mutations that inactivate tumour suppressor genes or activate oncogenes in cancer, epigenetic modifications can be reversed. Thus, the dynamism of the epigenome allows the correction of aberrant epigenetic profiles by therapeutic manipulation. Epigenetic therapies targeting some chromatin regulators have already been approved by the Food and Drug Administration (FDA). This is the case for 5-azacytidine and 5-aza-2′-deoxycytidine, nucleoside analogues that irreversibly inhibit the DNA methyltransferases DNMT1 and DNMT3B and which are currently used as first-line treatment for patients with myelodysplastic syndrome (MDS; Garcia-Manero and Fenaux, 2011; Wells et al., 2014). Not only drugs targeting epigenetic writers are used in clinics at present, but also drugs against epigenetic erasers. Vorinostat (SAHA) and romidepsin are inhibitors of histone deacetylases (HDAC) approved for the treatment of refractory cutaneous T-cell lymphoma (Foss et al., 2011; Khan and La Thangue, 2012). Although the introduction of these compounds into clinics has been a success for the field, their precise mechanism of action remains unclear, and no reliable biomarkers are available for the prediction of their clinical activity.
New generation epigenetic therapies are being designed that take into account the abnormal expression levels of chromatin-associated proteins present in tumours. In this review, we focus on the proteins involved in depositing, removing or binding to PTMs, as well as the latest advances in the development of specific inhibitors to these proteins associated to chromatin.
Targeting epigenetic writers
Epigenetic writers encompass enzymes such as histone acetylases (HAT), kinases, histone methyltransferases (HMT) and ubiquitin ligases. These chromatin-associated proteins catalyse the deposition of the PTMs on proteins and introduce dynamic modifications that respond rapidly to environmental changes, like histone acetylation.
HMTs
A clear association between histone methylation, transcriptional regulation and tumour phenotype has encouraged the design of specific low MW inhibitors of distinct histone arginine and lysine methyltransferases. The lysine residues can exist in monomethylated, dimethylated and trimethylated states, while arginine residues are either monomethylated or dimethylated. Demethylation of arginine residues can occur symmetrically (via monomethylation of both terminal guanidine nitrogens) or asymmetrically (via dimethylation of one of the terminal guanidine nitrogens; Chesworth et al., 2014).
Protein arginine methyltransferases (PRMTs) and protein lysine methyltransferases (KMTs) are able to transfer a methyl group from the cofactor S-adenosylmethionine (SAM) to arginine or lysine residues respectively. The catalytic domains of PRMT and KMT proteins are structurally different. All the enzymes of the KMT class, with the exception of DOT1L, share a conserved catalytic domain known as the SET domain. The catalytic domain of DOT1L shares structural homology with the PRMT class (Richon et al., 2011).
Histone methylation does not affect chromatin structure directly because this chemical modification does not change the charged state of an aminoacidic residue. Instead, each type of methyl mark represents a specific modification that is recognized as a docking site for chromatin-associated proteins that maintain chromatin architecture (Trojer et al., 2007) or regulate gene expression (Lee et al., 2007). Depending on each specific residue, methylation is associated with activated euchromatic genes (H3K4, H3K36 and H3K79) or with silenced heterochromatic genes (H3K9, H3K27 and H4K20; Barski et al., 2007; Bannister and Kouzarides, 2011).
Aberrant activity of HMTs, due to chromosomal translocation, amplification, deletion, overexpression or silencing of their corresponding genes, has been discovered in cancer (Ryan and Bernstein, 2012; Shih et al., 2012). The recent discovery of HMT disruption in cancer suggests a strategy for targeting patient populations with these alterations using low MW compounds designed to selectively inhibit oncogenic methyltransferases. Drug discovery efforts are giving rise to low MW inhibitors that can reduce HMT activity and reverse the abnormal transcription patterns of tumour cells (Figure 2).
Figure 2.

Inhibition of HMTs. (A) DOT1L is a HMT that specifically methylates the lysine H3K79, a histone modification that is associated with actively transcribed genes. Different subtypes of haematological malignancies involve MLL-fusion proteins (e.g. MLL-AF6) that recruit DOT1L to unusual localizations for the induction of leukaemogenic gene expression. (B) Reported DOT1L inhibitors are EPZ004777 and EPZ-5676. (C) EZH2, a protein of the PRC2, catalyses dimethylation and trimethylation of H3K27 to maintain transcription repression of target genes. EZH2, which is up-regulated in several tumours, induces cell migration, colony formation and genomic instability. (D) 3-deazaneplanocin (DZNeP), GSK126, EPZ-6438 and EI1 are specific inhibitors of EZH2. (E) SETDB1 is an HMT responsible for the methylation of H3K9, a mark associated with gene repression. This protein has been found amplified in cancer. (F) Mithramycin is a clinically approved antibiotic that represses SETDB1 function.
DOT1L
Over recent decades, an increasing number of non-random chromosomal abnormalities have been described in different subtypes of haematological malignancies. Approximately 10% of adult acute myeloid leukaemias (AMLs) and 70% of infant leukaemias involve rearrangements of the MLL (also known as KTM2A) gene, resulting in fusion of MLL with several different protein partners (De Boer et al., 2013). The artificial fusion complexes recruit DOT1L, a HMT that specifically methylates Lys79 of histone H3 (H3K79). After this abnormal recruitment to unusual localizations, DOT1L enhances the expression of genes required for leukaemia initiation (Okada et al., 2005; Deshpande et al., 2013).
As DOT1L is a key protein in the development of MLL-rearranged leukaemia, there is an increasing interest in its therapeutic targeting. EPZ004777 is selective inhibitor of DOT1L H3K79 methyltransferase activity, which acts by mimicking the cofactor SAM (Table 1; Daigle et al., 2011). In MLL-rearranged cell lines, EPZ004777 decreases global H3K79 methylation levels and has anti-proliferative effects after blocking the expression of MLL-fusion target genes (Daigle et al., 2011). The specificity of its mechanism of action ensures that the DOT1L inhibitor only affects cells with MLL gene fusion and preserves the non-rearranged cell lines. Preclinical experiments in mice show good EPZ004777 tolerance and efficacy (Daigle et al., 2011). Although this DOT1L inhibitor presents such attractive features, it still has poor pharmacokinetic properties, such as a short plasma half-life that entails continuous infusions. An attempt is being made to solve some of these deficiencies using second-generation DOT1L inhibitors like EPZ-5676, which is already undergoing clinical trials (Daigle et al., 2013; ClincalTrials.gov identifier: NCT01684150).
Table 1.
Inhibitors of epigenetic writers
| Category | Compound | Phase | Tumour type | References |
|---|---|---|---|---|
| HMT inhibitors | ||||
| DOT1L inhibitors | EPZ00477 | Preclinical | MLL-rearranged leukaemia | Daigle et al., 2011 |
| EPZ-5676 | Clinical trials | Haematological malignancies | Daigle et al., 2013; NCT01684150 | |
| SGC0946 | Preclinical | Leukaemia | Yu et al., 2012 | |
| EZH2 inhibitors | DZNeP | Preclinical | Breast, colon, prostate | Tan et al., 2007 |
| GSK126 | Preclinical | EZH2 mutant lymphomas | McCabe et al., 2012 | |
| GSK343 | Preclinical | Epithelial ovarian cancer | Amatangelo et al., 2013 | |
| EPZ005687 | Preclinical | EZH2 mutant lymphomas | Knutson et al., 2012 | |
| EI1 | Preclinical | Diffuse large B-cell lymphoma | Qi et al., 2012 | |
| EPZ-6438 | Clinical trials | Advanced solid tumours, B-cell lymphoma | Knutson et al., 2013; NCT01897571 | |
| UNC1999 | Preclinical | Diffuse large B-cell lymphoma | Konze et al., 2013 | |
| SUV39H1 inhibitors | Chaetocin | Preclinical | AML | Chaib et al., 2012 |
| G9A inhibitors | BIX-01294 | Preclinical | Neuroblastoma | Lu et al., 2013 |
| UNC0321 | Preclinical | (Enzymic inhibition of the target protein) | Liu et al., 2010 | |
| UNC0638 | Preclinical | Breast | Vedadi et al., 2011 | |
| BRD4770 | Preclinical | Pancreas | Yuan et al., 2012 | |
| SETDB1 inhibitors | Mithramycin | Preclinical | Non-small cell lung cancer | Rodriguez-Paredes et al., 2013 |
| HAT inhibitors | ||||
| Bisubstrate inhibitors | Acetyl-CoA derivatives | Preclinical | (Enzymic inhibition of the target protein) | Wu et al., 2009 |
| Natural products | Anacardic acid | Preclinical | Breast | Hemshekhar et al., 2012 |
| Garcinol | Preclinical | Hepatocellular carcinoma | Sethi et al., 2014 | |
| LTK14, LTK15 | Preclinical | (Enzymatic inhibition of the target protein) | Mantelingu et al., 2007 | |
| Curcumin | Preclinical | Head and neck, lung | Kumar et al., 2014; Malhotra et al., 2014 | |
| Low MW compounds | α-methylene-γ-butyrolactone 3 (MB-3) | Preclinical | Acute lymphoblastic leukaemia | Holmlund et al., 2013 |
| Isothiazolone | Preclinical | Colon | Stimson et al., 2005 | |
| C646 | Preclinical | Prostate | Santer et al., 2011 |
EZH2
The catalytic component of the polycomb repressive complex 2 (PRC2) is the enzyme EZH2, responsible for the methylation of H3K27. EZH2 and the whole PRC2 are critical for silencing a large number of genes involved in development and differentiation processes (Morey and Helin, 2010). This HMT is overexpressed in prostate, breast, kidney and lung cancers, solid tumours in which EZH2 up-regulation induces cell migration, colony formation and genomic instability (Varambally et al., 2002; Kleer et al., 2003; Wagener et al., 2010; Takawa et al., 2011). Additionally, somatic gain-of-function mutations in the SET domain of EZH2 have been discovered in follicular and diffuse large B-cell lymphomas (Morin et al., 2010; Pasqualucci et al., 2011). However, EZH2 loss-of-function mutations have also been identified in MDS (Nikoloski et al., 2010). The presence of both activating and inactivating mutations of EZH2 in cancer suggests a context-dependent role for these HMTs and both sides of the coin should be taken into account in the development of Polycomb-targeted therapies.
The high frequency of genetic changes affecting H3K27 has prompted the development of several histone methylation inhibitors. 3-deazaneplanocin is a SAM-derived molecule that leads to a decrease of H3K27 methylation together with apoptosis of cancer cells (Tan et al., 2007). However, in some cells, this compound decreases the methylation of other histone residues and does not seem to be solely a selective inhibitor of the repressive marks but also of the active histone methylation marks (Miranda et al., 2009). It is therefore useful to continue looking for more specific inhibitors of histone methylation, which could directly target the EZH2 enzyme.
More recently, several non-SAM-derived inhibitors of the catalytic activity of EZH2 have been discovered. All of them are highly potent selective inhibitors with in vivo anti-tumour activity. Some examples are GSK126 and EPZ005687, inhibitors effective against EZH2 mutant lymphomas, and EI1, a low MW inhibitor that blocks diffuse large B-cell lymphoma proliferation (Knutson et al., 2012; McCabe et al., 2012; Qi et al., 2012). Another EZH2 selective inhibitor is EPZ-6438, which has already entered clinical trials for the treatment of patients with B-cell lymphoma (ClincalTrials.gov identifier: NCT01897571). EPZ-6438 is the first EZH2 inhibitor with activity in solid tumours such as pediatric malignant rhabdoid cancer (Table 1; Knutson et al., 2013).
H3K9 methyltransferases
The di- or trimethylation of Lys9 on histone H3 (H3K9me2 and H3K9me3) are histone marks generally associated with a compact, closed chromatin state (heterochromatin) and gene repression (Barski et al., 2007). Several HMTs responsible for the deposition of these methyl groups are altered in cancer and some inhibitors of these enzymes have been developed. Chaetocin is a fungal mycotoxin capable of inhibiting SUV39H1 methyltransferase in vivo (Greiner et al., 2005) and exhibits anti-tumour effects in leukaemia cell lines in vitro and primary AML cells ex vivo (Chaib et al., 2012). However, the specificity of this compound for inhibiting the SUV39H1 enzyme has already been questioned (Cherblanc et al., 2013). The low MW compound BIX-01294 is also an example of an H3K9 methyltransferase inhibitor. It blocks G9A (EHMT2) and leads to a decrease of proliferation and induced apoptosis of neuroblastoma cells (Kubicek et al., 2007; Chang et al., 2009; Lu et al., 2013). A new generation analogue based on the BIX-01294 structure is 3-deazaneplanocin, a GLP and G9a inhibitor with higher in vitro potency and better cell membrane permeability than its precursors (Vedadi et al., 2011). The SETDB1 gene, which encodes another HMT for H3K9, has been found to be amplified in melanoma and lung cancer and its expression can be diminished by a clinically approved anti-tumour antibiotic, mithramycin, which binds to the SETDB1 promoter and inhibits the binding of Sp transcription factors (Figure 2; Ryu et al., 2006; Ceol et al., 2011; Rodriguez-Paredes et al., 2013).
Histone acetyltransferases (HATs)
Acetyltransferases mediate the transfer of an acetyl group from acetyl coenzyme A (acetyl-CoA) to the ε-amino group of lysine residues in histones and other proteins. Upon acetylation, the positive charge of lysines is neutralized and, in the case of histones, the interaction with the negatively charged DNA backbone is diminished, producing an open chromatin status. HATs have been classified into type A HATs, which are nuclear proteins that acetylate chromatin-associated proteins and histones, and type B HATs, which are located both in the nucleus and the cytoplasm and acetylate newly synthesized cytoplasmic histones to promote their nuclear localization and deposition onto nascent DNA chains (Ruiz-Carrillo et al., 1975). While type A HATs comprise three families of enzymes (GNATs, P300/CBP and MYST), KAT1 is the only HAT in the type B group. Despite showing little sequence homology, the catalytic domains of all HATs are organized around a recognizable acetyl-CoA binding site. This pocket that contains the acetyl-CoA cofactor seems to be a putative chemically tractable domain (Liu et al., 2008).
Knowing that the catalytic activity of HAT enzymes requires the presence of acetyl-CoA, HAT inhibitors are conjugates, derivatives or compounds related to this coenzyme (Figure 3). These compounds can be classified into bisubstrate HAT inhibitors, natural product HAT inhibitors and low MW HAT inhibitors. The first HAT inhibitor class to be identified was a bisubstrate inhibitor, whose mechanism of action is to imitate the acetyl-CoA-lysine intermediate complex in HAT reactions. The major limitation of this class of HAT inhibitors is that they do not have drug-like properties because of their lack of cell permeability (Lau et al., 2000). Unfortunately, most of the natural product HAT inhibitors also have a similar limitation. Anacardic acid, a bioactive phytochemical found in the shell of nuts from Anacardium occidentale, is a non-competitive inhibitor of p300, PCAF and Tip60 (Hemshekhar et al., 2012). Chemically, anacardic acid is a mixture of organic compounds structurally related to salicylic acid, each substituted with different alkyl groups of different length, from C15 to C17, and degrees of unsaturation, up to three double bonds (Paul and Yeddanapalli, 1956; Figure 3B). Although it has HAT inhibitor activity, this molecule has poor cell permeability, which limits its practical applications (Eliseeva et al., 2007). In contrast, garcinol, a compound extracted from the rinds of the Garcinia indica fruit, is a highly permeable but non-specific HAT inhibitor. Its non-specific nature makes it highly cytotoxic (Balasubramanyam et al., 2004), which inspired the synthesis of LTK14 and LTK15, two garcinol derivatives that are more specific and less cytotoxic (Mantelingu et al., 2007). One of the best characterized natural HAT inhibitors is curcumin, which is extracted from the rhizome of Curcuma longa and presents high efficacy in the prevention and treatment of several tumour types, such as those of head and neck and lung cancer (Kumar et al., 2014; Malhotra et al., 2014). The last group of HAT inhibitors include several low MW compounds with better cell permeability properties, like α-methylene-γ-butyrolactone 3, quinoline, isothiazolone and their derivatives, which have been linked to reduced cell proliferation in human colon cancer cell lines (Stimson et al., 2005). One of the most recently described low MW inhibitors is C646, a selective, potent and drug-like HAT inhibitor that, after binding to EP300, acts as a cofactor competitor inducing apoptosis in prostate cancer cells (Table 1; Bowers et al., 2010; Santer et al., 2011).
Figure 3.

Inhibition of HATs and HDACs. (A) HATs are responsible for the acetylation of certain lysine residues of histone tails, inducing a looser open state of chromatin structure (euchromatin) and, therefore, transcriptional activation of certain genes. A reverse process occurs when HDACs remove the acetyl group of lysine residues, the DNA–histone interaction is intensified and, thus, chromatin acquires a more condensed state (heterochromatin). Several compounds have been designed for the inhibition of HATs and HDACs (HATi and HDACi). (B) HAT inhibitors include natural products like anacardic acid, garcinol and curcumin, and small molecules like α-methylene-γ-butyrolactone 3 (MB-3), C646 and isothiazolone. (C) Romidepsin, vorinostat, panobinostat, belinostat, entinostat and valproic acid are some of the reported HDACis.
The lack of powerful HAT inhibitors reported to date suggests that many of the functions and interactions of these enzymes are yet to be fully investigated. There are several factors complicating the analysis of HAT functions. For example, it is noteworthy that HATs are part of large multi-protein complexes and the study of the whole reconstituted complexes may be necessary if we are to discover new inhibitors.
Targeting epigenetic erasers
Epigenetic modifications of the genome that are deposited by epigenetic writers also need to be removed by specific epigenetic erasers in order to regulate gene expression. Epigenetic erasers are classified in several groups of enzymes that target histones; these include, among others, histone lysine and arginine demethylases and HDACs.
Histone demethylases
Until a decade ago, histone methylation was considered a stable chemical modification that, together with DNA methylation, defined epigenetic programmes, but this view changed with the discovery of lysine-specific demethylase 1 (LSD1; Shi et al., 2004) and the identification of the JMJC-domain-containing lysine demethylase family (Tsukada et al., 2006; Whetstine et al., 2006). Therefore, histone lysine methylation is not a fixed modification and can be dynamically regulated at particular gene loci through the recruitment of methyltransferases and demethylases, as with acetylation (Barth and Imhof, 2010; Zee et al., 2010).
Several members of the histone demethylase family appear to be genetically amplified and overexpressed in some human tumours. Moreover, the active site of these epigenetic enzymes is well studied. All these characteristics have prompted the assessment of HMTs as putative drug targets and the development of selective and high-affinity low MW inhibitors of tumour growth.
LSD family
The LSD family comprises only two members: the histone demethylases LSD1 (KDM1A) and LSD2 (KDM1B). The oxidase-like domain of these enzymes is responsible for the catalytic activity and the removal of the methyl group from histone lysines is performed through an oxidation mechanism that depends on the cofactor FAD (Fitzpatrick, 2010). The LSD enzymes catalyse the demethylation of mono- and dimethylated lysines but not trimethylated lysines, in contrast to JMJC-domain histone demethylases. LSD1 protein presents high-level expression in prostate, oestrogen-negative breast, bladder and colorectal cancers (Kahl et al., 2006; Hayami et al., 2011; Kauffman et al., 2011), among others. Therefore, LSD1 could be a tumour biomarker to consider as a valuable therapeutic target.
As the sequence of the catalytic domain of the LSD proteins is highly homologous with that of the MAO A and MAO B enzymes, some existing MAO inhibitors, like tranylcypromine, can also inhibit LSD1 (Lee et al., 2006; Schenk et al., 2012). Given that non-selective amine oxidase inhibitors have several side effects, derivatives of tranylcypromine are being developed as more potent and selective LSD1 inhibitors. The biotech company Oryzon Genomics has already examined ORY-1001 in clinical trials for the treatment of relapsed or refractory acute leukaemia (EudraCT Number: 2013–002447-29; Table 2). Although tranylcypromine derivatives are the most promising compounds described so far, other molecules have been identified as LSD1 inhibitors, such as polyamines with no target selectivity and clear potential side effects (Huang et al., 2007; 2009,). Other studies have investigated a weak but selective LSD1 inhibitor which has in vitro and in vivo activity (Willmann et al., 2012), and an inhibitor that was designed to mimic peptide-based inhibitors (Wang et al., 2011).
Table 2.
Inhibitors of epigenetic erasers
| Category | Compound | Phase | Tumour type | References |
|---|---|---|---|---|
| Histone demethylase inhibitors | ||||
| LSD1 inhibitors | Tranylcypromine | Preclinical | AML | Schenk et al., 2012 |
| ORY-1001 | Clinical trials | Relapsed or refractory acute leukaemia | 2013-002447-29 | |
| JMJC domain-containing inhibitors | 8-hydroxyquinolines | Preclinical | (Enzymatic inhibition of the target protein) | King et al., 2010 |
| Pyridine hydrazones | Preclinical | (Enzymatic inhibition of the target protein) | Chang et al., 2011 | |
| GSK-J1 | Preclinical | (Enzymatic inhibition of the target protein) | Kruidenier et al., 2012 | |
| HDAC inhibitors | ||||
| Cyclic peptides | Romidepsin | Approved | Refractory cutaneous T-cell lymphoma | Whittaker et al., 2010 |
| Hydroxamic acids | TCA | Preclinical | Lung | Chan et al., 2013 |
| Vorinostat | Approved | Cutaneous T-cell lymphoma, lymphoid malignancies | Mann et al., 2007; NCT00691210 | |
| Panobinostat | Clinical trials | Haematological malignancies, solid tumours | Cassier et al., 2014; NCT01321346 | |
| Belinostat | Clinical trials | Ovary, colon, haematological malignancies | Plumb et al., 2003; NCT01686165 | |
| Pracinostat | Clinical trials | Colorectal, MDS | Novotny-Diermayr et al., 2010; NCT01873703 | |
| Quisinostat | Clinical trials | Advanced solid tumours, cutaneous T-cell lymphoma | Venugopal et al., 2013; NCT01486277 | |
| CHR-3996 | Clinical trials | Refractory solid tumours | Banerji et al., 2012; NCT00697879 | |
| Short-chain fatty acids | Phenylacetate | Clinical trials | Brain | NCT00003241 |
| Phenylbutirate | Clinical trials | Advanced colorectal cancer | NCT00002796 | |
| Valproic acid | Clinical trials | Leukaemia, solid tumours | Fredly et al., 2013; Coronel et al., 2011 | |
| Benzamides | Entinostat | Clinical trials | Refractory advanced non-small cell lung cancer | Juergens et al., 2011 |
| Mocetinostat | Clinical trials | Haematological malignancies, advanced solid tumours | Younes et al., 2011; Siu et al., 2008 | |
| CS055 | Clinical trials | Advanced non-small cell lung cancer | NCT01836679 |
JMJC domain-containing demethylases
The catalytic JMJC domain is characteristic of the second family of histone lysine demethylases. This demethylation reaction occurs through an oxidative mechanism that requires two cofactors, 2-oxoglutarate and iron, and it concludes with the hydroxylation of the methyl group (McDonough et al., 2010; Kooistra and Helin, 2012). Through this mechanism, JMJC proteins are able to remove methyl groups from mono-, di- and trimethylated lysines. Thus, the JMJC demethylases can demethylate trimethylated lysines, in contrast to the LSD demethylases (Cloos et al., 2006; Whetstine et al., 2006). Several results have associated this histone lysine demethylase family with cancer. For example, members of the JMJD2 subfamily are overexpressed by genomic amplification in breast cancer, squamous cell carcinoma and medulloblastoma (Yang et al., 2000; Ehrbrecht et al., 2006; Liu et al., 2009). Moreover, members of the JARID1 family are overexpressed in breast and bladder cancers (Lu et al., 1999; Hayami et al., 2010), and FBXL10 is also up-regulated in leukaemia (He et al., 2011). Given its strong association with cancer development, targeting the enzymic activity of JMJC proteins could have therapeutic potential.
Most of the current inhibitors of JMJC domain-containing demethylases are methyl chelators that bind to the catalytic pocket containing iron and compete with the 2-oxoglutarate cofactor for its binding (Lohse et al., 2011). This is the case of 8-hydroxyquinolines and pyridine hydrazones, which are potent and selective inhibitors with good drug-like properties, such as cell permeability (King et al., 2010; Chang et al., 2011). Another encouraging compound is GSK-J1, which is an inhibitor of the JMJD3 subfamily and works by binding competitively to the 2-oxoglutarate cofactor and chelating the metal in the active site (Table 2; Kruidenier et al., 2012). A recently discovered therapeutically promising compound is a highly specific and potent inhibitor of the JARID1 family developed by the biotech company EpiTherapeutics. Xenograft mouse models have already been treated with this drug and a reduction of the proliferation of cancer cells has been shown (L.-O. Gerlach, pers. comm.).
HDACs
HDACs are enzymes responsible for the removal of the acetyl group of lysine residues in histones. Upon histone deacetylation, the positive charge of lysines is restored and the intensified interaction with the DNA backbone brings about a transcriptional repression state within heterochromatin, a more condensed form of chromatin. HDACs are divided into five classes based on their phylogenetic comparison with yeast enzymes. Class I comprises HDAC1, HDAC2, HDAC3 and HDAC8; class IIa consists of HDAC4, HDAC5, HDAC7 and HDAC9; class IIb includes HDAC6 and HDAC10; class III comprises the sirtuins from SIRT1 to SIRT7; and class IV contains only HDAC11. Enzymes from classes I, II and IV require a zinc ion for catalysis, whereas sirtuins are NAD+-dependent enzymes with protein deacetylase and ADP-ribosylase activity (Sauve, 2010).
Many proteins other than histones have been recognized as substrates for HDACs (Choudhary et al., 2009). For example, HDAC6 is involved in the deacetylation of microtubules and the hsp90 chaperone (Hubbert et al., 2002; Kovacs et al., 2005). Other non-histone proteins like the tumour suppressor p53 are deacetylated by class I HDACs (Luo et al., 2000). HDACs are sometimes referred to as lysine deacetylases rather than HDACs because of the many non-histone targets.
More than 10 years ago, Lin et al. (2001) discovered that deregulation of HDAC activity in association with chromosomal translocation was involved in the stimulation of leukemogenesis. To date, several studies have provided evidence of aberrant acetylation and altered expression of HDACs in cancer cells and tumour tissues (Ropero and Esteller, 2013; West and Johnstone, 2014). Therefore, using HDAC inhibition to reverse epigenetic aberrancies in cancer cells is a powerful approach for the treatment of several tumour types (Figure 3).
HDAC inhibitors are compounds that bind to the catalytic pocket of HDACs and prevent substrate binding to the enzyme, leading to re-expression of particular genes. These inhibitors affect several biological processes: they cause cell cycle arrest in G1 and/or G2 phase, leading to inhibition of cell growth (Bolden et al., 2006); they induce cell differentiation and promote apoptosis (Nebbioso et al., 2005); they inhibit angiogenesis and even enhance sensitivity to chemotherapy (Geng et al., 2006; Qian et al., 2006). HDACi have been classified into four major structural classes with different HDAC subtype selectivity profiles: cyclic peptides, hydroxamates, short-chain fatty acids and benzamides (Table 2).
Romidepsin (Istodax®) is a member of the cyclic peptide group. This class I HDAC-selective inhibitor is a prodrug isolated from Chromobacterium violaceum, which is activated in the cell after a reduction reaction (Furumai et al., 2002). Romidepsin induces cell cycle arrest and apoptosis in several human cancer cells and it was approved by the US FDA in 2009 for the treatment of refractory cutaneous T-cell lymphoma (Piekarz et al., 2014; Whittaker et al., 2010) and in 2011 for the treatment of peripheral T-cell lymphoma (Piekarz et al., 2011; Coiffier et al., 2012).
The high potency of romidepsin, like most of the HDAC inhibitors, is due to a crucial functional group that binds strongly to the Zn2+ ion in the active site of the enzyme. Nevertheless, this feature of its inhibition seems also to be responsible for undesirable off-target interactions with other metalloenzymes. In order to diminish these consequences, cyclic peptides that exhibit HDAC inhibition in the nM range and lack the Zn2+ binding moiety have been described recently. They are active against human cancer cell lines in vitro and, thus, could be precursors for the development of new drugs (Vickers et al., 2012).
Another important structural group is the hydroxamic acids, which include the first compound found to inhibit HDACs: the natural product, trichostatin A (TSA; Yoshida et al., 1990). Although TSA has a wide range of anti-cancer effects (Hu and Colburn, 2005; Chan et al., 2013), it has negative side effects that have excluded this molecule from clinical trial. However, a new generation of compounds like vorinostat, panobinostat, belinostat, givinostat and pracinostat (SB939) are already under clinical investigation. Vorinostat (Zolinza®), which inhibits HDAC1, HDAC2, HDAC3 and HDAC6, can induce differentiation, cell growth arrest and apoptosis in various cancer cell lines at μM concentrations, and inhibits tumour growth with low toxicity in murine xenograft models (Marks, 2007). In 2006, vorinostat was approved by the FDA for the treatment of cutaneous T-cell lymphoma in patients with persistent, progressive or recurrent disease (Mann et al., 2007). Moreover, there is an ongoing clinical trial combining vorinostat with two other anti-cancer drugs for the treatment of lymphoid malignancies (Amengual et al., 2013; ClincalTrials.gov identifier: NCT00691210). Panobinostat, a non-selective HDAC inhibitor, exhibits cytotoxic and anti-proliferative activity in several cancer cell lines and induces apoptosis in human tumour xenografts (Vilas-Zornoza et al., 2012). This compound is currently undergoing clinical trials for the treatment not only of several myeloproliferative disorders like multiple myeloma, chronic myeloid leukaemia and Hodgkin's lymphoma (Younes et al., 2012), but also of solid tumours like prostate cancer and ovarian sex-cord tumours (Cassier et al., 2014). Belinostat is a potent HDAC inhibitor that exerts growth-inhibitory and proapoptotic effects in human cancer cell lines and xenografts at nM concentrations (Plumb et al., 2003). Pracinostat (SB939) is a novel HDACi with improved pharmacokinetic properties: it has higher bioavailability and a longer plasma half-life than vorinostat (Novotny-Diermayr et al., 2010). Quisinostat (JNJ-26481585) is a hydroxamic acid-containing inhibitor with potent anti-tumoural activity and encouraging pharmacodynamic properties that are currently investigated in clinical trial (Arts et al., 2009; Venugopal et al., 2013). CHR-3996 is a potent and promising class I HDAC inhibitor with good oral bioavailability and the ability to completely inhibit human tumour xenografts (Moffat et al., 2010). This compound is also already being used in the clinical setting (Banerji et al., 2012).
A third category of HDAC inhibitor is the short-chain fatty acids, including sodium phenylacetate, phenylbutyrate and valproic acid. These three compounds have weaker inhibitory effects than romidepsin or vorinostat, but they are already used clinically for other reasons (acute hyperammonemia, urea cycle disorders and epilepsy respectively). The well-characterized kinetic properties and side effects of these molecules has led to some of them, such as valproic acid, being investigated to determine their value as anti-leukaemic agents in combination with other drugs (Fredly et al., 2013). In addition, the only HDAC inhibitor that has achieved a phase III clinical trial in solid tumours is valproic acid. These trials shown a significant improvement in progression-free survival of cervical cancer patients treated with valproic acid added to a current standard combination chemotherapy (Coronel et al., 2011).
Benzamides like entinostat (MS-275), mocetinostat (MGCD-0103) and CS055 are another class of HDAC inhibitors. These compunds have an amino anilide group which is responsible for enzyme inhibition and the selectivity for class I HDACs (Bressi et al., 2010). Entinostat administered orally inhibits cell proliferation and growth in human cancer cell lines implanted into nude mice (Saito et al., 1999), and induces apoptosis of B-lymphocytic leukaemia cells (Lucas et al., 2004). It has already undergone clinical trials in combination with other existing anti-tumour drugs (Juergens et al., 2011). Mocetinostat, developed by the biotech company MethylGene, induces apoptosis and exhibits anti-proliferative activities against human cancer cell lines and xenografts (Fournel et al., 2008). It is currently in phase II clinical trials for the treatment of haematological malignancies (Younes et al., 2011) and in phase I/II trials with solid tumours (Siu et al., 2008). CS055, a compound structurally similar to entinostat, is a class I selective inhibitor with lower toxicity and better tolerance than other benzamides, and which induces growth arrest, apoptosis and differentiation of leukaemia cells (Ke et al., 2012).
Although HDAC inhibitors were initially included in clinical trials as single therapeutic agents, the tendency to use them in combination with other anti-cancer drugs is increasing. In haematological malignancies, for example, HDACs are aberrantly recruited to nuclear protein complexes and, for this reason, the current DNA-methyltransferase inhibitor therapy is now combined with HDAC inhibtion (Khan and La Thangue, 2012). As HDAC6 has a role in protein degradation, another combined regimen that can enhance the effects of HDAC inhibition involves proteasome inhibitors (Jagannath et al., 2010). Therefore, the synergistic effects achieved by the combination of HDAC inhibition and classical chemotherapy represent a further step forward in cancer therapy research.
Targeting epigenetic readers
Many epigenetic regulators contain specialized domains that allow them to ‘read’ the chromatin by recognizing specific covalent histone modifications, interpreting them and imposing structural changes (Taverna et al., 2007). Chromatin readers are able to identify different modified amino acids and also different modification states of the same amino acid. For instance, as mentioned above, lysine residues can be affected by several covalent modifications, such as methylation, acetylation, sumoylation and ubiquitination. Another level of complexity is achieved when each lysine residue can undergo several degrees of methylation: unmethylation, monomethylation, dimethylation or even trimethylation. Chromatin readers contain several types of methyl-lysine-recognition motifs like tudor domains, chromodomains, PWWP domains and plant homeodomain fingers (PHD). Each type of domain within a family of proteins can have variants that alter their preferred binding substrate. For example, the PHD finger of the protein ING2 binds to di- and trimethylated lysines (Pena et al., 2006; Shi et al., 2006), while the PHD fingers of DNMT3L and BHC80 prefer binding to unmethylated residues (Lan et al., 2007; Ooi et al., 2007). On the other hand, if a lysine residue undergoes acetylation instead of methylation, a different docking site is generated and it immediately recruits proteins with acetyl-lysine binding motifs, like bromodomains (Jacobson et al., 2000). Finally, adding a new layer of difficulty, many chromatin regulators include several types of reader domains in their structure and the binding at specific chromatin sites depends on the surrounding histone modification map (Ruthenburg et al., 2007).
Many epigenetic readers have been found to be disrupted in a variety of diseases, including cancer (Chi et al., 2010). These chromatin reader domains are novel targets for the development of new therapies against this malignancy. For example, low MW compounds that specifically inhibit the bromodomain family proteins are being studied (Table 3; Dawson et al., 2011).
Table 3.
Inhibitors of epigenetic readers
| Category | Compound | Phase | Tumour type | References |
|---|---|---|---|---|
| Bromodomain inhibitors | ||||
| BET inhibitors | JQ1 | Preclinical | NUT midline carcinoma, MLL-rearranged leukaemia, multiple myeloma | Filippakopoulos et al., 2010; Dawson et al., 2011; Delmore et al., 2011 |
| I-BET151 | Preclinical | Multiple myeloma | Chaidos et al., 2014 | |
| I-BET762 | Clinical trials | Haematological malignancies, NUT midline carcinoma, solid tumours | NCT01943851; NCT01587703 | |
| BAZ2B inhibitors | GSK2801 | Preclinical | (Enzymic inhibition of the target protein) | http://www.thesgc.org/chemical-probes/GSK2801 |
| Chromodomain inhibitors | ||||
| L3MBTL1 inhibitors | UNC669 | Preclinical | (Enzymic inhibition of the target protein) | Herold et al., 2011 |
| L3MBTL3 inhibitors | UNC1215 | Preclinical | (Enzymic inhibition of the target protein) | James et al., 2013 |
Bromodomain proteins
Bromodomains are highly conserved motifs which, after identifying and binding to acetylated lysines on histone tails, form a scaffold for the assembly of multi-protein macromolecular complexes that facilitate DNA-templated processes. More than 50 bromodomain proteins are encoded by the human genome and they can be clustered in nine subfamilies according to sequence homology (Filippakopoulos et al., 2012). Bromodomain proteins are physiologically important because experimental knockout of particular proteins of this family in mice results in embryonic lethality (Gyuris et al., 2009).
The bromodomain and extraterminal (BET) subfamily includes four protein members (BRD2, BRD3, BRD4 and BRDT), which contain a tandem bromodomain at the N-terminal. These proteins play a decisive role in the regulation of transcription and cell growth (Figure 4). For instance, BRD4 is associated with a coactivator complex of transcription (Malik and Roeder, 2010) and promotes transcriptional elongation by increasing the processivity of RNA polymerase II, leading to expression of growth-promoting genes (Jang et al., 2005; Yang et al., 2005). BET proteins are usually part of large nuclear complexes that are involved not only in transcription processes, but also in chromatin remodelling, replication and DNA damage (Dawson et al., 2011). Hence, dysregulation of BET proteins has been reported in several diseases such as cancer. BRD2, for example, is overexpressed in lymphocytes of B-cell lymphoma patients (Greenwald et al., 2004) and recurrent translocations of both BRD3 and BRD4 are drivers of proliferation in the lethal malignancy NUT midline carcinoma (French, 2010). Moreover, BRD4 has been discovered as a therapeutic target in AML (Zuber et al., 2011). All these discoveries have increased current interest in the therapeutic targeting of BET proteins.
Figure 4.
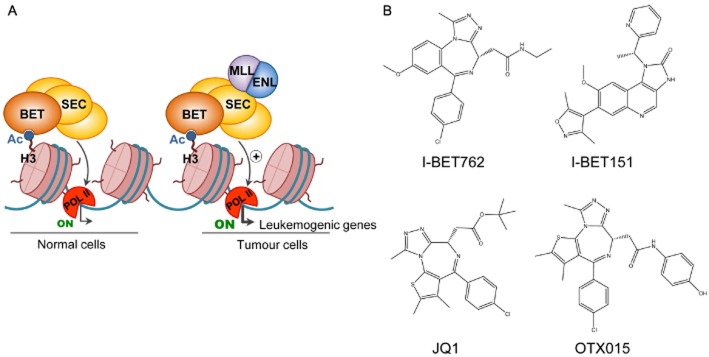
Inhibition of bromodomain proteins. (A) Bromodomains can recognize and bind to acetylated lysine residues on histone tails, recruiting macromolecular complexes that facilitate DNA-templated processes. For example, BET proteins are associated with the superelongation complex (SEC) for the regulation of gene transcription. However, in MLL-translocated leukaemia, MLL-fusion proteins are responsible for an abnormal activity of this complex, which leads to aberrant transcriptional programmes that culminate in disease. (B) The chemical structures of the BET inhibitors JQ1, I-BET762, I-BET151 and OTX015 are shown.
BET inhibitors are a new class of molecules designed to block the assembly of a functional protein complex at a particular gene locus by obstructing the interaction of the bromodomain with the acetylated residue (Table 3). To date, some BET inhibitors (JQ1, I-BET151, I-BET762, OTX015, TEN-010 and CPI-0610) have been developed for the treatment of haematopoietic malignancies and the rare NUT midline carcinoma disease (French, 2010; Dawson et al., 2011; Filippakopoulos and Knapp, 2014). The BET bromodomain inhibitor JQ1 induces differentiation and stops the proliferation of NUT midline carcinoma cell lines and murine xenografts because it is able to displace BRD4-NUT, one of the aberrant fusion proteins responsible for this disease (Filippakopoulos et al., 2010). JQ1 is also highly effective in vitro and in vivo against AML with MLL translocations, a scenario in which the inhibition of BET proteins reduces the transcriptional activity exerted by the leukaemic MLL fusions and, thus, the transcription of genes that are essential for the maintenance of leukaemia (Dawson et al., 2011). Additionally, JQ1 has a dramatic effect on multiple myeloma cell lines, preventing the binding of BRD4 in the upstream region of the MYC promoter and diminishing the transcription and expression of this potent oncogene (Delmore et al., 2011; Mertz et al., 2011). The BET bromodomain inhibitor I-BET151 is also responsible for reducing the expression of crucial oncogenes such as MYC in multiple myeloma (Chaidos et al., 2014) and the BET inhibitor I-BET762 is undergoing clinical trials for the treatment of haematological malignancies, NUT midline carcinoma and other solid tumours such as N-MYC-amplified lung and colorectal cancers (ClincalTrials.gov identifiers: NCT01943851 and NCT01587703).
OTX015 targets three of the four members of the BET subfamily: BRD2, 3 and 4. In preliminary results from an ongoing phase I trial, the drug shows tolerability and promising clinical responses in some patients with acute leukaemia and other haematological malignancies (ClinicalTrials.gov identifier: NCT01713582). TEN-010 is another BET inhibitor that is already in clinical trials for the treatment of NUT midline carcinoma patients and for the treatment of those cases with advanced solid tumours that do not respond to approved therapies (ClinicalTrials.gov identifier: NCT01987362). The BET inhibitor CPI-0610 is also undergoing clinical trials for the treatment of AML, MDS, multiple myeloma and lymphoma (ClinicalTrials.gov identifiers: NCT01949883, NCT02158858 and NCT02157636).
Concluding remarks
The ongoing research into cancer epigenetics is increasing general knowledge about the molecular bases of this disease and it is now definitely established as an important source for drug development. The epigenetic proteins described in this review represent several targets for the discovery of new active drugs. In fact, the scientific community already has at its disposal inhibitors of reading, writing or erasing of the histone code that have been discovered through different approaches. On the one hand, many studies have shown that amplifications, translocations and somatic mutations in genes that encode for chromatin-related proteins appear to be frequent in cancer, and the discovery of compounds that target the active domain of these epigenetic regulators has been fruitful. On the other hand, targeting protein–protein interactions that confine chromatin elements in particular locations has enabled the discovery of other novel anti-cancer drugs.
Although some of the existing inhibitors are already in clinical trials for the treatment of various tumour types, there is still a long way to go. Most of the current clinical trials have been based on genetic aberrations of the targeted protein in a specific cancer type but, in some tumours, the epigenetic therapeutic targets are not necessarily mutated. Thus, simple mutational screenings are not enough to enable responses to be predicted. They should be combined with drug sensitivity studies in which specific inhibitors are tested in large well-characterized cell line panels (Barretina et al., 2012; Garnett et al., 2012).
Chromatin proteins are mostly components of larger complexes in the cell, implying that the activity of inhibitors against individual proteins, outside their cellular context, could differ considerably from their activity inside the natural multifunctional complexes. Additionally, these multi-component functional units are linked with several genes and specific locations along the genome. Depending on the tissue type, the genetic scenery of each cell and the biological circumstances, the same chromatin protein could act as an oncogene or a tumour suppressor, thereby increasing the level of complexity. A full understanding of the biological functions of the target proteins and also a more detailed mechanism of action of chromatin protein inhibitors is still a challenge. In fact, the generation of new active chemical molecules with higher specificity will be decisive in revealing the biological function of new chromatin-associated proteins and discovering other pathways that could also be crucial for tumour development.
Currently, the best anti-tumour therapies responses are achieved by targeting several oncogenic pathways simultaneously. Epigenetic drugs are already used in combination with established cancer chemotherapy and, in some cases, targeting the epigenome could directly reverse transcriptional resistance mechanisms (Stronach et al., 2011). In fact, the regulation of gene expression at the transcriptional level is also the mechanism of the recently discovered bromodomain inhibitors for the silencing of crucial oncogenes in several tumour types. Although more detailed knowledge about how these BET inhibitors work is required, they are very attractive compounds for introduction into the clinical setting. Despite all the well-acknowledged limitations, current epigenetic-targeting molecules are already proving successful in cancer therapy and this is a good basis to motivate basic research scientists, clinicians and also the pharmaceutical industry to look for new weapons in the present and future fight against cancer.
Author contributions
Both authors contributed equally to this work.
Conflict of interest
The authors declare no conflict of interest.
Glossary
- AML
acute myeloid leukaemia
- FDA
Food and Drug Administration
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- HMT
histone methyltransferase
- KMT
protein lysine methyltransferase
- LSD1
lysine-specific demethylase 1
- MDS
myelodysplastic syndrome
- PHD
plant homeodomain fingers
- PRC2
polycomb repressive complex 2
- PRMT
protein arginine methyltransferase
- PTM
post-translational modification
- SAM
S-adenosylmethionine
- TSA
trichostatin A
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatangelo MD, Garipov A, Li H, Conejo-Garcia JR, Speicher DW, Zhang R. Three-dimensional culture sensitizes epithelial ovarian cancer cells to EZH2 methyltransferase inhibition. Cell Cycle. 2013;12:2113–2119. doi: 10.4161/cc.25163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amengual JE, Clark-Garvey S, Kalac M, Scotto L, Marchi E, Neylon E, et al. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 2013;122:2104–2113. doi: 10.1182/blood-2013-02-485441. [DOI] [PubMed] [Google Scholar]
- Arts J, King P, Mariën A, Floren W, Beliën A, Janssen L, et al. JNJ-26481585, a novel ‘second-generation’ oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res. 2009;15:6841–6851. doi: 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279:33716–33726. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- Banerji U, van Doorn L, Papadatos-Pastos D, Kristeleit R, Debnam P, Tall M, et al. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin Cancer Res. 2012;18:2687–2694. doi: 10.1158/1078-0432.CCR-11-3165. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci. 2010;35:618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, Jong RD, et al. Exploration of the HDAC2 foot pocket: synthesis and SAR of substituted N-(2-aminophenyl) benzamides. Bioorg Med Chem Lett. 2010;20:3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- Cassier P, Floquet A, Penel N, Derbel O, N'guyen BB, Guastalla J-P, et al. The histone deacetylase inhibitor panobinostat is active in patients with advanced pretreated ovarian sex-cord tumors. Ann Oncol. 2014;25:1074–1075. doi: 10.1093/annonc/mdu045. [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaib H, Nebbioso A, Prebet T, Castellano R, Garbit S, Restouin A, et al. Anti-leukemia activity of chaetocin via death receptor-dependent apoptosis and dual modulation of the histone methyl-transferase SUV39H1. Leukemia. 2012;26:662–674. doi: 10.1038/leu.2011.271. [DOI] [PubMed] [Google Scholar]
- Chaidos A, Caputo V, Gouvedenou K, Liu B, Marigo I, Chaudhry MS, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014;123:697–705. doi: 10.1182/blood-2013-01-478420. [DOI] [PubMed] [Google Scholar]
- Chan S-T, Yang N-C, Huang C-S, Liao J-W, Yeh S-L. Quercetin enhances the antitumor activity of trichostatin A through upregulation of p53 protein expression in vitro and in vivo. PLoS ONE. 2013;8:e54255. doi: 10.1371/journal.pone.0054255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, King ON, Tumber A, Woon EC, Heightman TD, McDonough MA, et al. Inhibition of histone demethylases by 4-carboxy-2, 2′-bipyridyl compounds. ChemMedChem. 2011;6:759–764. doi: 10.1002/cmdc.201100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat Chem Biol. 2013;9:136–137. doi: 10.1038/nchembio.1187. [DOI] [PubMed] [Google Scholar]
- Chesworth R, Wigle TJ, Kuntz KW, Smith JJ, Richon VM. Histone methyltransferases: opportunities in cancer drug discovery. In: Lübbert M, Jones PA, editors. Epigenetic Therapy of Cancer. Berlin, Heidelberg: Springer-Verlag; 2014. pp. 189–226. [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications – miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The putative oncogene GASC1 demethylates tri-and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30:631–636. doi: 10.1200/JCO.2011.37.4223. [DOI] [PubMed] [Google Scholar]
- Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med Oncol. 2011;28:540–546. doi: 10.1007/s12032-010-9700-3. [DOI] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer J, Walf-Vorderwülbecke V, Williams O. In focus: MLL-rearranged leukemia. Leukemia. 2013;27:1224–1228. doi: 10.1038/leu.2013.78. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121:2533–2541. doi: 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrbrecht A, Müller U, Wolter M, Hoischen A, Koch A, Radlwimmer B, et al. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol. 2006;208:554–563. doi: 10.1002/path.1925. [DOI] [PubMed] [Google Scholar]
- Eliseeva ED, Valkov V, Jung M, Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Mol Cancer Ther. 2007;6:2391–2398. doi: 10.1158/1535-7163.MCT-07-0159. [DOI] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. NEJM. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick PF. Oxidation of amines by flavoproteins. Arch Biochem Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117:6756–6767. doi: 10.1182/blood-2010-05-231548. [DOI] [PubMed] [Google Scholar]
- Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget M-C, Kalita A, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- Fredly H, Gjertsen BT, Bruserud O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia-the effects of valproic acid on leukemic cells and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin Epigenetics. 2013;5:e12. doi: 10.1186/1868-7083-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA. Demystified molecular pathology of NUT midline carcinomas. J Clin Pathol. 2010;63:492–496. doi: 10.1136/jcp.2007.052902. [DOI] [PubMed] [Google Scholar]
- Furumai R, Matsuyama A, Kobashi N, Lee K-H, Nishiyama M, Nakajima H, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- Garcia-Manero G, Fenaux P. Hypomethylating agents and other novel strategies in myelodysplastic syndromes. J Clin Oncol. 2011;29:516–523. doi: 10.1200/JCO.2010.31.0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of γ-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66:11298–11304. doi: 10.1158/0008-5472.CAN-06-0049. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, et al. Eμ-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood. 2004;103:1475–1484. doi: 10.1182/blood-2003-06-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU (VAR) 3–9. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- Gyuris A, Donovan DJ, Seymour KA, Lovasco LA, Smilowitz NR, Halperin AL, et al. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim Biophys Acta. 2009;1789:413–421. doi: 10.1016/j.bbagrm.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayami S, Yoshimatsu M, Veerakumarasivam A, Unoki M, Iwai Y, Tsunoda T, et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer. 2010;9:e59. doi: 10.1186/1476-4598-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011;128:574–586. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117:3869–3880. doi: 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemshekhar M, Sebastin Santhosh M, Kemparaju K, Girish KS. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Pharmacol Toxicol. 2012;110:122–132. doi: 10.1111/j.1742-7843.2011.00833.x. [DOI] [PubMed] [Google Scholar]
- Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, et al. Small-molecule ligands of methyl-lysine binding proteins. J Med Chem. 2011;54:2504–2511. doi: 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmlund T, Lindberg M, Grander D, Wallberg A. GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia. 2013;27:578–585. doi: 10.1038/leu.2012.265. [DOI] [PubMed] [Google Scholar]
- Hu J, Colburn NH. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-κB/p65 DNA binding. Mol Cancer Res. 2005;3:100–109. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- Huang Y, Greene E, Stewart TM, Goodwin AC, Baylin SB, Woster PM, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. PNAS. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, et al. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- Jagannath S, Dimopoulos MA, Lonial S. Combined proteasome and histone deacetylase inhibition: a promising synergy for patients with relapsed/refractory multiple myeloma. Leuk Res. 2010;34:1111–1118. doi: 10.1016/j.leukres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol. 2013;9:184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong H-S, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- Kauffman EC, Robinson BD, Downes MJ, Powell LG, Lee MM, Scherr DS, et al. Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog. 2011;50:931–944. doi: 10.1002/mc.20758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke G, Jia X, Hong Y, Wenhua L. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukemia cells. Biochem J. 2012;443:735–746. doi: 10.1042/BJ20111685. [DOI] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS ONE. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. PNAS. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. PNAS. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu J-T, Nicchitta CV, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Kruidenier L, Chung C-W, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Kumar B, Yadav A, Hideg K, Kuppusamy P, Teknos TN, Kumar P. A novel curcumin analog (H-4073) enhances the therapeutic efficacy of cisplatin treatment in head and neck cancer. PLoS ONE. 2014;9:e93208. doi: 10.1371/journal.pone.0093208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan K-P, Reinberg D, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene. 2001;20:7204–7215. doi: 10.1038/sj.onc.1204853. [DOI] [PubMed] [Google Scholar]
- Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, et al. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2, 4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, et al. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28:4491–4500. doi: 10.1038/onc.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–850. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- Lohse B, Kristensen JL, Kristensen LH, Agger K, Helin K, Gajhede M, et al. Inhibitors of histone demethylases. Bioorg Med Chem. 2011;19:3625–3636. doi: 10.1016/j.bmc.2011.01.046. [DOI] [PubMed] [Google Scholar]
- Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S, et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–15645. doi: 10.1074/jbc.274.22.15633. [DOI] [PubMed] [Google Scholar]
- Lu Z, Tian Y, Salwen HR, Chlenski A, Godley LA, Raj JU, et al. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs. 2013;24:484–493. doi: 10.1097/CAD.0b013e32835ffdbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas D, Davis M, Parthun M, Mone A, Kitada S, Cunningham K, et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18:1207–1214. doi: 10.1038/sj.leu.2403388. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Malhotra A, Nair P, Dhawan DK. Study to evaluate molecular mechanics behind synergistic chemo-preventive effects of curcumin and resveratrol during lung carcinogenesis. PLoS ONE. 2014;9:e93820. doi: 10.1371/journal.pone.0093820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Mantelingu K, Reddy B, Swaminathan V, Kishore AH, Siddappa NB, Kumar G, et al. Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chem Biol. 2007;14:645–657. doi: 10.1016/j.chembiol.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Marks P. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–672. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. PNAS. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat D, Patel S, Day F, Belfield A, Donald A, Rowlands M, et al. Discovery of 2-(6-{[(6-Fluoroquinolin-2-yl) methyl] amino} bicyclo [3.1. 0] hex-3-yl)-N-hydroxypyrimidine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J Med Chem. 2010;53:8663–8678. doi: 10.1021/jm101177s. [DOI] [PubMed] [Google Scholar]
- Morey L, Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem Sci. 2010;35:323–332. doi: 10.1016/j.tibs.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- Novotny-Diermayr V, Sangthongpitag K, Hu CY, Wu X, Sausgruber N, Yeo P, et al. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther. 2010;9:642–652. doi: 10.1158/1535-7163.MCT-09-0689. [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul VJ, Yeddanapalli LM. On the olefinic nature of anacardic acid from Indian cashew nut shell liquid. J Am Chem Soc. 1956;78:5675–5678. [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D, et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol. 2014;10:574–581. doi: 10.1038/nchembio.1532. [DOI] [PubMed] [Google Scholar]
- Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117:5827–5834. doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2003;2:721–728. [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. PNAS. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–642. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, et al. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, de Paz AM, Simó-Riudalbas L, Sayols S, Moutinho C, Moran S, et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2013;33:2807–2813. doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Carrillo A, Wangh LJ, Allfrey VG. Processing of newly synthesized histone molecules. Science. 1975;190:117–128. doi: 10.1126/science.1166303. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RJ, Bernstein BE. Genetic events that shape the cancer epigenome. Science. 2012;336:1513–1514. doi: 10.1126/science.1223730. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, et al. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. PNAS. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, et al. A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors. PNAS. 1999;96:4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer FR, Höschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644–1655. doi: 10.1158/1535-7163.MCT-11-0182. [DOI] [PubMed] [Google Scholar]
- Sauve AA. Sirtuin chemical mechanisms. Biochim Biophys Acta. 2010;1804:1591–1603. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. What controls nucleosome positions? Trends Genet. 2009;25:335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi G, Chatterjee S, Rajendran P, Li F, Shanmugam MK, Wong KF, et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol Cancer. 2014;13:e66. doi: 10.1186/1476-4598-13-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- Simó-Riudalbas L, Esteller M. Cancer genomics identifies disrupted epigenetic genes. Hum Genet. 2013;133:713–725. doi: 10.1007/s00439-013-1373-5. [DOI] [PubMed] [Google Scholar]
- Siu LL, Pili R, Duran I, Messersmith WA, Chen EX, Sullivan R, et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol. 2008;26:1940–1947. doi: 10.1200/JCO.2007.14.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P, et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Mol Cancer Ther. 2005;4:1521–1532. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- Stronach EA, Alfraidi A, Rama N, Datler C, Studd JB, Agarwal R, et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011;71:4412–4422. doi: 10.1158/0008-5472.CAN-10-4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takawa M, Masuda K, Kunizaki M, Daigo Y, Takagi K, Iwai Y, et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011;102:1298–1305. doi: 10.1111/j.1349-7006.2011.01958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Li G, Sims IIIRJ, Vaquero A, Kalakonda N, Boccuni P, et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- Tsukada Y-I, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal B, Baird R, Kristeleit RS, Plummer R, Cowan R, Stewart A, et al. A Phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:4262–4272. doi: 10.1158/1078-0432.CCR-13-0312. [DOI] [PubMed] [Google Scholar]
- Vickers CJ, Olsen CA, Leman LJ, Ghadiri MR. Discovery of HDAC inhibitors that lack an active site Zn2+-binding functional group. ACS Med Chem Lett. 2012;3:505–508. doi: 10.1021/ml300081u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas-Zornoza A, Agirre X, Abizanda G, Moreno C, Segura V, De Martino Rodriguez A, et al. Preclinical activity of LBH589 alone or in combination with chemotherapy in a xenogeneic mouse model of human acute lymphoblastic leukemia. Leukemia. 2012;26:1517–1526. doi: 10.1038/leu.2012.31. [DOI] [PubMed] [Google Scholar]
- Waddington CH. Selection of the genetic basis for an acquired character. Nature. 1952;169:625–626. doi: 10.1038/169625b0. [DOI] [PubMed] [Google Scholar]
- Wagener N, Macher-Goeppinger S, Pritsch M, Hüsing J, Hoppe-Seyler K, Schirmacher P, et al. Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer. 2010;10:524. doi: 10.1186/1471-2407-10-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011;71:7238–7249. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells R, Leber B, Zhu N, Storring J. Optimizing outcomes with azacitidine: recommendations from Canadian centres of excellence. Curr Oncol. 2014;21:44–50. doi: 10.3747/co.21.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Whittaker SJ, Demierre M-F, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- Willmann D, Lim S, Wetzel S, Metzger E, Jandausch A, Wilk W, et al. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int J Cancer. 2012;131:2704–2709. doi: 10.1002/ijc.27555. [DOI] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xie N, Wu Z, Zhang Y, Zheng YG. Bisubstrate inhibitors of the MYST HATs Esa1 and Tip60. Bioorg Med Chem. 2009;17:1381–1386. doi: 10.1016/j.bmc.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Yang Z-Q, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, et al. Identification of a novel gene, GASC1, within an amplicon at 9p23–24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60:4735–4739. [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- Younes A, Oki Y, Bociek RG, Kuruvilla J, Fanale M, Neelapu S, et al. Mocetinostat for relapsed classical Hodgkin's lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12:1222–1228. doi: 10.1016/S1470-2045(11)70265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong T-C, Prince HM, et al. Panobinostat in patients with relapsed/refractory Hodgkin's lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol. 2012;30:2197–2203. doi: 10.1200/JCO.2011.38.1350. [DOI] [PubMed] [Google Scholar]
- Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:e1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, et al. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152–1157. doi: 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Levin RS, Xu B, LeRoy G, Wingreen NS, Garcia BA. In vivo residue-specific histone methylation dynamics. J Biol Chem. 2010;285:3341–3350. doi: 10.1074/jbc.M109.063784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]