

Abstract
Background and Purpose
The effects of ketamine in attenuating morphine tolerance have been suggested to result from a pharmacodynamic interaction. We studied whether ketamine might increase brain morphine concentrations in acute coadministration, in morphine tolerance and morphine withdrawal.
Experimental Approach
Morphine minipumps (6 mg·day–1) induced tolerance during 5 days in Sprague–Dawley rats, after which s.c. ketamine (10 mg·kg–1) was administered. Tail flick, hot plate and rotarod tests were used for behavioural testing. Serum levels and whole tissue brain and liver concentrations of morphine, morphine-3-glucuronide, ketamine and norketamine were measured using HPLC-tandem mass spectrometry.
Key Results
In morphine-naïve rats, ketamine caused no antinociception whereas in morphine-tolerant rats there was significant antinociception (57% maximum possible effect in the tail flick test 90 min after administration) lasting up to 150 min. In the brain of morphine-tolerant ketamine-treated rats, the morphine, ketamine and norketamine concentrations were 2.1-, 1.4- and 3.4-fold, respectively, compared with the rats treated with morphine or ketamine only. In the liver of morphine-tolerant ketamine-treated rats, ketamine concentration was sixfold compared with morphine-naïve rats. After a 2 day morphine withdrawal period, smaller but parallel concentration changes were observed. In acute coadministration, ketamine increased the brain morphine concentration by 20%, but no increase in ketamine concentrations or increased antinociception was observed.
Conclusions and Implications
The ability of ketamine to induce antinociception in rats made tolerant to morphine may also be due to increased brain concentrations of morphine, ketamine and norketamine. The relevance of these findings needs to be assessed in humans.
Tables of Links
| TARGETS | |
|---|---|
| Ligand-gated ion channela | Transportersc |
| NMDA receptors | MRP, multidrug resistance protein, ABCC1 |
| Enzymesb | P-gp, P-glycoprotein, ABCB1 |
| CYP2B6 | |
| CYP3A4 |
| LIGANDS |
|---|
| Ketamine |
| Morphine |
| Normorphine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guideto PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c).
Introduction
The treatment of neuropathic pain and severe cancer pain remains a major challenge. Opioids such as morphine and oxycodone are used for severe and moderate cancer pain (Caraceni et al., 2012) and also as second-line pharmacological management of neuropathic pain (Dworkin et al., 2007). Long-term use of opioids can, however, cause tolerance and numerous adverse effects, including opioid-induced hyperalgesia, that restrict their use.
Since the first report by Trujillo and Akil (1991), the effects of NMDA receptor antagonists have attracted great interest in studies on opioid tolerance. Ketamine, a non-competitive antagonist of the NMDA receptor, is highly lipophilic and easily crosses the blood–brain barrier (BBB) (White, 1988). Its main clinical use is as an anaesthetic agent, but it is increasingly studied for the treatment of chronic pain and also depression (Kavalali and Monteggia, 2012). As with opioids, ketamine has abuse potential (Morgan et al., 2012). At low doses, it has been shown to be efficacious in perioperative analgesia and in chronic pain (Bell et al., 2006; Visser and Schug, 2006; Laskowski et al., 2011). In humans, the elimination t1/2 of ketamine is 2–3 h and approximately 80% of the drug undergoes hepatic metabolism to the active metabolite norketamine after oral administration (Ebert et al., 1997; Shimoyama et al., 1999; Sinner and Graf, 2008).
There is some evidence that ketamine coadministered with morphine can significantly increase the efficacy of morphine in cancer pain management (Bell et al., 2012). The majority of preclinical (Trujillo and Akil, 1994; Shimoyama et al., 1996; González et al., 1997; Miyamoto et al., 2000) and human (Bell, 1999; Nesher et al., 2009; Arroyo-Novoa et al., 2011; Hardy et al., 2012; Honarmand et al., 2012; Mathews et al., 2012; Salas et al., 2012; Suppa et al., 2012) studies have focused on the pharmacodynamic properties of ketamine as an antagonist of the NMDA receptor leading to decreased opioid tolerance and hyperalgesia. However, very little information is available of any possible pharmacokinetic interactions between opioids and ketamine. Morphine is glucuronidated to morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) by uridine diphosphate (UDP)-glucuronosyl transferase 2B7 (UGT2B7) in humans (Coffman et al., 1997) and mostly to M3G by UGT2b1 in rodents (Pritchard et al., 1994; Coffman et al., 1996; King et al., 1997). Ketamine has been shown to inhibit UGT2B7 and metabolism of morphine in vitro (Qi et al., 2010; Uchaipichat et al., 2011). In the rat, infusion of the opioid alfentanil has been reported to increase the distribution volume and brain concentration of ketamine, whereas the brain alfentanil concentration decreased during coadministration (Edwards et al., 2002). The effects of ketamine on opioid tissue concentrations and vice versa have not been studied during chronic treatment. Here, we studied the ability of ketamine to restore or augment morphine antinociception during chronic opioid treatment and tolerance, opioid withdrawal and acute coadministration. The brain, serum and liver morphine and ketamine concentrations were assessed at the time point of maximum antinociception to study a possible pharmacokinetic interaction behind the phenomenon.
This study shows that ketamine coadministration to rats under chronic morphine treatment resulted in increased brain morphine, ketamine and norketamine concentrations compared with rats treated with morphine or ketamine only. This drug interaction should be considered when studying the interactions between opioids and ketamine.
Methods
Animals
All animal care and experimental procedures were in accordance with the guidelines of the International Association for the Study of Pain (Zimmermann, 1983) and approved by the provincial government of Southern Finland (Etelä-Suomen aluehallintovirasto, Hämeenlinna, Finland, ESAVI/5684/04.10.03/2011). Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 72 rats were used in the experiments described here.
Male Sprague–Dawley rats (Scanbur, Sollentuna, Sweden; n = 8 per group; weight, 300–350 g) were housed in groups of four in plastic cages in standard light- and temperature-controlled rooms (lights on at 0700 h and off at 1900 h, temperature 22 ± 2°C). Every effort was made to minimize the stress and suffering of the experimental animals. Tap water and standard laboratory chow were available ad libitum. Before the tests, the animals were habituated to the testing environment 60 min·day–1 for 3 days. All behavioural testing was randomized and blinded. After the experiments, the animals were killed by decapitation and whole brain, liver and serum samples were collected.
Drugs for acute nociceptive tests
Morphine hydrochloride and racemic ketamine hydrochloride (Ketaminol® vet, 50 mg·mL–1, Boxmeer, The Netherlands) were purchased from the University Pharmacy, Helsinki, Finland. Morphine was dissolved and ketamine was diluted in isotonic saline and administered s.c. in a volume of 2 mL·kg–1. All drug concentrations are given as free base amounts.
Induction of morphine tolerance
Opioid tolerance was induced with continuous administration of morphine via osmotic minipumps (Alzet 2ML1, Durect, Cupertino, CA, USA). The pumps were filled with 25 mg·mL–1 morphine in isotonic saline to deliver a constant 6 mg dose of morphine daily. In pilot experiments, this steady dose of daily morphine induced tolerance after 5 days. For the control animals, isotonic saline was used as a vehicle. The pumps were implanted s.c. in the back under brief isoflurane (3.0%) anaesthesia. After 6 days from implantation, the pumps were removed under similar brief anaesthesia.
Nociceptive tests
Tail flick latencies were measured with a Ugo Basile 37360 (Comerio, Italy) radiant heat tail flick device. The rats were restrained in hard plastic tubes covered with a dark cloth. The light was directed in turn at three different points of the middle third of the tail. The mean of these three values was used as the result. The intensity was adjusted to produce a baseline latency of approximately 4 s. If an individual measurement reached cut-off (10 s), no further tests were performed for that particular time point.
Hot plate tests were performed with a Harvard Apparatus Ltd. apparatus (Edenbridge, Kent, UK). The rats were kept inside a circular transparent plastic cage on the hot plate (52 ± 0.2°C). Licking or brisk shaking of the hind paw or jumping was considered as a sign of thermal nociception. The cut-off time was set to 60 s.
The predrug (baseline) latencies were measured separately for each experimental day immediately before the administration of any drugs.
Motor coordination
A rotarod apparatus (Palmer electric recording drum, United Kingdom, diameter 80 mm, speed 12 r.p.m.) was used to assess the effects of the test drugs on motor coordination. The rat was placed on the rotating rod and the time the rat stayed on it was measured. Animals that survived at least 60 s on the rotating rod before drug administration were accepted into the test and 60 s was also used as a cut-off time in the test proper.
Determination of morphine, M3G, M6G, normorphine, ketamine and norketamine
The measurements were carried out as previously described (Zheng et al., 1998), with some modifications using an Agilent 1100 series HPLC system (Agilent Technologies, Waldbronn, Germany) coupled to an API 3000 tandem mass spectrometry (AB Sciex, Toronto, Ontario, Canada) and the chosen method was validated also for the analysis of ketamine and norketamine (data not shown). The chromatographic separation of morphine, M3G, M6G, normorphine, ketamine and norketamine was achieved on Atlantis HILIC Silica column (3 μm particle size, 2.1 × 100 mm I.D.) (Waters, Milford, MA, USA) using a gradient elution of mobile phase consisting of acetonitrile and 20 mmol·L–1 ammonium acetate (pH 3.0, adjusted with formic acid). An aliquot (5 μL) was injected at a flow rate of 200 μL·min–1 to give a total chromatographic run time of 24 min. Oxycodone served as an internal standard for morphine and its metabolites, and deuterium-labelled internal standards were used for ketamine and norketamine. The target ion transitions monitored were as follows: morphine m/z 286–152, M3G and M6G m/z 462–286, normorphine m/z 272–152, ketamine m/z 238–125 and norketamine m/z 224–125. The limit of quantification was 1.0 ng·mL–1 for morphine, M3G and M6G, and 0.5 ng·mL–1 for ketamine and norketamine. A signal-to-noise ratio of 20:1 was used as limit of detection for normorphine, and the quantities were given in arbitrary units (U·mL–1) relative to the ratio of the peak height of normorphine to that of the internal standard. The calibration curves were linear over the concentration range of LOQ-250 ng·mL–1, and day-to-day coefficients of variation were below 15% at relevant concentrations for all analytes. None of the measured compounds interfered with the mass spectrometric assay.
Experimental design
The experimental design is described briefly in Figure 1. We started by investigating the effects of an acute subanaesthetic dose of ketamine (10 mg·kg–1 s.c.) in morphine-tolerant rats under chronic morphine treatment. On day 0, a total of 48 rats were implanted with minipumps delivering morphine 6 mg·day–1 or vehicle. On day 5, after confirming the development of morphine tolerance with tail flick and hot plate nociceptive tests, rats received 10 mg·kg–1 s.c. racemic ketamine or vehicle, and antinociception was monitored up to 210 min with tail flick and hot plate tests. The rotarod test was used at 30 and 90 min to monitor the effect of the drugs on motor coordination. No extra doses of morphine were given on day 5.
Figure 1.
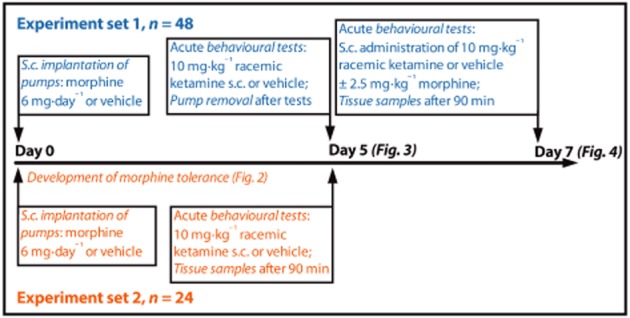
The design of the morphine tolerance and withdrawal experiments. In experiment sets 1 (blue) and 2 (red), 48 and 24 Sprague–Dawley rats were used respectively.
As significant antinociception was noticed in the morphine–ketamine cotreatment group, we continued the experiment to assess the persistence of the effects of ketamine during the morphine withdrawal period. In the evening of day 5, the minipumps were removed. On day 7, the rats were administered ketamine 10 mg·kg–1 s.c. alone or in combination with 2.5 mg·kg–1 s.c. morphine. Morphine was administered 15 min before ketamine to permit its potential effects on ketamine pharmacokinetics.
We also assessed the acute effects of morphine and ketamine coadministration in drug-naïve rats. For the tissue concentration measurements at 90 min after acute ketamine or vehicle administration, the rats were decapitated and whole brain, blood and liver samples were collected. The whole brain and part of the right lateral lobe of the liver (mean weight 400 mg) samples were covered in aluminium foil and snap-frozen in liquid nitrogen, after which they were stored at −80°C. The blood samples collected in decapitation were allowed to coagulate at room temperature for 60 min, after which they were stirred with a glass rod and centrifuged at 4900× g for 20 min at +23°C. While on ice, serum was collected and stored at −20°C.
To obtain tissue samples from day 5, a separate group of rats (n = 24) was implanted with minipumps in a similar fashion on day 0 and followed up to day 5. The development of tolerance to morphine antinociception was characterized with tail flick and hot plate tests after 1, 3 and 5 days of pump implantation. On day 5, similar to the previously described group of rats, acute ketamine or vehicle was administered and samples were collected at 90 min after drug administration. Behavioural tests were in a similar manner before the killing, and similar behavioural results were observed as with the previous group of animals.
Data analysis
The results of the hot plate and tail flick experiments are expressed as percentage of the maximum possible effect (MPE%), calculated as MPE% = [(postdrug latency – baseline latency)/(cut-off time – baseline latency)] × 100%, which takes into account the differences in baseline nociceptive latencies. The results are presented as means of the groups (±SEM). The behavioural data were tested for statistically significant differences in the mean values by two-way anova. To detect the differences in the presence of a significant two-way anova, a Holm–Sidak post hoc analysis was performed. For the concentration data, an unpaired two-tailed t-test or one-way anova followed by the Holm–Sidak post hoc analysis was used. For the nonparametric rotarod test data, the Kruskal–Wallis test followed by Dunn's multiple comparison was used. The difference was considered significant at P < 0.05 in both the analysis of variance and the post hoc test. The data were analysed using GraphPad Prism, version 6.0c for Mac OS X (GraphPad Software, La Jolla, CA, USA).
Results
Tolerance develops after 5 days of continuous morphine administration
Following 24 h of continuous morphine treatment, significant antinociception was observed both in the tail flick (Figure 2A) and hot plate (Figure 2B) tests compared with the group that had received the vehicle. On day 5, antinociceptive tolerance to morphine had developed in both tests, but no morphine-induced hyperalgesia was observed in either of the tests.
Figure 2.
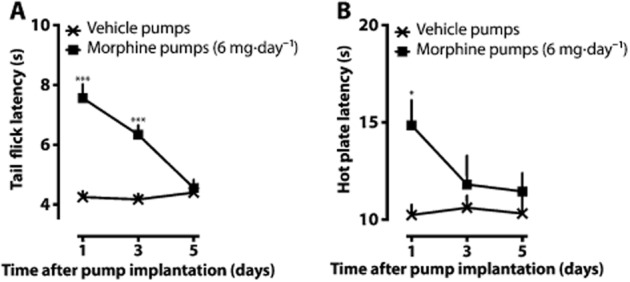
Development of antinociceptive tolerance after morphine minipump implantations. On day 0, minipumps delivering morphine 6 mg·day–1 or vehicle were implanted s.c. under brief isoflurane anaesthesia. The means (±SEM) of the tail flick (A) and hot plate (B) latencies are shown after 1, 3 and 5 days of pump implantation. The mean raw nociceptive latencies are presented to allow assessment of the data quality. No extra morphine doses were given before the behavioural measurements. *P < 0.05, ***P < 0.001; significantly different from the vehicle control group; n = 8 in the vehicle group and n = 16 in the morphine group.
A low dose of ketamine induces antinociception in opioid-tolerant rats under chronic morphine administration but not in morphine-naïve rats
On day 5, a low dose of ketamine or vehicle was administered to rats under chronic minipump administration of morphine or vehicle. Acute ketamine (10 mg·kg–1) administered to morphine-naïve rats caused no significant antinociception compared with vehicle only. However, ketamine administered to morphine-tolerant rats caused significant antinociception compared with the morphine-naïve ketamine-treated rats both in the tail flick (Figure 3A) and hot plate (Figure 3B) tests. In both tests, the effect was at its peak at 90 min after acute drug administration. In the hot plate test, a statistically significant difference compared with the morphine-naïve ketamine-treated group was reached already at 30 min, whereas in the tail flick test the effect started later (90 min) and lasted up to 150 min. Ketamine administered both to morphine-naïve and morphine-tolerant rats decreased motor performance in the rotarod test (Figure 3C) at 30 min when compared with morphine-tolerant or vehicle-treated rats, but at 90 min the motor performance had returned to normal. Ketamine administered to morphine-tolerant rats decreased the performance significantly more compared with the morphine-naïve rats that were administered ketamine.
Figure 3.
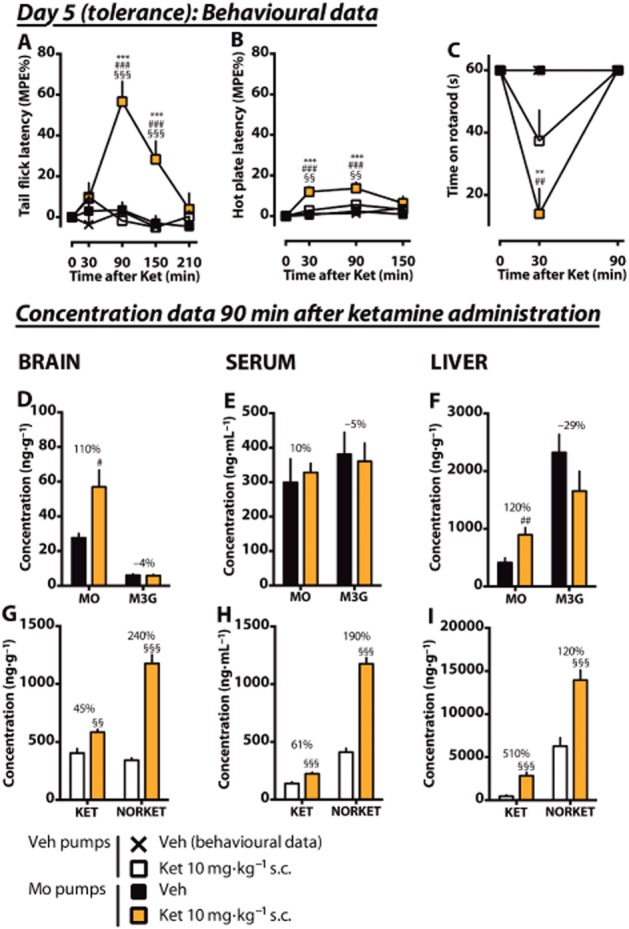
Effects of an acute low dose of ketamine on antinociception in morphine-tolerant rats under chronic morphine treatment. Rats under an ongoing morphine (6 mg·day–1, Mo) or vehicle (Veh) pump treatment received an acute s.c. dose of ketamine (10 mg·kg–1, Ket) or vehicle (Veh) on day 5. Antinociception was measured using tail flick (A) and hot plate (B) tests. The mean of the maximum possible effect (MPE%) ±SEM is plotted. In the rotarod test (C), the mean (±SEM) survival time (seconds) is plotted. From separate animals having had the same pretreatments, whole brain, serum and liver samples were collected after 90 min of ketamine administration. The tissue concentrations and serum levels of the experimental drugs and their main metabolites (morphine, MO; M3G; ketamine, KET; norketamine, NORKET) in the tissues were quantified and the means of the groups (±SEM) are presented in graphs D–I. The percentage difference between the treatment groups is shown for each compound. n = 8. **P < 0.01, ***P < 0.001; significantly different from the vehicle control group. #P < 0.05, ##P < 0.01, ###P < 0.001; significantly different from the morphine pump treated group that received acute vehicle. §§P < 0.01, §§§P < 0.001; significantly different from the vehicle-pretreated group that received acute ketamine.
A low dose of ketamine administered to rats under chronic morphine treatment increases the concentration of morphine in the brain and the liver
To study the mechanisms behind the antinociceptive effect of ketamine in morphine-tolerant rats, we measured serum levels and brain and liver whole-tissue concentrations of morphine, M3G, ketamine and norketamine at the 90 min time point (peak antinociception) of the previously described experiment. No significant amounts of M6G were detected in any sample.
In morphine-treated rats that were administered 10 mg·kg–1 s.c. ketamine, the brain concentration of morphine (Figure 3D) doubled compared with the rats that were not given acute ketamine (110% increase, P < 0.05). Meanwhile, the serum level was increased only by 10% in ketamine coadministration (Figure 3E), indicating an increased brain : serum ratio of morphine (0.17 ± 0.024 vs. 0.11 ± 0.018, P < 0.05, Table 1). The M3G concentrations or levels in the brain or serum were not significantly changed. In the liver (Figure 3F), the morphine concentration was significantly elevated after ketamine (120% increase, P < 0.05). However, the liver : serum concentration ratio was not significantly increased (Table 1). The M3G concentrations or liver : serum concentration ratios did not significantly change. The monitored normorphine peak was elevated (230% increase, P < 0.05, data not shown).
Table 1.
Tissue : serum concentration ratios for studied drugs on different study days
| Day 5 (tolerance) | Day 7 (withdrawal) | Acute administration | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Studied substance | Chronic morphine treatment (days 0–4) | Acute morphine | Acute ketamine | Brain : serum ratio | P | Liver : serum ratio | P | Brain : serum ratio | P | Liver : serum ratio | p | Brain : serum ratio | P | Liver : serum ratio | P |
| Morphine | + | – | – | 0.11 ± 0.018 | * | 1.9 ± 0.38 | n.s. | – | – | – | – | ||||
| + | – | + | 0.17 ± 0.024 | 2.8 ± 0.36 | |||||||||||
| + | + | – | – | – | 1.2 ± 0.11 | n.s. | 2.9 ± 0.53 | n.s. | |||||||
| + | + | + | 0.99 ± 0.091 | 2.8 ± 0.23 | |||||||||||
| – | + | – | – | – | 0.87 ± 0.13 | n.s. | 1.3 ± 0.13 | * | |||||||
| – | + | + | 0.95 ± 0.091 | 1.9 ± 0.22 | |||||||||||
| Ketamine | – | – | + | 2.9 ± 0.090 | n.s. | 3.0 ± 0.66 | *** | 4.0 ± 0.47 | n.s. | 4.7 ± 1.5 | n.s. | 4.0 ± 0.47 | n.s. | 4.7 ± 1.5 | n.s. |
| + | – | + | 2.7 ± 0.18 | 13 ± 2.1 | 3.9 ± 0.10 | 6.2 ± 0.69 | – | – | |||||||
| + | + | + | – | – | 4.3 ± 0.23 | 5.3 ± 0.58 | |||||||||
| – | + | + | – | – | 3.7 ± 0.21 | 5.2 ± 1.4 | |||||||||
| Norketamine | – | – | + | 0.85 ± 0.042 | * | 15 ± 1.2 | n.s. | 0.99 ± 0.15 | n.s. | 19 ± 3.9 | n.s. | 0.99 ± 0.15 | n.s. | 19 ± 3.9 | n.s. |
| + | – | + | 1.0 ± 0.059 | 12 ± 1.1 | 1.2 ± 0.083 | 15 ± 1.3 | – | – | |||||||
| + | + | + | – | – | 1.17 ± 0.080 | 15 ± 0.90 | |||||||||
| – | + | + | – | – | 1.0 ± 0.77 | 16 ± 1.7 | |||||||||
Tissue : serum (brain : serum and liver : serum) ratios (mean ± SEM, n = 8) for morphine, ketamine and norketamine shown for different experiment days. *, ***P < 0.05 and P < 0.001, respectively, between the two groups. n.s., non-significant.
Previous chronic treatment with morphine increases the concentrations of ketamine and norketamine in the brain, serum and liver
Chronic morphine treatment caused a greater disposition of ketamine into the brain compared with morphine-naïve rats that were given ketamine (45% increase, P < 0.01, Figure 3G). The increase of the norketamine concentration in the brain after drug coadministration was even more pronounced (240% increase, P < 0.001). In serum, however, ketamine and norketamine levels were elevated as well (Figure 3H), and thus only the brain : serum concentration ratio of norketamine was significantly increased (P < 0.05, Table 1). However, the most pronounced difference related to the morphine pretreatment was seen in the liver ketamine concentration (510% increase, P < 0.001, Figure 3I). The norketamine concentration was roughly doubled. The liver : serum concentration ratio of ketamine was increased fourfold (P < 0.001, Table 1), but the corresponding norketamine ratio was not altered.
The synergy in the antinociceptive effect by morphine and ketamine cotreatment continues in the withdrawal phase
In the evening of day 5, after the acute tests, the morphine and vehicle pumps were removed and on day 7, the rats were given the same dose of ketamine (10 mg·kg–1 s.c.) with or without 2.5 mg·kg–1 s.c. morphine. In the baseline values of nociceptive tests, no significant differences were detected before drug administrations (data not shown). In the tail flick test (Figure 4A), acute ketamine did not cause any antinociception in the morphine-naïve or rats under morphine withdrawal. Morphine 2.5 mg·kg–1 s.c. given to rats under morphine withdrawal caused only minor antinociception at 30 min compared with vehicle (14 ± 6.6% MPE), whereas a statistically significantly greater response was measured in drug-naïve animals (71 ± 13% MPE, data shown in Figure 5A), indicating that the morphine tolerance had persisted. During morphine withdrawal, when ketamine was coadministered with morphine, significantly greater antinociception was achieved compared when morphine was administered alone (28 ± 9.1% MPE, P < 0.01) at 30 min but no more at 90 min. In the hot plate test (Figure 4B), statistically significant but minor antinociception was observed at 30 min in three groups (morphine-tolerant rats that were acutely administered morphine, ketamine, or both) compared with vehicle but no more at 90 min. In the rotarod test (Figure 4C), motor impairment was noticed in the morphine withdrawal group that was acutely treated with both morphine and ketamine (38 ± 11 s, P < 0.05 compared with ketamine only).
Figure 4.
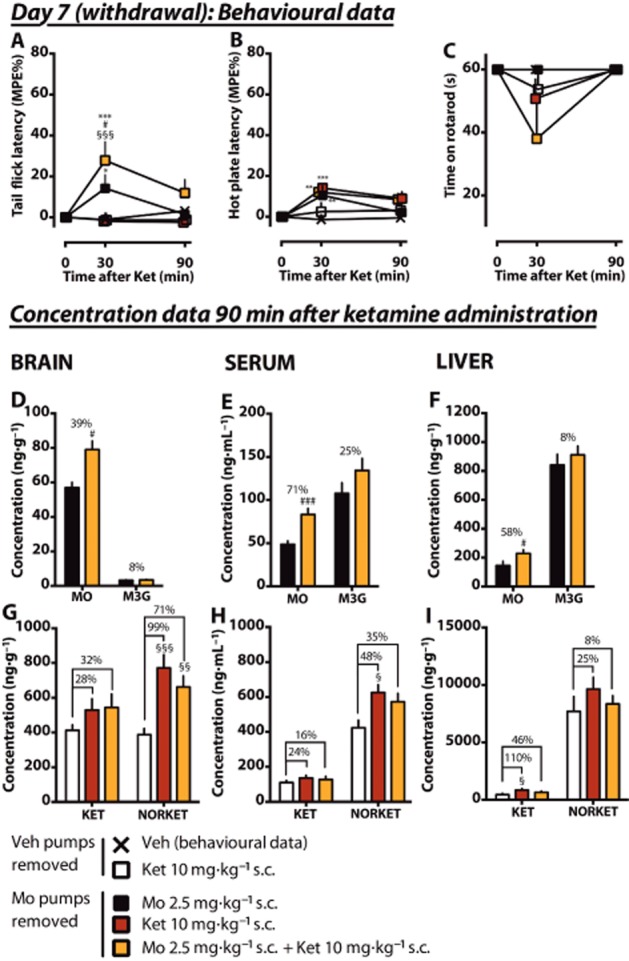
Effects of an acute low dose of ketamine, morphine and their combination on antinociception in morphine-tolerant rats under morphine withdrawal. Rats had morphine pumps that delivered morphine (Mo) or vehicle (Veh) 6 mg·day–1 for 5 days in total. On the evening of day 5, the morphine pumps were removed. On day 7, rats in morphine withdrawal received s.c. doses of morphine, ketamine or a combination. Control animals received vehicle or ketamine. Morphine was administered 15 min before ketamine. Antinociception was measured using tail flick (A) and hot plate (B) tests. The mean of the maximum possible effect (MPE%) ±SEM is plotted. In the rotarod test (C), the mean (±SEM) survival time (seconds) is plotted. Whole brain, serum and liver samples were collected after 90 min of ketamine administration. The tissue concentrations and serum levels of the experimental drugs and their main metabolites (morphine, MO; M3G; ketamine, KET; norketamine, NORKET) in the tissues were quantified and the means of the groups (±SEM) are presented in graphs D–I. The percentage difference between the treatment groups is shown for each compound. n = 8. *P < 0.05, **P < 0.01, ***P < 0.001; significantly different from the vehicle control group. #P < 0.05, ###P < 0.001; significantly different from the morphine pump treated group that received acute morphine only. §P < 0.05, §§P < 0.01, §§§P < 0.001; significantly different from the vehicle-pretreated group that received acute ketamine.
Figure 5.
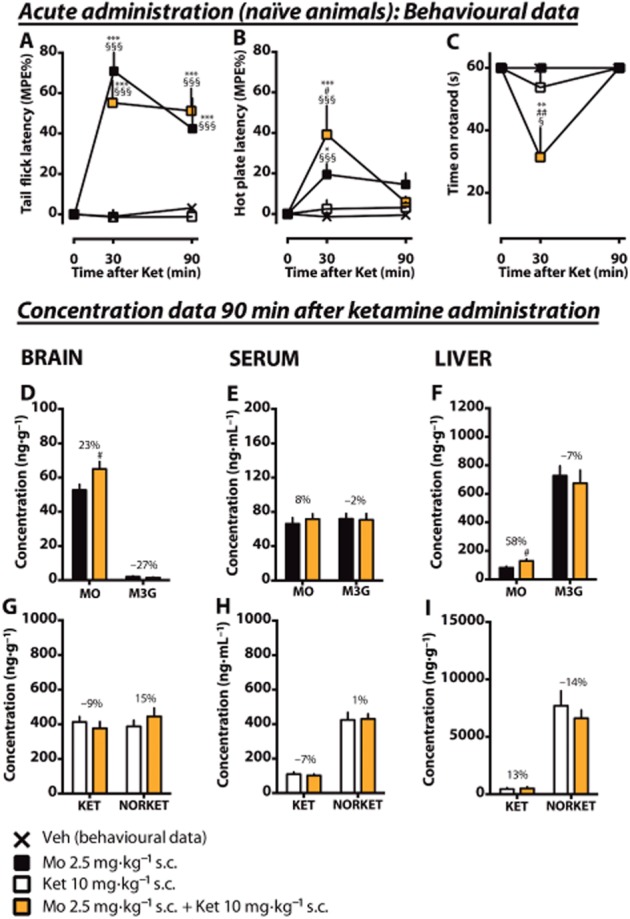
Effects of an acute low dose of ketamine on antinociception in acute coadministration with morphine. Rats received an acute s.c. dose of ketamine (10 mg·kg–1, Ket) or vehicle (Veh). Morphine was given 15 min before ketamine. Antinociception was measured using tail flick (A) and hot plate (B) tests. The mean of the maximum possible effect (MPE%) ±SEM is plotted. In the rotarod test (C), the mean (±SEM) survival time (seconds) is plotted. Whole brain, serum and liver samples were collected after 90 min of ketamine administration. The tissue concentrations and serum levels of the experimental drugs and their main metabolites (morphine, MO; M3G; ketamine, KET; norketamine, NORKET) in the tissues were quantified and the means of the groups (±SEM) are presented in graphs D–I. The percentage difference between the treatment groups is shown for each compound. n = 8, except n = 7 in the morphine-treated group. *P < 0.05, **P < 0.01, ***P < 0.001; significantly different from the vehicle control group. #P < 0.05, ##P < 0.01, ###P < 0.001; significantly different from morphine only. §P < 0.05, §§§P < 0.001; significantly different from acute ketamine only.
The increased accumulation of ketamine continues in rats under morphine withdrawal
Two days after the removal of morphine minipumps, the measured morphine serum levels and brain and liver concentrations were 2.2 ± 0.65 ng·mL–1, 0.31 ± 0.31 ng·g–1 and 34 ± 8.3 ng·g–1 respectively (n = 8). The corresponding M3G concentrations were 7.8 ± 3.0 ng·mL–1, 0.38 ± 0.27 ng·g–1 and 13 ± 4.8 ng·g–1, indicating almost complete washout of the substances. As noticed during chronic morphine administration, the coadministration of ketamine with morphine increased the concentration of morphine in the brain (39% increase, P < 0.05, Figure 4D) also during withdrawal. A significant increase of morphine was observed also in the serum (71% increase, P < 0.001, Figure 4E) and liver (58% increase, P < 0.05, Figure 4F). The tissue : serum concentration ratios of morphine did not significantly differ between groups (Table 1).
The effect of morphine on ketamine concentrations during withdrawal was assessed in three groups. The first (control) morphine-naïve group received ketamine only. The two groups under morphine withdrawal received ketamine with or without acute morphine. Previous morphine pump treatment increased the brain norketamine concentration (99% increase, P < 0.001, Figure 4G) but not the ketamine concentration. Norketamine levels were significantly greater in the serum as well (Figure 4H). The coadministration of acute morphine did not significantly alter the increase. In the liver, an increase in the ketamine concentration (110%, P < 0.05) was observed in the withdrawal group that was administered ketamine only (Figure 4I). When acute morphine was combined to ketamine, no significant difference was found. During withdrawal, no morphine-induced alterations in the tissue : serum concentration ratios of ketamine and norketamine were detected (Table 1).
Acute coadministration of morphine and ketamine increases the brain and liver morphine concentrations in naïve animals
Finally, we assessed the effects of acute morphine and ketamine coadministration in morphine-naïve rats previously treated with vehicle pumps. The experiment was performed concomitantly with the withdrawal experiment to allow statistical comparisons. Morphine (2.5 mg·kg–1, s.c.) was administered 15 min before ketamine (10 mg·kg–1, s.c.).
In the tail flick test (Figure 5A), ketamine or vehicle administrations alone did not cause any antinociception. Compared with vehicle, morphine alone caused significant antinociception at 30 and 90 min. When ketamine was coadministered with morphine, no changes in antinociception were observed when compared with the morphine only group.
In the hot plate test (Figure 5B), no antinociception by ketamine was observed. Morphine alone caused minor antinociception at 30 min (19 ± 5.3% MPE, P < 0.05 compared with vehicle). Ketamine coadministration had an additive effect at 30 min (39 ± 5.7% MPE, P < 0.05 compared with morphine only). In the rotarod test (Figure 5C), morphine or ketamine alone caused no motor impairment, but the combination of morphine and ketamine significantly impaired it at 30 min (31 ± 9.4 s, P < 0.05 compared with ketamine only).
In the brain (Figure 5D), a 23% increase (P < 0.05) of the morphine concentration was observed in the morphine- and ketamine-cotreated rats when compared with the morphine-treated group. In the serum (Figure 5E), no significant changes were observed, but in the liver morphine concentration was significantly elevated after coadministration. An increased liver : serum concentration ratio of morphine (P < 0.05, Table 1) was observed, whereas the increase in brain : serum concentration ratio was not significant. Morphine coadministration did not cause any significant changes in the ketamine or norketamine concentrations (Figures 5G–I) or tissue : serum concentration ratios (Table 1).
Metabolic ratios of the studied drugs in chronic administration of morphine, morphine withdrawal and acute coadministration
The metabolic ratios (M3G : morphine and norketamine : ketamine) on different study days in serum and liver are presented in Table 2. Acute ketamine treatment significantly decreased the M3G : morphine metabolic ratio in the liver, but not in serum, on all experimental days. Acute coadministration of morphine did not have an effect on the metabolic ratio of ketamine. However, chronic morphine treatment induced an increase in the serum norketamine : ketamine metabolic ratio on day 5 and a significant decrease in the liver on days 5 and 7.
Table 2.
Metabolic ratios for the studied drugs and their main metabolites
| Day 5 (tolerance) | Day 7 (withdrawal) | Acute administration | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Studied ratio | Chronic morphine treatment (days 0–4) | Acute morphine | Acute ketamine | Serum | P | Liver | P | Serum | P | Liver | P | Serum | P | Liver | P |
| M3G:Mo | + | – | – | 1.4 ± 0.30 | n.s. | 6.8 ± 1.2 | ** | – | – | – | – | ||||
| + | – | + | 1.1 ± 0.15 | 1.8 ± 0.24 | |||||||||||
| + | + | – | – | – | 2.3 ± 0.32 | n.s. | 7.2 ± 1.2 | * | |||||||
| + | + | + | 1.6 ± 0.14 | 4.3 ± 0.50 | |||||||||||
| – | + | – | – | – | 1.2 ± 0.14 | n.s. | 9.4 ± 1.1 | * | |||||||
| – | + | + | 1.0 ± 0.14 | 5.5 ± 0.80 | |||||||||||
| Norket:Ket | – | – | + | 3.1 ± 0.23 | *** | 18 ± 2.3 | *** | 4.2 ± 0.6 | n.s. | 22 ± 2.8 | # | 4.2 ± 0.6 | n.s. | 22 ± 2.8 | n.s. |
| + | – | + | 5.3 ± 0.23 | 5.4 ± 0.67 | 4.9 ± 0.52 | 12 ± 1.6 | – | – | |||||||
| + | + | + | – | – | 4.8 ± 0.49 | 14 ± 1.7 | |||||||||
| – | + | + | – | – | 4.4 ± 0.39 | 18 ± 3.1 | |||||||||
Metabolite : parent drug ratios (mean ± SEM; n = 8) in serum and liver shown for different experiment days. *, **, ***P < 0.05, P < 0.01, and P < 0.001, respectively, between the two groups. #P < 0.05 as compared with the two other groups. Ket, ketamine; Mo, morphine; Norket, norketamine; n.s., non-significant.
Discussion
The main finding of the study was that a single small dose of ketamine caused long-lasting antinociception in rats under chronic morphine treatment, whereas administration to naïve animals had no antinociceptive effect. The time point of maximum antinociception cotreatment caused a large increase in the brain concentrations of morphine, ketamine and norketamine, suggesting that morphine and ketamine may have pharmacokinetic interactions in addition to the previously reported pharmacodynamic interactions during chronic morphine treatment. Chronic morphine administration apparently decreased elimination of ketamine, as it radically increased the concentrations of ketamine and norketamine in the brain, serum and liver during the elimination phase. During morphine withdrawal, the synergistic antinociceptive effect of ketamine and morphine in coadministration was still present but to a smaller extent as was the accumulation of morphine and norketamine to the brain. Acute morphine and ketamine cotreatment in naïve rats did not have such effects, suggesting that chronic morphine treatment may modify the expression of proteins affecting ketamine pharmacokinetics.
Possible connection between increased antinociception and increased drug brain concentrations during morphine tolerance
A summary of the changes in the brain drug concentrations is shown in Table 3. The most prominent cotreatment-induced increases in morphine, ketamine and norketamine brain concentrations were observed during chronic morphine administration, during which the increase of antinociception was also remarkable in the morphine-tolerant rats. The brain concentration of morphine was increased by 110%, and those of ketamine and norketamine by 40 and 240% respectively.
Table 3.
A summary of the changes of the mean brain drug concentrations (%) and tail flick latencies (MPE%) during coadministration of morphine and ketamine compared with the groups that received only morphine or ketamine
| Variable | Tolerance (day 5) | P | Withdrawal with acute morphine (day 7) | P | Withdrawal without acute morphine (day 7) | P | Acute coadm | P |
|---|---|---|---|---|---|---|---|---|
| Morphine concentration | +110 | * | +39 | * | n/a | n/a | +23 | * |
| Ketamine concentration | +45 | ** | +32 | n.s. | +28 | n.s. | –9 | n.s. |
| Norketamine concentration | +240 | *** | +71 | ** | +99 | *** | +15 | n.s. |
| Tail-flick latency | +54 | *** | +13 | * | 0 | n.s. | +9 | n.s. |
The maximal change of the tail flick response in percentage units is shown for each experiment (morphine and ketamine-cotreated group vs. the groups that received only morphine or ketamine). For the exact treatment schemes of every experiment, please see the Methods section. *, **, ***P < 0.05, P < 0.01, P < 0.005 respectively. Coadm, coadministration; n.s., nonsignificant; n/a, not available (no morphine was administered to these rats).
It is important to note that brain morphine concentrations 90 min after acute ketamine treatment on day 7 were higher (Figure 4D, measurement was made 105 min after acute s.c. morphine treatment during the elimination phase) compared with day 5 (Figure 3D, the morphine concentrations were at steady state as a result of the 5 day pump treatment before acute ketamine that increased morphine concentrations by 110%). In spite of this, acute ketamine combined with morphine did not cause antinociception (1.4% MPE, Figure 4A) on day 7 but caused a time-dependent, maximal 57% MPE (Figure 3A) increase in the tail flick latency on day 5. Therefore, surprisingly, acute ketamine treatment did not restore morphine antinociception at 90 min in morphine-tolerant rats on day 7 as it did on day 5. This result suggests that a pharmacodynamic interaction alone does not explain the higher potency of ketamine during morphine tolerance. In addition, the considerably increased antinociception in morphine-tolerant, ketamine-treated rats may not be solely due to the increased brain concentrations of morphine or ketamine because their concentrations were at the same level on days 5 and 7. However, a notable (doubled) increase in the brain norketamine concentration was observed. Thus, the increased brain norketamine concentration may be an important factor contributing to the increased antinociception on day 5.
At the peak antinociception on day 5, the brain molar concentration of norketamine exceeded that of ketamine by 110%, supporting the idea that the increase is significant. In binding studies, norketamine has been identified as a potent NMDA receptor antagonist: Ebert et al. (1997) reported that S-norketamine is approximately five times weaker than S-ketamine. In rats, norketamine has been shown to have antinociceptive properties (Shimoyama et al., 1999; Holtman et al., 2008a) and it potentiated morphine antinociception in thermal nociception, peripheral neuropathy and tonic inflammatory pain models (Holtman et al., 2008b). In humans, the peak analgesic effect after oral ketamine administration occurred at the peak plasma norketamine, not ketamine, concentration (Grant et al., 1981). Taken together, our results showed that a single low dose of ketamine, given during opioid tolerance, induced important brain drug concentration changes that may have a role in augmenting the pharmacodynamic effects of ketamine in morphine tolerance.
Effects of ketamine administration on morphine pharmacokinetics
After ketamine administration to rats under chronic morphine treatment, the brain concentration of morphine was doubled at maximum antinociception (Figure 3D) and the brain : serum concentration ratio was increased (Table 1) implicating increased disposition of morphine in the CNS. A similar, although smaller, effect was seen under morphine withdrawal (39% increase, Figure 4D) and in acute coadministration (23% increase, Figure 5D). In these situations, however, the brain : serum concentration ratios were not significantly increased. Morphine is a substrate for the transporters, P-glycoprotein (P-gp) and the multidrug resistance protein (MRP), both of which are efflux proteins at the BBB (Schinkel et al., 1995; Letrent et al., 1999; Xie et al., 1999; Su and Pasternak, 2013). The inhibition of such efflux proteins by ketamine could account for decreased efflux and concomitantly increased disposition of morphine in the brain. However, there no published reports of ketamine as an inhibitor or substrate of P-gp (Doan, 2002; Varma et al., 2005) or MRP. Morphine may also be a substrate of the rat organic cation transporter proteins (Amphoux et al., 2006), influx proteins at the BBB. Inhibition of these proteins by ketamine (Tzvetkov et al., 2013) would lead to decreased CNS morphine concentrations, contrary to the changes observed. Thus, the mechanism behind the increased brain : serum concentration ratio of morphine on day 5 remains unclear.
Ketamine increased morphine concentrations in the liver in all experiments (Figures 3C, 4C and 5C) and decreased the M3G : morphine metabolic ratio (Table 2), indicating decreased hepatic metabolism of morphine to its glucuronide M3G. Previous in vitro studies (Qi et al., 2010; Uchaipichat et al., 2011) have shown that ketamine may inhibit the metabolism of morphine by UGT enzymes and our present in vivo results support this finding. However, decreased hepatic metabolism of morphine leads to increased serum morphine concentrations only on day 7 whereas serum M3G levels were not decreased in any experiment. Thus, the inhibition of efflux proteins rather than decreased metabolism might explain the increased brain concentration of morphine after ketamine treatment.
Effects of chronic morphine treatment on ketamine and norketamine distribution
Interestingly, acute coadministration of ketamine during chronic morphine treatment caused a large accumulation of ketamine and norketamine in liver, serum and brain (Figure 3G–I). Moreover, this effect persisted, even though to a lesser extent, during the withdrawal period and it was independent of an acute dose of morphine (Figure 4G–I). Similar accumulation could not be seen after acute coadministration in morphine-naïve animals (Figure 5G–I), strongly suggesting chronic morphine treatment-induced changes in the function of metabolic enzymes or transporter proteins affecting ketamine pharmacokinetics. At high (overdose) concentrations, ketamine is metabolized to norketamine mainly by cytochrome P (CYP)3A4, but at lower therapeutic concentrations also CYP2B6 has a profound role in the metabolism (Hijazi, 2002; Li et al., 2013). Importantly, however, also the norketamine concentrations were increased during coadministration. This result indicates that CYP enzyme inhibition is not the key factor explaining ketamine accumulation.
One possible explanation of the morphine tolerance-induced ketamine accumulation would be the down-regulation of a ketamine (or its metabolite) efflux transporter or the up-regulation of a reuptake transporter. The effects of chronic morphine treatment on the induction of xenobiotic receptors, transporters and drug-metabolizing enzymes is not thoroughly known, but chronic oxycodone has been reported to significantly down- or up-regulate 27 different drug transporters in the rat liver (Hassan et al., 2013). However, in the light of current knowledge about transporter proteins for ketamine, the demonstrated change in ketamine pharmacokinetics during chronic morphine treatment remains unexplained. Of clinical interest, the accumulation of ketamine during chronic morphine administration could potentially increase the risk for hepatic toxicity (Bell, 2012).
Effects on the BBB
Acute lipopolysaccharide treatment has been reported to lower plasma ketamine AUC and increase its elimination t1/2, indicating increased BBB permeability and access of ketamine into the brain (Vachon et al., 2012). In repeated administration, morphine may also increase the permeability of the BBB (Sharma and Ali, 2006). This provides another interesting theory for the altered pharmacokinetics of ketamine in morphine tolerance.
Conclusion
The results of the present study indicate a novel pharmacokinetic mechanism behind the interaction between chronic morphine administration and ketamine. The coadministration resulted in greatly increased concentrations of both parent drugs and also norketamine in the CNS. The results also suggest a probable decrease in the elimination of ketamine, as the concentrations of ketamine and its main metabolite norketamine were increased in all observed tissues at the elimination phase. This phenomenon was observed also during the morphine withdrawal period. Based on these results, it is difficult to define the exact contribution of the pharmacokinetic interaction to the increased antinociception. However, these results may help in explaining the profound ketamine analgesia often observed in morphine-tolerant patients. More research is needed to clarify the mechanisms of morphine-induced changes in ketamine pharmacokinetics. These findings also imply that possible changes in ketamine and morphine brain concentrations should be taken into account in future studies that involve coadministration of these drugs.
Acknowledgments
The technical help of Mr. Jouko Laitila is acknowledged. The study was funded by The Finnish Medical Society (Finska Läkaresällskapet), Helsinki, Finland; Orion-Farmos research foundation, Espoo, Finland; Biomedicum Helsinki Foundation, Helsinki, Finland; Emil Aaltonen Foundation, Tampere, Finland; Sohlberg Foundation, Helsinki, Finland; The Maud Kuistila Memorial Foundation, Helsinki, Finland; The Finnish Dental Society Apollonia, Helsinki, Finland; and European Union Seventh Framework Programme (FP7/2007 – 2013) under grant agreement no 602919.
Glossary
- BBB
blood–brain barrier
- CYP
cytochrome P
- M3G
morphine-3-glucuronide
- M6G
morphine-6-glucuronide
- MPE%
percentage of the maximum possible effect
- MRP
multidrug resistance protein
- P-gp
P-glycoprotein
- UGT
UDP-glucuronosyl transferase
Author contributions
T. O. L. designed and conducted the study, analysed the data and prepared the manuscript. V. J. designed and conducted the study and critically commented on the manuscript. M. S. N. conducted the study and prepared and critically commented on the manuscript. M. N. designed the study and critically commented on the manuscript. E. A. K. designed the study and critically commented on the manuscript. P. V. R. designed and conducted the study, analysed the data, and prepared the manuscript.
Conflict of interest
The authors of this manuscript have no conflicts of interest to declare.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, CGTP Collaborators et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-gated ion channels. Br J Pharmacol. 2013a;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013c;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amphoux A, Vialou V, Drescher E, Brüss M, La Cour CM, Rochat C, et al. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology. 2006;50:941–952. doi: 10.1016/j.neuropharm.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Arroyo-Novoa CM, Figueroa-Ramos MI, Miaskowski C, Padilla G, Paul SM, Rodríguez-Ortiz P, et al. Efficacy of small doses of ketamine with morphine to decrease procedural pain responses during open wound care. Clin J Pain. 2011;27:561–566. doi: 10.1097/AJP.0b013e318211936a. [DOI] [PubMed] [Google Scholar]
- Bell RF. Low-dose subcutaneous ketamine infusion and morphine tolerance. Pain. 1999;83:101–103. doi: 10.1016/s0304-3959(99)00096-2. [DOI] [PubMed] [Google Scholar]
- Bell RF. Ketamine for chronic noncancer pain: concerns regarding toxicity. Curr Opin Support Palliat Care. 2012;6:183–187. doi: 10.1097/SPC.0b013e328352812c. [DOI] [PubMed] [Google Scholar]
- Bell RF, Dahl JB, Moore RA, Kalso EA. Perioperative ketamine for acute postoperative pain. Cochrane Database Syst Rev. 2006;(1) doi: 10.1002/14651858.CD004603.pub2. CD004603. [DOI] [PubMed] [Google Scholar]
- Bell RF, Eccleston C, Kalso EA. Ketamine as an adjuvant to opioids for cancer pain. Cochrane Database Syst Rev. 2012;(11) doi: 10.1002/14651858.CD003351.pub2. CD003351. [DOI] [PubMed] [Google Scholar]
- Caraceni A, Hanks G, Kaasa S, Bennett MI, Brunelli C, Cherny N, et al. Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. Lancet Oncol. 2012;13:e58–e68. doi: 10.1016/S1470-2045(12)70040-2. [DOI] [PubMed] [Google Scholar]
- Coffman BL, Rios GR, Tephly TR. Purification and properties of two rat liver phenobarbital-inducible UDP-glucuronosyltransferases that catalyze the glucuronidation of opioids. Drug Metab Dispos. 1996;24:329–333. [PubMed] [Google Scholar]
- Coffman BL, Rios GR, King CD, Tephly TR. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos. 1997;25:1–4. [PubMed] [Google Scholar]
- Doan KMM. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237–251. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur J Pharmacol. 1997;333:99–104. doi: 10.1016/s0014-2999(97)01116-3. [DOI] [PubMed] [Google Scholar]
- Edwards SR, Minto CF, Mather LE. Concurrent ketamine and alfentanil administration: pharmacokinetic considerations. Br J Anaesth. 2002;88:94–100. doi: 10.1093/bja/88.1.94. [DOI] [PubMed] [Google Scholar]
- González P, Cabello P, Germany A, Norris B, Contreras E. Decrease of tolerance to physical dependence on morphine by, glutamate receptor antagonists. Eur J Pharmacol. 1997;332:257–262. doi: 10.1016/s0014-2999(97)01099-6. [DOI] [PubMed] [Google Scholar]
- Grant IS, Nimmo WS, Clements JA. Pharmacokinetics and analgesic effects of i.m. and oral ketamine. Br J Anaesth. 1981;53:805–810. doi: 10.1093/bja/53.8.805. [DOI] [PubMed] [Google Scholar]
- Hardy J, Quinn S, Fazekas B, Plummer J, Eckermann S, Agar M, et al. Randomized, double-blind, placebo-controlled study to assess the efficacy and toxicity of subcutaneous ketamine in the management of cancer pain. J Clin Oncol. 2012;30:3611–3617. doi: 10.1200/JCO.2012.42.1081. [DOI] [PubMed] [Google Scholar]
- Hassan HE, Myers AL, Lee IJ, Mason CW, Wang D, Sinz MW, et al. Induction of xenobiotic receptors, transporters, and drug metabolizing enzymes by oxycodone. Drug Metab Dispos. 2013;41:1060–1069. doi: 10.1124/dmd.112.050401. [DOI] [PubMed] [Google Scholar]
- Hijazi Y. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–858. doi: 10.1124/dmd.30.7.853. [DOI] [PubMed] [Google Scholar]
- Holtman JR, Crooks PA, Johnson-Hardy JK, Hojomat M, Kleven M, Wala EP. Effects of norketamine enantiomers in rodent models of persistent pain. Pharmacol Biochem Behav. 2008a;90:676–685. doi: 10.1016/j.pbb.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Holtman JR, Crooks P, Johnson-Hardy JK, Wala E. Interaction between morphine and norketamine enantiomers in rodent models of nociception. Pharmacol Biochem Behav. 2008b;90:769–777. doi: 10.1016/j.pbb.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Honarmand A, Safavi M, Karaky H. Preincisional administration of intravenous or subcutaneous infiltration of low-dose ketamine suppresses postoperative pain after appendectomy. J Pain Res. 2012;5:1–6. doi: 10.2147/JPR.S26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. 2012;169:1150–1156. doi: 10.1176/appi.ajp.2012.12040531. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CD, Rios GR, Green MD, MacKenzie PI, Tephly TR. Comparison of stably expressed rat UGT1.1 and UGT2B1 in the glucuronidation of opioid compounds. Drug Metab Dispos. 1997;25:251–255. [PubMed] [Google Scholar]
- Laskowski K, Stirling A, McKay WP, Lim HJ. A systematic review of intravenous ketamine for postoperative analgesia. Can J Anesth. 2011;58:911–923. doi: 10.1007/s12630-011-9560-0. [DOI] [PubMed] [Google Scholar]
- Letrent SP, Pollack GM, Brouwer KR, Brouwer KL. Effects of a potent and specific P-glycoprotein inhibitor on the blood-brain barrier distribution and antinociceptive effect of morphine in the rat. Drug Metab Dispos. 1999;27:827–834. [PubMed] [Google Scholar]
- Li Y, Coller JK, Hutchinson MR, Klein K, Zanger UM, Stanley NJ, et al. The CYP2B6*6 allele significantly alters the N-demethylation of ketamine enantiomers in vitro. Drug Metab Dispos. 2013;41:1264–1272. doi: 10.1124/dmd.113.051631. [DOI] [PubMed] [Google Scholar]
- Mathews TJ, Churchhouse AMD, Housden T, Dunning J. Does adding ketamine to morphine patient-controlled analgesia safely improve post-thoracotomy pain? Interact Cardiovasc Thorac Surg. 2012;14:194–199. doi: 10.1093/icvts/ivr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto H, Saito Y, Kirihara Y, Hara K, Sakura S, Kosaka Y. Spinal coadministration of ketamine reduces the development of tolerance to visceral as well as somatic antinociception during spinal morphine infusion. Anesth Analg. 2000;90:136–141. doi: 10.1097/00000539-200001000-00030. [DOI] [PubMed] [Google Scholar]
- Morgan CJA, Curran HV Independent Scientific Committee on Drugs. Ketamine use: a review. Addiction. 2012;107:27–38. doi: 10.1111/j.1360-0443.2011.03576.x. [DOI] [PubMed] [Google Scholar]
- Nesher N, Ekstein MP, Paz Y, Marouani N, Chazan S, Weinbroum AA. Morphine with adjuvant ketamine vs higher dose of morphine alone for immediate postthoracotomy analgesia. Chest. 2009;136:245–252. doi: 10.1378/chest.08-0246. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard M, Fournel-Gigleux S, Siest G, Mackenzie P, Magdalou J. A recombinant phenobarbital-inducible rat liver UDP-glucuronosyltransferase (UDP-glucuronosyltransferase 2B1) stably expressed in V79 cells catalyzes the glucuronidation of morphine, phenols, and carboxylic acids. Mol Pharmacol. 1994;45:42–50. [PubMed] [Google Scholar]
- Qi X, Evans AM, Wang J, Miners JO, Upton RN, Milne RW. Inhibition of morphine metabolism by ketamine. Drug Metab Dispos. 2010;38:728–731. doi: 10.1124/dmd.109.030957. [DOI] [PubMed] [Google Scholar]
- Salas S, Frasca M, Planchet-Barraud B, Burucoa B, Pascal M, Lapiana J-M, et al. Ketamine analgesic effect by continuous intravenous infusion in refractory cancer pain: considerations about the clinical research in palliative care. J Palliat Med. 2012;15:287–293. doi: 10.1089/jpm.2011.0353. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96:1698–1705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma HS, Ali SF. Alterations in blood-brain barrier function by morphine and methamphetamine. Ann N Y Acad Sci. 2006;1074:198–224. doi: 10.1196/annals.1369.020. [DOI] [PubMed] [Google Scholar]
- Shimoyama M, Shimoyama N, Gorman AL, Elliott KJ, Inturrisi CE. Oral ketamine is antinociceptive in the rat formalin test: role of the metabolite, norketamine. Pain. 1999;81:85–93. doi: 10.1016/s0304-3959(98)00269-3. [DOI] [PubMed] [Google Scholar]
- Shimoyama N, Shimoyama M, Inturrisi CE, Elliott KJ. Ketamine attenuates and reverses morphine tolerance in rodents. Anesthesiology. 1996;85:1357–1366. doi: 10.1097/00000542-199612000-00017. [DOI] [PubMed] [Google Scholar]
- Sinner B, Graf BM. Ketamine. In: Schuttler J, Schwilden H, editors. Modern Anesthetics. Berlin Heidelberg: Springer; 2008. pp. 313–333. . In: (eds). [Google Scholar]
- Su W, Pasternak GW. The role of multidrug resistance-associated protein in the blood-brain barrier and opioid analgesia. Synapse. 2013;67:609–619. doi: 10.1002/syn.21667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suppa E, Valente A, Catarci S, Zanfini B, Draisci G. A study of low-dose S-Ketamine infusion as ‘preventive’ pain treatment for cesarean section with spinal anesthesia: benefits and side effects. Minerva Anestesiol. 2012;7:774–781. [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of opiate tolerance by non-competitive N-d-aspartate receptor antagonists. Brain Res. 1994;633:178–188. doi: 10.1016/0006-8993(94)91538-5. [DOI] [PubMed] [Google Scholar]
- Tzvetkov MV, dos Santos Pereira JN, Meineke I, Saadatmand AR, Stingl JC, Brockmöller J. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem Pharmacol. 2013;86:666–678. doi: 10.1016/j.bcp.2013.06.019. [DOI] [PubMed] [Google Scholar]
- Uchaipichat V, Raungrut P, Chau N, Janchawee B, Evans AM, Miners JO. Effects of ketamine on human UDP-glucuronosyltransferases in vitro predict potential drug-drug interactions arising from ketamine inhibition of codeine and morphine glucuronidation. Drug Metab Dispos. 2011;39:1324–1328. doi: 10.1124/dmd.111.039727. [DOI] [PubMed] [Google Scholar]
- Vachon P, Hélie P, Veilleux-Lemieux D, Beaudry F. Effects of endotoxemia on the pharmacodynamics and pharmacokinetics of ketamine and xylazine anesthesia in Sprague–Dawley rats. Vet Med Res Rep. 2012;3:99–109. doi: 10.2147/VMRR.S35666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma MVS, Sateesh K, Panchagnula R. Functional role of p-glycoprotein in limiting intestinal absorption of drugs: contribution of passive permeability to P-glycoprotein mediated efflux transport. Mol Pharmaceutics. 2005;2:12–21. doi: 10.1021/mp0499196. [DOI] [PubMed] [Google Scholar]
- Visser E, Schug SA. The role of ketamine in pain management. Biomed Pharmacother. 2006;60:341–348. doi: 10.1016/j.biopha.2006.06.021. [DOI] [PubMed] [Google Scholar]
- White PF. Clinical pharmacology of intravenous induction drugs. Int Anesthesiol Clin. 1988;26:98–104. doi: 10.1097/00004311-198802620-00003. [DOI] [PubMed] [Google Scholar]
- Xie R, Hammarlund-Udenaes M, de Boer AG, de Lange EC. The role of P-glycoprotein in blood-brain barrier transport of morphine: transcortical microdialysis studies in mdr1a (–/–) and mdr1a (+/+) mice. Br J Pharmacol. 1999;128:563–568. doi: 10.1038/sj.bjp.0702804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, McErlane KM, Ong MC. High-performance liquid chromatography-mass spectrometry-mass spectrometry analysis of morphine and morphine metabolites and its application to a pharmacokinetic study in male Sprague-Dawley rats. J Pharm Biomed Anal. 1998;16:971–980. doi: 10.1016/s0731-7085(97)00094-0. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]